
Abstract
The ultimate strategy to prevent species extinction is captive breeding followed by reintroduction of individuals into the wild. Unfortunately, overall success of reintroductions is poor and in most cases conservation breeding is applied for species where individual numbers, population numbers and genetic diversity is strongly reduced. In addition, reintroductions inevitably result in small populations with poor genetic status. Systematic demographic and genetic monitoring is needed to optimize conservation actions. Here we show how genetic monitoring was useful and informative in a reintroduction project for the highly endangered Common hamster (Cricetus cricetus) in the Netherlands and Belgium. Using well defined breeding lines of original relict populations combined with a systematic reintroduction scheme including consecutive supplementations made it possible to infer success of reintroduction, supplementation and following admixture of populations. An initial loss of genetic diversity could be detected in some of the reintroduced populations, but it could be shown that due to following supplementation of populations, genetic diversity and also effective population size in the wild stabilized or even increased. Multivariate (DAPC) and Bayesian inference (STRUCTURE) revealed admixture of supplemented individuals with wild-born individuals. Although population size estimates differed strongly between populations, a link between census size, breeding lines, effective population size and genetic diversity could not be proven. This study highlights that genetic monitoring is not only descriptive but also reveals detailed information on reintroduction success, admixture and population development. We recommend that genetic monitoring should be a basic element of reintroductions and should be used to optimize reintroduction attempts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The current global biodiversity crisis affects many groups of species, including a wide range of mammalian species (Di Marco et al. 2014). A global conservation strategy for mammalian species is therefore urgently needed (Rondinini et al. 2011), but the success of conservation strategies is uncertain as many factors influence the result of conservation projects (Crees et al. 2016). The ultimate strategy to prevent extinction or to increase population numbers of mammalian species is captive breeding followed by reintroduction of captive-bred individuals into the wild (IUCN 2013). However, captive breeding and reintroduction have severe disadvantages. Reintroductions can be difficult and expensive (Tenhumberg et al. 2004), and the release of captive-bred individuals is often accompanied by high initial losses of released individuals (Villemey et al. 2013; McCleery et al. 2014). Moreover, captive breeding is mostly started in species with already highly threatened populations with low numbers (Frankham et al. 2002; Allendorf et al. 2013), which may result in ex-situ populations with small genetic variation. Together with limitations in the number of individuals available for reintroduction (Van Houtan et al. 2009; Brekke et al. 2011; Tracy et al. 2011) it seems almost inevitable that reintroductions initially result in small populations, with a poor genetic status (Spielman et al. 2004; Tracy et al. 2011; Pacioni et al. 2013). Despite these disadvantages, reintroduction has become an important and frequently applied strategy for the conservation of a broad variety of mammals, ranging from small mammals (Ottewell et al. 2014; McCleery et al. 2014) till large carnivores (Hayward et al. 2007). Unfortunately, the overall success of reintroductions is low (Fischer and Lindenmayer 2000; Tenhumberg et al. 2004; Armstrong and Seddon 2008) and several authors (Robert 2009; Weeks et al. 2011) have stressed the need of giving attention to the genetic adaptive potential of reintroduced populations (Carlson et al. 2014; Jamieson 2015) as a low genetic adaptive potential may hamper a successful recovery of the species (Madsen et al. 1999; Westemeier et al. 1998; Carlson et al. 2014; Whiteley et al. 2015). By implementing a systematic genetic monitoring, which we define as ‘quantifying temporal changes in population genetic metrics’ (following Schwartz et al. 2007), when starting a reintroduction project, very important information of both the genetic status of the population and population demographic parameters can be gained, while the results of such a monitoring can be used to optimize conservation actions (Schwartz et al. 2007).
Using genetic monitoring as a diagnostic tool, information on population processes such as founder effects, success of supplementation, effective population size, population admixture and the genetic relatedness of individuals can be revealed (DeBarba et al. 2010; Koelewijn et al. 2010; Reiners et al. 2014; Seignobosc et al. 2011; Frosch et al. 2014; Jamieson 2015). Genetic monitoring can also provide estimates of genetic diversity in time, thereby indicating the loss or maintenance of genetic diversity in reintroduced or managed populations (Ozer et al. 2011; Ottewell et al. 2014; Jamieson 2015). Furthermore temporal genetic population sampling can provide estimates on effective population size, which can span several generations and give further insights into population dynamics (Luikart et al. 2010).
However, the number of published studies using genetic monitoring to follow genetic diversity of populations before, during and after reintroduction is still limited, although growing during the last years (DeBarba et al. 2010; Ozer et al. 2011; Diefenbach et al. 2015). In this paper we show how genetic monitoring was a useful and informative tool in a large conservation project, including reintroduction and supplementation attempts, of the Common hamster (Cricetus cricetus) in the Netherlands and Belgium. Genetic monitoring was used to study and analyze three important topics:
-
1)
Differences in genetic diversity indices of captive, reintroduced and supplemented populations to access the effect of management decisions i.e. test if release of different breeding lines at the same time versus ongoing supplementation resulted in different outcomes regarding genetic diversity estimates.
-
2)
Magnitude of genetic admixture when releasing animals of different breeding lines or supplementing established wild populations with individuals from different genetic origins.
-
3)
Temporal changes in genetic diversity indices and population size, i.e. testing whether there are significant shifts in genetic indices and if effective population size inferred using the temporal method coincides with changes in census population size.
