
Abstract
Circadian rhythms influence physiological processes from sleep–wake cycles to body temperature and are controlled by highly conserved cycling molecules. Although the mechanistic basis of the circadian clock has been known for decades, the extent to which circadian rhythms vary in nature and the underlying genetic basis for that variation is not well understood. We measured circadian period (Ʈ) and rhythmicity index in the Drosophila Genetic Reference Panel (DGRP) and observed extensive genetic variation in both. Seven DGRP lines had sexually dimorphic arrhythmicity and one line had an exceptionally long Ʈ. Genome-wide analyses identified 584 polymorphisms in 268 genes. We observed differences among transcripts for nine genes predicted to interact among themselves and canonical clock genes in the long period line and a control. Mutations/RNAi knockdown targeting these genes also affected circadian behavior. Our observations reveal that complex genetic interactions influence high levels of variation in circadian phenotypes.
Similar content being viewed by others
Background
Circadian rhythms are endogenous cycles present in almost all living organisms. They affect myriads of biological processes in humans, such as sleep/wake cycles, body temperature, hormone levels, heart rate, and even cognitive performance (Van Dongen et al. 2004). Circadian rhythms may thus play a fundamental role in human health (Zee et al. 2014). Disruption to circadian rhythms has been associated with detrimental neurobehavioral consequences. For example, rotating shift workers, people experiencing chronic jet lag, and those living in areas with extreme long or short photoperiods have increased risk of psychiatric and mood disorders (Bunney and Bunney 2000; Boivin 2000; Grandin et al. 2006; Magnusson and Boivin 2003). Furthermore, circadian disruptions often precede the development of neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease, though whether they have a causal role in these conditions is unknown (Mattis and Sehgal 2016; Videnovic and Zee 2015). Disruption of circadian rhythms may also contribute to metabolic disease. For instance, circadian misalignment, often manifested as rotating or shifting work schedules, has been associated with increased hypertension, Type 2 diabetes, total cholesterol, and cardiovascular disease (Reutrakul and Knutson 2015; McHill and Wright 2017; Reutrakul and Van Cauter 2014). Profound effects on metabolic processes such as glucose tolerance, body mass index (BMI) and cortisol levels result from the disruption of sleep and circadian cycles combined with poor dietary choices (Shi and Zheng 2013; McHill et al. 2014). Thus, disrupted circadian rhythms are an important consideration in the etiology of disease.
The circadian period in humans is tightly regulated and close to 24 h (Czeisler et al. 1999). Human clocks have a built-in mechanism to adjust endogenous period and phase in response to a moderate light stimulus (Scheer et al. 2007). Human studies focus on chronotype, which is the preference for morning or evening activity (Kalmbach et al. 2016). Chronotype has been shown to have a genetic component (Jones et al. 2016; Lane et al. 2016; Kalmbach et al. 2016; Toomey et al. 2015). Candidate gene studies show associations between core canonical clock genes and chronotype (Kalmbach et al. 2016). For instance, a hereditary form of delayed sleep phase disorder (DSPD) is associated with a dominant coding variation in the core circadian clock gene CRY1 (Patke et al. 2017). Recently, three large-scale genome-wide association studies in humans revealed that, in addition to canonical clock genes, novel genes may contribute to differences in human chronotype (Jones et al. 2016; Lane et al. 2016; Hu et al. 2016).
Core clock mechanisms regulating circadian behavior are remarkably similar between mammals and Drosophila melanogaster, even though they diverged approximately 600 million years ago (Yu and Hardin 2006; Panda et al. 2002; Nitta et al. 2015). In fact, the genes and processes involved in complex mammalian circadian rhythms were first identified in flies (Konopka and Benzer 1971; Hardin et al. 1990). Rhythmic rest and activity behavior in Drosophila is one of the most reliable phenotypes for the identification of novel genes regulating circadian rhythms (Hardin 2011; Helfrich-Forster et al. 2011). The use of flies for rest and activity studies offers several advantages over mammalian models. For example, the availability of an extensive collection of stocks with mapped mutations, chromosomal deletions, and transgenic constructs have facilitated forward genetics approaches in circadian rhythm studies. This approach has identified numerous genes of the circadian clock (Hardin et al. 1990; Takahashi 1993; Reppert and Weaver 2002; Sehgal et al. 1995; Dunlap et al. 2007).