Materials and methods
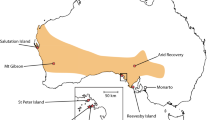
The Common hamster is a rodent inhabiting farmland on loess and loamy soils across Europe (Meinig et al. 2014), with adults weighing 200–500 g. Most hamsters only survive 1 or 2 years in the wild (Kuiters et al. 2010). The species is strongly declining all over Europe (Ziomek and Banaszek 2007; Tkadlec et al. 2012; Meinig et al. 2014; O’Brien 2015; Surov et al. 2016). In the Netherlands and Belgium the species has declined dramatically over the last 30 years (Kuiters et al. 2010). In 2000 only three small relict populations were left in the Netherlands and Belgium (Fig. 1, NL1, B1 and B2) with strongly reduced genetic diversity (La Haye et al. 2012a). In 1999 a breeding program was set up in The Netherlands and ten out of 14 wild-trapped individuals from the last population in the Netherlands (NL1) produced offspring (La Haye et al. 2012b). Some years later, two males from the Belgian population of Bertem (B1) and one male from the nearby German population of Neuss (G1) were successfully crossed into the breeding stock. Within the breeding program different breeding lines were maintained (La Haye et al. 2012b):

a Historic range of the Common hamster in Belgium, the Netherlands and nearby North Rhine-Westphalia (Germany). The original wild relict populations, Bertem (B1), Tongeren (B2), Heer (NL1), Neuss (G1) and Zülpich (G2), are indicated with squares. Reintroduced and/or supplemented populations, Sibbe (R1), Amby (R2), Heer (R3), Sittard (R4), Puth (R5), Bertem (B1) and Tongeren (B2) are represented by dots. b Captive breeding, reintroduction and supplementation history of all hamster populations. On the left the original relict populations. Individuals from these populations founded the breeding program and were the basis of the different breeding lines (middle). Solid lines represent reintroductions, dashed lines indicate supplementations, dotted lines represent wild-trapped individuals translocated to other populations. c Overview of estimated population sizes (black lines and dots) with sizes of reintroduction and supplementation cohorts indicated by bars. The color of the bars indicate the breeding lines used for reintroduction and supplementation
-
NL, with individuals from the last Dutch population (NL1),
-
B/NL, offspring of the two males from Belgium (B1) and NL line females,
-
G/NL, offspring of the male from Germany (G1) and NL line females.
The G/NL breeding line had an increased litter size (7.1 vs. 5.3, respectively 5.7 offspring, in the NL and B/NL breeding line), which can be interpreted as an increased fitness with a probably positive effect on population persistence or success of reintroduction (La Haye et al. 2012b). Surplus stock of the breeding program was used for reintroduction and supplementation within the Netherlands (R1-R5) and Belgium (B1-B2) to establish new populations, to increase levels of genetic diversity and boost the size of local populations.
Reintroduction and supplementation strategy
The first reintroduction in the Netherlands started in 2002 and was followed by multiple reintroduction (R) and supplementation (S) attempts (Fig. 1b, c). Attempts were always done at the start of the breeding season in May/June to optimize reproduction. The NL breeding line was used for reintroductions in all areas, while in R4 Sittard and R5 Puth also individuals from the G/NL breeding line were used for reintroduction. Supplementation in the Netherlands was mostly done with captive-bred individuals from the G/NL and B/NL breeding line (Fig. 1b, c). Supplementation in Belgium (populations B1 and B2) was always done with individuals with NL or G/NL genetic profiles, either from captivity (in 2007) or a mix of captive-bred and wild-trapped individuals (in 2008).
Population development
Population development and local dynamics were considered to be independent in most of the studied populations (B1-B2, and R1-R3). Migration of individuals was only possible between the nearby populations of R4 Sittard and R5 Puth, but very unlikely. Most populations are separated by physical barriers like rivers, cities, highways and unsuitable habitat (Fig. 1a), which result in geographic distances between populations that by far exceed the migration distance of 1.5–2 km for this species (Kuiters et al. 2010). The two Belgian populations are even 60 km apart. The population size and development of all reintroduced hamster populations in the Netherlands (R1-R5) was monitored with yearly burrow surveys in late summer/autumn (Fig. 1c). Each burrow, fresh and abandoned, accounts for one hamster (Kuiters et al. 2010). These surveys, although being estimates, give a good indication of census population size (Kuiters et al. 2010). No yearly population surveys were done in Belgium (B1-B2) and population sizes at these locations are rough estimates based on non-exhaustive inventories, but population sizes in Belgium were assumed to be low in all years (pers. obs. V. Verbist).
Sampling and genotyping
Samples for DNA analysis were collected within the conservation and breeding program on a regular basis since 1999 and in the wild whenever possible, including tissue from dead hamsters (no hamsters were killed for sampling). High quality hair samples were collected when hamsters were trapped (Kuiters et al. 2010). In Belgium, special hair-traps (Reiners et al. 2011) were used in 2010 for collecting hair-samples in the wild. Hair samples from the wild were genotyped at least two or three times (details in Reiners et al. 2014).
Genotyping was done at the laboratory of Alterra, the Netherlands and at the Conservation Genetics Group, Senckenberg Research Institute and Natural History Museum Frankfurt, Germany. The technical analysis is described in Reiners et al. (2014). Scoring of alleles was harmonized between institutes by analyzing and scoring the same samples at both laboratories. All samples were genotyped for a maximum of eight polymorphic microsatellite loci: Ccrμ10, Ccrμ11, Ccrμ12, Ccrμ15, Ccrμ17, Ccrμ19, Ccrμ20 (Neumann and Jansman 2004) and Cricri-IPK02 (Jakob and Mammen 2006). However, all loci had only two different alleles, which results in a maximum of 16 different alleles. Some of these loci were polymorphic in one founder population (B1, NL1 of G1), but monomorphic in other populations, resulting in several private alleles per founder population. Samples with more than two missing loci were excluded from further analysis.
In our study we used 818 genetic profiles, of which 268 profiles are from wild-sampled individuals and 550 from individuals of the captive breeding program that were released into the wild. The individuals that were used for reintroduction were exactly known due to the Studbook of the European hamster which is managed by Rotterdam Zoo (de Boer 2014). However, not all genetic profiles of released individuals were available and missing genetic profiles were inferred by reconstructing the total pedigree, with known genotypes from founders and their descendants as input. The DNA samples of the original wild populations in Belgium, B1 and B2 before supplementation, were genetically indistinguishable and sampled individuals, at least individuals sampled later than 1999 resp. 2001, had lost all of their genetic diversity; individuals of both populations were genetically monomorphic for all eight microsatellites, but carried unique alleles in comparison with nearby populations from the Netherlands and Germany (La Haye et al. 2012a).
Data analysis
Several studies have shown that populations of reintroduced species may lose genetic diversity (Ozer et al. 2011; Pacioni et al. 2013) or may shift towards a specific source population when using multiple breeding lines or sources (Kennington et al. 2012). Such an effect can be detected by monitoring the genetic diversity of the released cohort and comparing the genetic diversity with that of the established population.
First, genetic diversity indices were calculated using GENALEX 6.5002 (Peakall and Smouse 2006) and FSTAT (Goudet 1995). If genetic diversity is lost or shifted towards a specific source, this may result in changes of genetic diversity indices: the number of alleles (N A ), allelic richness (A R ), the effective number of alleles (A E ), changes in observed heterozygosity (H O ), expected heterozygosity (H E ) and inbreeding coefficient (F IS ).
Second, genetic monitoring was used to evaluate the success of admixture between released breeding lines. A successful supplementation leads to admixture between cohorts. For admixture analysis and interference of population ancestries (NL1, B1, G1) we used discriminant analysis of principal components (DAPC) method in the adegenet package (Jombart et al. 2010) as well as STRUCTURE 2.3.4 (Pritchard et al. 2000; Falush et al. 2003). DAPC was used assuming three clusters and retaining the first six PCA axes, estimated by optim.a.score function, which predicts the optimal number of principal components. We ran STRUCTURE using the admixture model with correlated allele frequencies with no prior population or location information with a K of three (= number of clusters/ancestral populations assumed) with a burn-in of 200,000 runs, following 500,000 MCMC runs. For comparative purpose we also run with a K (= assumed number of ancestral populations) from two to five (Appendix Fig. 4). We repeated STRUCTURE runs 20 times and used CLUMPP 1.1.2 (Jakobsson and Rosenberg 2007) to match runs.
Third, reintroduction and supplementation attempts aimed at maintenance of genetic diversity (N A , A R , A E , H O , H E ) and higher effective population sizes (N e ) through time. Therefore, effective population size and genetic diversity was followed in populations from the start of the reintroductions at t0 (differs in each population), after the first phase of reintroductions t1 and until the last year of sampling t2 in 2010 (Fig. 1c; Tables 2 and 3 in Apppendix). Note that t0 is the genetic diversity of captive bred individuals that were reintroduced in R1-R5, but in B1-B2 it is the genetic diversity of the wild relict population before supplementation. The genetic diversity of populations was measured a second time at t1 a few years after establishment or supplementation. A final measurement of genetic diversity in R1-R5, B1 and B2 was done in 2010 at t2. Using FSTAT we tested for an increased pair-wise population-differentiation (increased F ST between measurements). Beside this we also tested group-wise differences in genetic diversity statistics (A R , H O , H E , F IS and F ST ) between t0 and t1 for all Dutch populations (R1-R5), between t0 and t2 for only Dutch as well as a combination of Dutch and Belgian (B1 and B2) populations. The group-wise comparisons were carried out to test significance of general observations.
Finally, we used the temporal samples of populations t0, t1 and t2 to estimate effective population size N e using NEEstimator V2 (Do et al. 2014) and MLNE (Wang and Whitlock 2003) which both are recommended superior to other programs and methods (Gilbert and Whitlock 2015). NEEstimator V2 was only used for calculations from t0 to t1 to compare estimates with MLNE. For our study MLNE is more appropriate because three temporal samples (t0, t1 and t2) can be used for calculations. We assumed 1.5 generations per year. As we are aware that assumptions may be violated (e.g. no immigration) we included MLNE models with estimation of migration m in populations which were supplemented. Allele frequency data of released cohorts was included in analysis. Successful supplementation can be considered as ‘migration’ of supplemented individuals into the local gene pool. For comparative purpose to N e , we also included minimum and maximum as well as the harmonic mean of population size counts.
Results
Differences in genetic diversity indices of captive, reintroduced and supplemented populations
A comparison of genetic diversity in established populations in the Netherlands (R1-R5) with that of the cohort used for reintroduction showed a small decline in genetic diversity after a few years in three out of five populations (R1-R3), while no decline was detected in the other two populations (R4-R5). Populations R1-R3 showed lower values for allelic richness (A R ), the effective number of alleles (A E ) and lowered heterozygosity rates (H O and H E ). No loss of alleles was detected in any of the populations. Inbreeding coefficients (F IS ) of all populations increased. Whereas most of the cohorts used for release had a negative F IS , all established populations had a positive F IS (Table 3 in Appendix).
Populations R4 and R5 were established by releasing individuals of the NL and the G/NL breeding line during the same reintroduction event. The genetic diversity of these breeding lines differ, with the NL breeding line having lower values for all indices (N A , A R , A E , H O , H E ) in comparison with the G/NL breeding line. However, the combined genetic diversity of the released cohorts (NL plus G/NL) was very similar to the genetic diversity of the established populations in R4 and R5 (Table 3 in Appendix).