Variability in circadian period and rhythmicity parameters among wild-derived isochromosomal lines of flies has previously been noted (Emery et al. 1994, 1995), and this variation has a genetic component (Emery et al. 1995). Heritability has been estimated at 0.14 for circadian period (Emery et al. 1995). Therefore, naturally occurring polymorphisms likely influence the fly’s circadian clock. Historically, most work has focused on polymorphic variation within the canonical clock genes in natural populations. Studies of a threonine-glycine-encoding repeat region in the period gene revealed a clinal distribution of these alleles in natural populations in Europe and Australia (Kyriacou et al. 2008; Sawyer et al. 1997). Likewise, an insertion in the 5′ coding region of timeless varies with latitude in European flies (Kyriacou et al. 2008). Furthermore, a polymorphic variant in the genomic region containing shaggy is clinal in North America, though not in Australia (Rand et al. 2010). In contrast, a non-synonymous polymorphism in cryptochrome does not vary with latitude but persists in nearly equal frequencies in European populations (Pegoraro et al. 2014). These polymorphisms are associated with differences in temperature-compensated period length (Kyriacou et al. 2008; Sawyer et al. 1997), the frequency of diapause (Kyriacou et al. 2008; Tauber et al. 2007), the timing of eclosion (Pegoraro et al. 2014), and thermal tolerance (Rand et al. 2010; Sawyer et al. 1997). These experiments demonstrate the importance of polymorphic variation in canonical clock genes to circadian behavior in flies; however, the contribution of polymorphic variants in the rest of the genome to circadian behavior in natural populations remains largely unexplored.
Recently, several efforts have been made to study genomic differences for a variety of behaviors, diseases and life history traits using genetic reference populations of Drosophila (King et al. 2012a, b; Mackay et al. 2012; Huang et al. 2014; Grenier et al. 2015). Here, we performed a genome-wide association study (GWAS) using the D. melanogaster Genetic Reference Panel (DGRP), a unique resource created by mating full siblings of wild-caught iso-female lines for 20 generations (Mackay et al. 2012; Huang et al. 2014), to explore the range of polymorphic variants contributing to genetic variation in Drosophila circadian phenotypes. Some of the salient features of the DGRP collection are (i) availability of full sequence data, (ii) rapid decay in linkage disequilibrium (LD) with physical distance, (iii) and a lack of population structure (Mackay et al. 2012; Huang et al. 2014). Another important feature of the DGRP is the ability to perform screening of identical genotypes in a controlled environment, thus elucidating the role of genes in micro-environmental plasticity (Morgante et al. 2015; Lin et al. 2016a). Many studies ranging from natural variation in physiological processes (food intake and nutrient stores) (Garlapow et al. 2015; Unkless et al. 2015), to variation in behavior (olfactory avoidance and aggression) (Arya et al. 2015; Shorter et al. 2015), to anatomical features (mushroom body size and abdominal pigmentation) (Dembeck et al. 2015; Zwarts et al. 2015), and to environmental and drug responses (radiation resistance and paraquat) (Vaisnav et al. 2014; Weber et al. 2012) have exploited the DGRP.
In this study, we examined two circadian phenotypes, circadian period (Ʈ) and rhythmicity index (RI), a measure of how consistent the fly’s daily activity patterns are, in 167 DGRP lines. We observed high levels of sex-specific genetic variation for both Ʈ and RI. Several lines had sex-specific arrhythmic behavior and one line (DGRP_892) had a very long average circadian period of 31 h. The GWAS identified 584 molecular polymorphisms in 268 candidate genes associated with Ʈ and RI. Thus, the association mapping strategy enabled us to identify candidate genes affecting variation in circadian behavior.