Supplementation of populations, adding individuals to a population to increase genetic diversity and/or population size, started in 2006 and was performed six times in five populations. Three populations in the Netherlands were supplemented: R1-R3, and two populations in Belgium: B1 and B2 (Fig. 1b).
Population R1 was supplemented with a small number of individuals of the NL breeding line, which had the same genetic diversity as the recipient population (Fig. 1b; Table 3 in Appendix). In all other supplementations individuals of non-resident breeding lines were used (Fig. 1b; Table 3 in Appendix). The supplementation of R1 resulted in a slightly increased A E , H O and H E .
The population of R2 was supplemented several times with individuals of the B/NL breeding line, but the first attempt had no effect on genetic diversity and A E and H O even declined (Fig. 3; Table 3 in Appendix). The following supplementations in R2 were more successful and resulted in increased A E , H O and H E (Fig. 3; Table 3 in Appendix). Supplementation in R3 with individuals having G/NL genetic origin resulted in increased N A , A E , H O and H E (Fig. 3; Table 3 in Appendix).
The relict populations in Belgium, B1 and B2, were supplemented with individuals having a NL and G/NL genetic background. In both areas the number of alleles (N A ) almost doubled and A E, H O and H E increased after supplementation (Fig. 3; Table 3 in Appendix).
Magnitude of genetic admixture
Both STRUCTURE and DAPC were used to assign individuals of founder populations to clusters according to their ancestral origin (NL-cluster, B-cluster, G-cluster, Fig. 2). STRUCTURE assigned the German and Belgian autochthonous individuals to the respective cluster with a high assignment rate of 0.99. The Dutch founders had also a high mean assignment of 0.94, where the pure NL-breeding line showed reduced mean assignment of 0.92 to the NL-cluster. Mixed lines as B/NL (0.86 to B-cluster and 0.11 to NL-cluster) and G/NL (0.84 to G-cluster and 0.13 to NL-cluster) showed reduced ancestry values compared to their founder populations, confirming admixture.

Comparison of genetic structure and diversity of reintroduced populations in R4 Sittard and R5 Puth. In DAPC scatterplots all genotypes are referenced with the relict populations of Belgium (B1, blue triangles), the Netherlands (NL1, orange squares) and Germany (G1, green diamonds). The STRUCTURE assignment, based on the membership proportion of each individual and population for each of the reference populations, is placed directly under the DAPC plots. Genetic diversity of wild populations in specific years is shown on the right as H O (vertical axis) and A E (size of each dot). (Color figure online)
Visualization using the DAPC loadings (position in plots) clearly separated autochthonous individuals. Individuals originating from the pure NL-line have similar loadings as the Dutch founders (Fig. 3—R1 Sibbe). Individuals of mixed lines B/NL and G/NL have intermediate loadings and are plotted between Dutch and German relict individuals (Fig. 2—R4, R5) or Dutch and Belgian relict individuals (Fig. 3—R2 Amby), clearly indicating admixture of individuals.

Comparison of genetic structure and diversity of reintroduced populations, before and after the release of cohorts used for supplementation (S1, S2, S3). In DAPC scatterplots all genotypes are referenced with the relict populations of Belgium (B1, blue triangles), the Netherlands (NL1, orange squares) and Germany (G1, green diamonds). The STRUCTURE assignment, based on the membership proportion of each population for each of the reference populations, is placed directly under the DAPC. Genetic diversity of wild populations in specific years is shown on the right as H O (vertical axis) and A E (size of each black dot), with dashed lines indicating the supplementation events. (Color figure online)
Using both approaches, DAPC and STRCUTURE, enabled to confirm long term survival of admixed individuals in R4 and R5, where released pure Dutch individuals could only be rarely detected in the final sampling. In R2 Amby where pure NL-individuals were initially released and later supplemented with B/NL-animals, only one admixed individual could be detected. In R3 Heer several admixed individuals were detected after supplementation with G/NL-individuals, but also individuals which show high assignment to the NL-cluster.
In Belgium where both pure NL- and mixed G/NL-Individuals were released in B1 and B2, individuals with intermediate German, Belgian and Dutch ancestry were found, confirming admixture of supplemented individuals with wild Belgian relict individuals (Fig. 3).
Temporal change of genetic diversity
Monitoring genetic diversity in the period 2002–2010 in the populations in the Netherlands (R1-R5) showed fluctuating levels of genetic diversity (Figs. 2, 3). Genetic diversity was stable (t0 vs. t1) and slightly increasing (t2) in populations that were established with individuals of the G/NL breeding line (R4-R5), while the other populations (R1, R2, R3) first lost genetic diversity (t0 vs. t1) and genetic diversity only increased after supplementation (t2) (Fig. 3). The genetic diversity of populations in Belgium (B1, B2) strongly increased after supplementation (t0 vs. t2) (Fig. 3). The F ST -values of all five Dutch populations (Table 1) showed slightly increased values in the first phase of the reintroductions between t0 and t1, but without significance. Supplementation in the secondary phase (t0 vs. t2) resulted in significantly increased F ST values in R3 Amby, B1 Bertem and B2 Tongeren (Table 1).
Group wise comparison between all Dutch populations (R1-R5) in the first phase of reintroductions revealed no significant difference in any genetic diversity estimate [one-sided p values after 1000 permutations for A R (p = 0.42), H O (p = 0.28), H E (p = 0.6), F IS (p = 0.98) or F ST (p = 0.63)]. Group comparisons between t0 and t2 of R1-R5 revealed higher diversity at t2 for heterozygosity values at marginal significance (H O with p = 0.057, H E with p = 0.062) and no difference for other indices (A R with p = 0.22, F IS with p = 0.47 and F ST with p = 0.49). Including the Belgian populations B1 and B2 in group wise comparisons between t0 and t2 resulted in significantly higher A R (p = 0.001) and heterozygosity (H O = 0.034, H E = 0.02), but still no significance for F IS and F ST .
Census versus effective population size
Reintroduced populations in the Netherlands showed stable population sizes between t0 and t1 with a moderate growth till 2007, but most populations severely declined in 2008 (Table 1; Fig. 1c). N e estimates derived from genetic data at t0 and t1 were in high concordance with census population size, where the highest numbers for both estimates were found in R1 and R4 and the lowest values in R2 (Table 1). Even though R2 was supplemented, MLNE indicated that only a very small number (m = 0.002) of individuals ‘immigrated’ in the population. In the years thereafter at t2 only the population of R4, somewhat recovered in census size, while the other populations further declined and harmonic mean population size was strongly reduced. In these populations peak estimates where never reached again and populations reached minimum census sizes in 2010.
N e estimates until t2 were in less concordance with census sizes. All N e estimates in Dutch populations were higher at t2 than at t1. Highest N e values were estimated for the supplemented populations R1 and R2 at t2, where in both cases ‘migration’ from supplemented individuals was confirmed. In R2, MLNE estimated a low N e with high recruitment of supplemented individuals, whereas in R1, N e was not strongly affected by ‘immigration’. Same situation was confirmed for R3 where N e was higher due to ‘migration’ from supplemented individuals. Only in R4, N e was lowered compared to t1 although population size of this population was highest. In R5 population size and effective population size were more or less stable over the whole period compared to other populations. The populations in Belgium (B1 and B2) were not regularly monitored, but population sizes were very small in all years. Anyhow N e estimation showed that due to a high ‘migration’ of supplemented individuals, effective population size was increased with B1 showing a higher recruitment than B2 (95% CI of N e estimation and comparison between NEEstimator V2 and MLNE, see Table 3 in Appendix).
Discussion
It is expected that in the coming decades reintroductions and other conservation activities will be necessary to prevent endangered species to become extinct (Armstrong and Seddon 2008; IUCN 2013), but the success of reintroductions is poor (Fischer and Lindenmayer 2000) and more systematic monitoring is needed to get a better understanding of the processes during and after reintroductions (Ewen and Armstrong 2007). Here, we applied systematic genetic monitoring to evaluate several aspects of reintroduction and supplementation attempts of the Common hamster in the Netherlands and Belgium in the period 2002 till 2010.
Effects of reintroduction on genetic diversity
Reintroductions of Common hamsters are often accompanied by high initial mortalities (Kuiters et al. 2010; Villemey et al. 2013), which may reduce the already impoverished genetic diversity of the hamsters used in reintroductions and supplementations in the Netherlands and Belgium (Robert 2009; Kuiters et al. 2010; La Haye et al. 2012a). The decline in genetic diversity in populations R1-R3 as observed in this study is an important warning, as the reintroductions in these areas were performed by releasing individuals from the NL breeding line only, having already a lower genetic diversity compared with other breeding lines. Even a small decline in genetic diversity may affect population persistence of R1-R3 in the long term as a lowered genetic diversity is associated with endangered and failing populations (Madsen et al. 1999; Westemeier et al. 1998; Carlson et al. 2014; Whiteley et al. 2015), although habitat quality and habitat management often plays a more dominant role in population persistence (Spielman et al. 2004; Bouzat et al. 2009; La Haye et al. 2014).
The populations of R4-R5 were established by releasing individuals of two breeding lines (NL and G/NL). This strategy was followed to maximize levels of genetic diversity in the new populations. Genetic monitoring of these populations showed that individuals of both breeding lines had clearly mixed, which is also proven by multivariate analysis (DAPC), and did not result in a dominance of one of the breeding lines as genetic diversity was stable (Fig. 2; Table 3 in Appendix). However, the STRUCTURE analysis showed a little different result: a few years after the initial release more individuals were assigned to the cluster which also had highest assignment to the German population compared with the released cohort. This result might be an effect of assuming a K of three when running STRUCTURE. We choose to use K = 3, because the breeding program was founded with individuals from three relict populations: B1, NL1 and G1. However, all breeding lines have NL1 founders (La Haye et al. 2012b), but individuals of mixed breeding lines (B/NL and G/NL) have unique private alleles from the original B1 and G1 population. Admixed Individuals with private alleles from the B1 or G1 population, are therefore more likely assigned to the original relict populations of B1 and G1 by STRUCTURE.
Success of supplementation on genetic diversity
A good strategy to optimize genetic diversity in reintroduced populations is releasing individuals from different source populations or breeding lines at the same time, because all reintroduced individuals have to adapt to the new area or wild environment (Villemey et al. 2013). In a later phase of a reintroduction some individuals or their offspring may have established a territory, which makes it much harder to integrate into the population (Koelewijn et al. 2010; Frosch et al. 2014). Releasing individuals of different breeding lines (NL and G/NL breeding line) at the same moment in R4 and R5 resulted in populations with a maximized genetic diversity. However, reintroductions in R1, R2 and R3 started before genetically more diverse individuals became available from the breeding program (La Haye et al. 2012b). It was therefore unavoidable that populations R1-R3 had to be supplemented in later years to increase genetic diversity.