Results
Quantitative genetic analyses
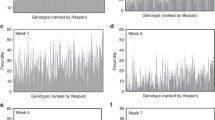
Circadian phenotypes were highly variable in the DGRP. We observed high levels of genetic variation among lines (all PLine(Block) < 0.0001, four-way nested ANOVA) for RI and for Ʈ, whether Ʈ was calculated using the Maximum Entropy Spectral Analysis method (MESA period) or by the Chi square Periodogram method (χ2 period) (Fig. 1; Table S1). These traits were also highly sexually dimorphic among lines (all PSex×Line(Block) < 0.001, four-way nested ANOVA). Broad-sense heritability was high for RI (H2 = 0.43), indicating that the genetic contribution to this trait is relatively large (Fig. 1a). Heritability was relatively low for MESA period (H2 = 0.17), consistent with previous estimates of heritability in wild-derived populations (Emery et al. 1995), but higher for χ2 period (H2 = 0.39). We observed that ~ 12% of the flies were arrhythmic, a figure consistent with previous reports of rhythmicity in natural populations (Emery et al. 1994; Kumar et al. 2005). Females tended to have a lower RI than males and were more likely than males to be arrhythmic (Fig. 1b). Female RI averages for lines DGRP_42, DGRP_153, DGRP_375, DGRP_509, DGRP_810, and DGRP_908 were below the threshold value for rhythmicity (\({r_k}\) = 0.0772). Females from these lines were therefore classified as arrhythmic. Males of one line, DGRP_101, were also classified as arrhythmic. Representative examples of autocorrelation plots for rhythmic and arrhythmic flies are shown in Fig. 1c, d. The distributions of MESA and χ2 period across the DGRP were quite different from one another (Fig. 1e–h). The range of values for the period as calculated by Chi square periodogram was smaller than the range of values seen using MESA, particularly on the low end of the scale. Ʈ estimates of line means varied from 15 to 31.3 h when calculated using MESA and from 23.2 to 31.3 h when calculated using the χ2 periodogram method. Interestingly, both methods identified a single line with a very long period, line DGRP_892. This line had an average period of 31.76 h in males and 30.92 h in females as calculated by the MESA and 31.78 (30.88) hours as calculated by χ2 for males (females) (Table S2; for a representative actogram, see Fig. S1A). DGRP_892 had long sleep in LD but was not an outlier (Harbison et al. 2013), nor was there a phase shift in the activity of these flies under LD conditions (Fig. S1B). Overall, circadian phenotypes were both variable and heritable in this natural population of flies.

Distribution of circadian rhythm phenotypes in the DGRP. Male line means are shown in dark blue; female line means are shown in pink. The difference in line means (male–female) is shown by purple bars. a Rhythmicity index. b Difference in rhythmicity index. c Autocorrelation plot for a representative male of a rhythmic line (DGRP_383; RI = 0.667) and an arrhythmic line (DGRP_101; RI = 0.066). d Autocorrelation plot for a representative female of a rhythmic line (DGRP_861; RI = 0.748) and an arrhythmic line (DGRP_375; RI = 0.037). e MESA period. f Difference in MESA period. g χ2 period. h Difference in χ2 period
We calculated the genetic correlations among all circadian parameters (Table S3). It has been noted previously that rhythmicity index does not predict circadian period; rather, it is an indicator of strength or robustness of the circadian rhythm (Grierson et al. 2016; Dowse 2007). Not surprisingly, the genetic correlation rG between RI and Ʈ was also low, though statistically significant, in the DGRP: rG was only 0.18 between RI and MESA period, and − 0.15 between RI and χ2 period. Thus, we expect that few genes will be common to RI and Ʈ. The genetic correlation between MESA period and χ2 period was 0.71 if line DGRP_892 was included, and 0.61 without this line. A perfect overlap between the two period calculations would not be expected as the distributions of the two traits are quite distinct; many more estimates of period occur in the low range for MESA period than for χ2 period (Fig. 1e, g; see also “Discussion”). These correlations suggest that some, but not all genes will be common among circadian traits.
Sleep and circadian rhythm behaviors are related as the timing of sleep is regulated by the circadian clock (Borbely 1982). We therefore correlated the circadian rhythm phenotypes with sleep phenotypes measured on the DGRP in a previous study (Harbison et al. 2013). The sleep study measured duration (minutes spent sleeping), bout number (number of naps), and average bout length (average nap length) during the day and night. It also measured waking activity, the number of activity counts per minute spent awake. In addition, the study measured the environmental sensitivity (coefficient of environmental variation, or CVE) (Mackay and Lyman 2005) for each sleep and activity trait (Harbison et al. 2013). Both MESA and χ2 period had significant genetic correlations with night average bout length (rG = 0.27 and 0.28, respectively). However, only MESA period was significantly genetically correlated with night bout number (rG = − 0.25) and night bout number CVE (rG = 0.41) (Table S4). RI had significant genetic correlations night and day bout number (rG = − 0.32 and − 0.52, respectively), night average bout length (rG = 0.32), night and day bout number CVE (rG = 0.37 and 0.28, respectively), and day average bout length CVE (rG = 0.26) (Table S4). While the genetic correlations between sleep and circadian phenotypes were statistically significant, inspection of Table S4 reveals that the correlations are not high. In addition, we computed sleep phenotypes from the rest and activity data collected in constant darkness in this study and used the data to calculate the phenotypic and genetic correlations with the circadian rhythm parameters. For RI, significant correlations were observed in the same traits (day and night bout number, and night average bout length), but the magnitudes of the correlations were higher (Table S4). A few additional sleep traits were correlated with the circadian rhythm phenotypes: day sleep duration and waking activity with RI, night bout number with MESA, and day average bout length and day sleep with χ2 period. Therefore, some common genetic architecture exists between sleep and circadian phenotypes, but it is far from being a complete overlap.