Most of the supplementations in the Netherlands were successful and genetic diversity increased in R1, R2 and R3 (Fig. 3; Table 3 in Appendix). The only supplementation that failed was the first supplementation in R2 Amby in 2006. It is not clear why this supplementation failed, but a miscalculation of the habitat quality of the release-plot is likely.
In Belgium, the populations of B1 Bertem and B2 Tongeren were supplemented in 2007 and 2008 and supplementation had a large positive effect on genetic diversity (Fig. 3; Table 4 in Appendix). Genetic diversity was completely lacking before supplementation, but afterwards genetic diversity of B1 and B2 was comparable with that of hamster populations in the Netherlands (Table 3 in Appendix).
The success or failure of supplementations is also proven with DAPC and STRUCTURE analysis (Fig. 3) and this figure show at a glance the success of supplementation through a change in DAPC loadings (position in plots) of individuals and/or changes in the population assignment by STRUCTURE.
Overall the success of supplementation, as detected through genetic monitoring, was promising, even when a small number of individuals were released.
Genetic diversity and effective population size in the future
The Common hamster has been reintroduced in five areas in the Netherlands since 2002 (Fig. 1b, R1-R5) and most populations increased in size in consecutive years (Fig. 1c). However, most hamster populations sharply declined in 2008 due to an increased predation rate (Table 2 in Appendix). In 2008 the population of common vole (Microtus arvalis) also crashed in the Netherlands, which resulted in an increased predation rate of hamsters by all kind of predators normally relying on common voles (Kuiters et al. 2010). Such a population crash is a real threat for the highly endangered hamster populations in Belgium and the Netherlands, as all hamster populations already have an impoverished genetic diversity (La Haye et al. 2012a) and a crash may result in a further loss of genetic variation and a reduction of the effective population size (Allendorf et al. 2013; Keller et al. 2012; Luikart et al. 2010; McEachern et al. 2011). Effective population sizes (Table 1) of established hamster populations in the Netherlands and Belgium are already small compared with other threatened hamster populations like in France, in the Netherlands and Belgium N e ranged from 25 till 147, while the French population had an N e of 427 (Reiners et al. 2014), showing the absolute need to prevent declines in N e .
On the other hand, large population fluctuations are typical for rodents like the Common hamster (Krebs 2013) and genetic diversity and effective population size can be maintained in rodents through migration (Rikalainen et al. 2012). Unfortunately, the current populations of Common hamster in Belgium and the Netherlands are highly isolated, which reduces the change of maintenance of genetic diversity through natural immigration drastically (La Haye et al. 2012a). As the isolation of hamster populations in Belgium and the Netherlands will also be problematic in the next decades (Kuiters et al. 2010), it is necessary that populations of the Common hamster consist of at least hundreds of individuals to prevent a loss of genetic variation and to maintain a stable effective population size (Keller et al. 2012; Frankham et al. 2014; Reiners et al. 2014; Waller 2015; Melosik et al. 2017). Unfortunately, it is very uncertain if population sizes in Belgium and the Netherlands can be kept large enough, because the area of suitable habitat is limited and population densities do not exceed more than two individuals per hectare (Kuiters et al. 2010) and realization of much more suitable habitat for the species is challenging because of agriculture regulations (Pe’er et al. 2014). An alternative conservation measure to prevent a loss of genetic diversity is regular supplementation of hamster populations as a substitute of natural immigration. Supplementation is indeed effective as a conservation measure, Table 1 shows that in several hamster populations, R1-R3, B1 and B2, ‘immigration’ rate as detected by MLNE is quite strong between t1 and t2, which results in larger effective population sizes (N e ), than it should have been the case without supplementation. The divergence in population B2 Tongeren, with N e -m being larger than N e , is opposite to what is expected and may be a result of a wrong census estimate, which was probably too low for B2.
The populations of R4 and R5, established with individuals of the G/NL and NL breeding line, show that it is possible to establish wild populations with a more or less stable genetic diversity during several years without supplementation. It is attractive to link the relative positive maintenance of genetic diversity and population size in these populations to the genetic advantages of individuals with a G/NL genetic background as individuals of the G/NL line produce larger litters (La Haye et al. 2012b), but many other factors may have played a role as well. The short distance between R4 and R5 may have promoted natural migration and demographic and/or genetic rescue and 20% of all arable fields in these areas have a suitable agriculture management (Kuiters et al. 2010). This makes it very difficult to determine the sole effect of an increased litter size on population development and maintenance of genetic diversity in R4 and R5.
Conclusion
To summarize, genetic monitoring can be an effective tool to gather information of management decisions and of data on relevant population-ecological processes. In the case of the Common hamster, it is clearly demonstrated that genetic monitoring was useful in monitoring genetic diversity after reintroduction and to show in which population supplementation was successful. By using multivariate (DAPC) and Bayesian (STRUCTURE) statistical inference and calculating effective population sizes it was possible to reveal changes in genetic diversity in an intuitive and easy interpretable way and to optimize conservation actions.
References
Allendorf FW, Luikart G, Aitken SN (2013) Conservation and the genetics of populations. Blackwell, London
Armstrong DP, Seddon PJ (2008) Directions in reintroduction biology. Trends Ecol Evol 23:20–25
Boer M de (2014) Regional studbook for the Common hamster Cricetus cricetus. Publication Royal Rotterdam Zoological & Botanical Gardens, Rotterdam
Bouzat JL, Johnson JA, Toepfer JE, Simpson SA, Esker TL, Westemeier RL (2009) Beyond the beneficial effects of translocations as an effective tool for the genetic restoration of isolated populations. Conserv Genet 10:191–201
Brekke P, Bennett PM, Santure AW, Ewen JG (2011) High genetic diversity in the remnant island population of hihi and the genetic consequences of re-introduction. Mol Ecol 20:29–45
Carlson SM, Cunningham CJ, Westley PAH (2014) Evolutionary rescue in a changing world. Trends Ecol Evol 29:521–530
Crees JJ, Collins AC, Stephenson PJ, Meredith HMR, Young RP, Howe C, Price MRS, Turvey ST (2016) A comparative approach to assess drivers of success in mammalian conservation recovery programs. Conserv Biol 30:694–705
DeBarba M, Waits LP, Garton EO, Genovesi P, Randi E, Mustoni A, Groff C (2010) The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Mol Ecol 19:3938–3951
Di Marco M, Boitani L, Mallon D, Hoffmann M, Iacucci A, Meijaard E, Visconti P, Schipper J, Rondinini C (2014) A retrospective evaluation of the global decline of carnivores and ungulates. Conserv Biol 28:1109–1118
Diefenbach D, Hansen L, Bohling J, Miller-Butterworth C (2015) Population and genetic outcomes 20 years after reintroducing bobcats (Lynx rufus) to Cumberland Island, Georgia USA. Ecol Evol 5:4885–4895
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Ewen JG, Armstrong DP (2007) Strategic monitoring of reintroductions in ecological restoration programmes. Ecoscience 14:401–409
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fischer J, Lindenmayer DB (2000) An assessment of the published results of animal relocations. Biol Conserv 96:1–11
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, red List criteria and population viability analyses. Biol Conserv 170:56–63
Frosch C, Kraus RHS, Angst C, Allgöwer R, Michaux J, Teubner J, Nowak C (2014) The genetic legacy of multiple beaver reintroductions in Central Europe. PloS One 9(5):e97619
Gilbert KJ, Whitlock MC (2015) Evaluating methods for estimating local effective population size with and without migration. Evol Int J Org Evol 69:2154–2166
Goudet J (1995) FSTAT: a computer program to calculate F-statistics. J Hered 86:485–486
Hayward MW, Adendorff J, O’Brien J, Sholto-Douglas A, Bissett C, Moolman LC, Bean P, Fogarty A, Howarth D, Slater R, Kerley GIH (2007) The reintroduction of large carnivores to the Eastern Cape, South Africa: an assessment. Oryx 41:205–214
IUCN/SSC (2013) Guidelines for reintroductions and other conservation translocations. Version 1.0. IUCN Species Survival Commission, Gland
Jakob SS, Mammen K (2006) Eight new polymorphic microsatellite loci for genetic analyses in the endangered Common hamster (Cricetus cricetus L.). Mol Ecol Notes 6:511–513
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jamieson AG (2015) Significance of population genetics for managing small natural and reintroduced populations in New Zealand. N Z J Ecol 39:1–18
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Keller LF, Biebach I, Ewing SR, Hoeck PEA (2012) The genetics of reintroductions: inbreeding and genetic drift. In: Reintroduction biology: integrating science and management. Blackwell, London
Kennington WJ, Hevroy TH, Johnson MS (2012) Long-term genetic monitoring reveals contrasting changes in the genetic composition of newly established populations of the intertidal snail Bembicium vittatum. Mol Ecol 21:3489–3500
Koelewijn HP, Pérez-Haro M, Jansman HAH, Boerwinkel MC, Bovenschen J, Lammertsma DR, Niewold FJJ, Kuiters AT (2010) The reintroduction of the otter (Lutra lutra) into the Netherlands: hidden life revealed by noninvasive genetic monitoring. Conserv Genet 11:601–614
Krebs CJ (2013) Population fluctuations in rodents. The University of Chicago Press, Chicago
Kuiters L, La Haye M, Müskens G, van Kats R (2010) Perspectieven voor een duurzame bescherming van de hamster in Nederland (in Dutch with English summary). Rapport 2022. Published by Alterra. Wageningen, The Netherlands
La Haye MJJ, Koelewijn HP, Siepel H, Verwimp N, Windig J (2012a) Genetic rescue and the increase of litter size in the recovery breeding program of the Common hamster (Cricetus cricetus) in the Netherlands. Relatedness, inbreeding and heritability of litter size in a breeding program of an endangered rodent. Hereditas 149:207–216
La Haye MJJ, Neumann K, Koelewijn HP (2012b) Strong decline of gene diversity in local populations of the highly endangered Common hamster (Cricetus cricetus) in the western part of its European range. Conserv Genet 13:311–322
La Haye MJJ, Swinnen KRR, Kuiters AT, Leirs H, Siepel H (2014) Modelling population dynamics of the Common hamster (Cricetus cricetus): timing of harvest as a critical aspect in the conservation of a highly endangered rodent. Biol Conserv 180:53–61
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11(2):355–373
Madsen T, Shine R, Olsson M, Wittzell H (1999) Conservation biology—restoration of an inbred adder population. Nature 402:34–35
Melosik I, Ziomek J, Winnicka K, Reiners TE, Banaszek A, Mammen K, Mammen U, Marciszak A (2017) The genetic characterization of an isolated remnant population of an endangered rodent (Cricetus cricetus L.) using comparative data: implications for conservation. Conserv Genet. doi:10.1007/s10592-017-0925-y
McCleery R, Hostetler JA, Oli MK (2014) Better off in the wild? Evaluating a captive breeding and release program for the recovery of an endangered rodent. Biol Conserv 169:198–205
McEachern MB, Van Vuren DH, Floyd CH, May B, Eadie JM (2011) Bottlenecks and rescue effects in a fluctuating population of golden-mantled ground squirrels (Spermophilus lateralis). Conserv Genet 12:285–296
Meinig H., Buschmann A, Reiners TE, Neukirchen M, Balzer S, Petermann R (2014) The status of the Common Hamster (Cricetus cricetus) in Germany. Natur und Landschaft 89:338–343
Neumann K, Jansman H (2004) Polymorphic microsatellites for the analysis of endangered Common hamster populations (Cricetus cricetus L.). Conserv Genet 5:127–130
O’Brien J (2015) Saving the Common hamster (Cricetus cricetus) from extinction in Alsace (France): potential flagship conservation or an exercise in futility? Hystrix 26:89–94
Ottewell K, Dunlop J, Thoma N, Morris K, Coates D, Byrne M (2014) Evaluating success of translocations in maintaining genetic diversity in a threatened mammal. Biol Conserv 171:209–219
Ozer F, Gellerman H, Ashley MV (2011) Genetic impacts of Anacapa deer mice reintroductions following rat eradication. Mol Ecol 20:3525–3539
Pacioni C, Wayne AF, Spencer PBS (2013) Genetic outcomes from the translocations of the critically endangered woylie. Curr Zool 59:294–310
Pe’er G et al (2014) EU agricultural reform fails on biodiversity. Science 344:1090–1092
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reiners TE, Encarnação JA, Wolters V (2011) An optimized hair trap for non-invasive genetic studies of small cryptic mammals. Eur J Wildlife Res 57:991–995
Reiners TE, Eidenschenk J, Neumann K, Nowak C (2014) Preservation of genetic diversity in a wild and captive population of a rapidly declining mammal, the Common hamster of the French Alsace region. Mamm Biol 79:240–246
Rikalainen K, Aspi J, Galarza JA, Koskela E, Mappes T (2012) Maintenance of genetic diversity in cyclic populations—a longitudinal analysis in Myodes glareolus. Ecol Evol 2(7):1491–1502
Robert A (2009) Captive breeding genetics and reintroduction success. Biol Conserv 142:2915–2922
Rondinini C, Rodrigues ASL, Boitani L (2011) The key elements of a comprehensive global mammal conservation strategy. Philos T R Soc B 366:2591–2597
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Seignobosc M, Hemerik L, Koelewijn HP (2011) A demo-genetic analysis of a small reintroduced carnivore population: the Otter (Lutra lutra) in the Netherlands. Int J Ecol Article ID 870853
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. PNAS 101:15261–15264
Surov A, Banaszek A, Bogomolov P, Feoktistova N, Monecke S (2016) Dramatic global decrease in the range and reproduction rate of the European hamster Cricetus cricetus. Endang Species Res 31:119–145
Tenhumberg B, Tyre AJ, Shea K, Possingham HP (2004) Linking wild and captive populations to maximize species persistence: optimal translocation strategies. Conserv Biol 18:1304–1314
Tkadlec E, Heroldová M, Veronika Víśková, Bednář M, Zejda J (2012) Distribution of the Common hamster in the Czech Republic after 2000: retreating to optimum lowland habitats. Folia Zool 61:246–253
Tracy LN, Wallis GP, Efford MG, Jamieson IG (2011) Preserving genetic diversity in threatened species reintroductions: how many individuals should be released? Anim Conserv 14:439–446
Van Houtan KS, Halley JM, Aarde R van, Pimm SL (2009) Achieving success with small, translocated mammal populations. Conserv Lett 2:254–262
Villemey A, Besnard A, Grandadam J, Eidenschenck J (2013) Testing restocking methods for an endangered species: effects of predator exclusion and vegetation cover on Common hamster (Cricetus cricetus) survival and reproduction. Biol Conserv 158:147–154
Waller DM (2015) Genetic rescue: a safe or risky bet? Mol Ecol 24:2595–2597
Wang J, Whitlock MC (2003) Estimating effective population size and migration rates from genetic samples over space and time. Genetics 163:429–446
Weeks AR et al (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725
Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, Walk JW, Kershner EL, Bouzat JL, Paige KN (1998) Tracking the long-term decline and recovery of an isolated population. Science 282:1695–1698
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon AD (2015) Genetic rescue to the rescue. Trends Ecol Evol 30:42–49
Ziomek J, Banaszek A (2007) The Common hamster, Cricetus cricetus in Poland: status and current range. Folia Zool 56:1–9
Acknowledgements
This study was supported by the Dutch Ministry of Economic Affairs, Program BO-02-013: Active policy on species management. Thanks to G. Müskens and R. van Kats for field assistance, colleagues from Rotterdam Zoo and Gaiazoo for collecting DNA samples and C. Nowak, L. Kuiters and H. Siepel for constructive comments on earlier drafts. We also thank two anonymous reviewers for their constructive and important comments to improve this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. J. J. La Haye and T. E. Reiners have contributed equally to this work.
Appendix
Appendix
See Fig. 4 and Tables 2, 3 and 4.

Posterior assignment propabilities to different number of assumed ancestral populatons/clusters (K 2 to 4) for each analyzed individual common hamster shown as vertical bar. Colors indicate assignment proability to corresponding clusters for each K. Vertical lines indicate the different cohorts (t0-reintroduction or initial assessment, r1, r2 and r3-restocking/supplementation, t1 and t2-assessment) and populations in this study
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
La Haye, M.J.J., Reiners, T.E., Raedts, R. et al. Genetic monitoring to evaluate reintroduction attempts of a highly endangered rodent. Conserv Genet 18, 877–892 (2017). https://doi.org/10.1007/s10592-017-0940-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0940-z