Genotype–phenotype associations
The DGRP is fully sequenced (Mackay et al. 2012; Huang et al. 2014), and all single nucleotide polymorphisms (SNPs), non-SNP variants (indels, tandem duplications, and complex variants), and chromosomal inversions have been mapped (Huang et al. 2014). We used the DGRP2 web-based analysis to associate these 1,920,276 genetic variants with the circadian phenotypes (Table S5) (Huang et al. 2014). Circadian phenotypes were not impacted by Wolbachia infection status, but some phenotypes were significantly associated with chromosomal inversions. RI in males was associated with In(2R)NS and In(3R)Mo; combined-sex RI was associated with In(2R)NS. In addition, the unusually long period observed in DGRP_892 was likely to impact the results of genome-wide association on circadian period as its inclusion would violate the assumption of independently and identically distributed residuals (Mackay et al. 2012). We therefore conducted the genome-wide analyses for circadian period without DGRP_892. We found 142 significant polymorphisms in 73 genes for RI; and 292 polymorphisms in 139 genes for MESA and 150 in 65 genes for χ2 period (Table S5) at a threshold P-value of 1 × 10−5. This threshold P-value is consistent with previous studies of quantitative traits in the DGRP (Arya et al. 2015; Dembeck et al. 2015a, b; Garlapow et al. 2015; Hunter et al. 2016; Shorter et al. 2015; Zwarts et al. 2015). Quantile–quantile (Q–Q) plots showed an excess of P-values below 10−5 for all traits, suggesting that variants below this threshold are enriched for true positive associations (Fig. S2–S4). At this threshold, the false-discovery rates range from 0.0092 to 0.52 for RI, 0.0033–0.25 for MESA period; and < 0.0001–0.28 for χ2 period (Table S5). A total of thirty-nine variants had P-values that were less than a Bonferroni-corrected P-value of 1.6 × 10− 8. Variants associated with larger effect sizes had lower minor allele frequencies, consistent with previous observations of complex traits in the DGRP (Fig. S5) (Jordan et al. 2012; Weber et al. 2012). We hypothesized that polymorphisms lying within a gene or ± 1000 bp from a gene affect that gene as linkage disequilibrium decays over an average 30–200 bp in the DGRP (Mackay et al. 2012).
Plots of significant variants for each trait plotted against minor allele frequency, − log10(P-value), and normalized effect size reveal the complex genetic architecture of circadian behavior. Both low-frequency and common polymorphisms affect RI (Fig. 2a and S4). Effect sizes were relatively small, and virtually no linkage disequilibrium was observed between the top variants. In contrast, significant polymorphisms for period tended to have low minor allele frequencies, higher P-values, and larger effect sizes (Fig. 2b, c). Some linkage disequilibrium is present on chromosomes 2L and 2R among variants associated with MESA period, and on chromosomes X and 2L among variants associated with χ2 period. Five genes overlapped between the two period measures: CG31676, Mob2, nAcRalpha-30D, Oamb, and shep. Variants in canonical clock genes were not implicated in circadian period, but a single intronic SNP in Pdp1 was significantly associated with RI. Several other candidate genes or their homologs in other species have known or predicted effects on circadian behavior: 5-HT1A (Yuan et al. 2006), CG14618 (Ueda et al. 2002), CG42321 (Abruzzi et al. 2011), Gga (Hughes et al. 2010), Rh5 (Szular et al. 2012), slo (Ceriani et al. 2002; Fernandez et al. 2007), Tep4 (Hughes et al. 2010; Hu et al. 2016), tinc (Mizrak et al. 2012), tnc (Hughes et al. 2010), and wap (Wu et al. 2008). Overall, we identified 584 unique polymorphisms that implicated 268 genes in circadian behavior.

Genome-wide association results for circadian rhythm phenotypes. Polymorphisms with P-values for association ≤ 1 × 10−5 are plotted. The top panel shows the P-values plotted as − log10(P-value). The middle panel shows the minor allele frequency (MAF) of each variant in the DGRP; only variants with a MAF of 0.05 or greater were considered in the analysis. Effect sizes are plotted as a/σG in the bottom panel, where σG is the among-line standard deviation. The triangle shows the linkage disequilibrium among significant variants as r2, in chromosomal order. a Rhythmicity index. b MESA period. c χ2 period
Epistatic associations
To search for epistatic interactions between pairs of variants that are associated with the traits, we performed a full genome-wide pair-wise search for all variants with a minor allele frequency (MAF) greater than 0.15. Variants in linkage disequilibrium (r2 ≥ 0.8) were pruned. We found 2,295,186 epistatic interactions for circadian period at a nominal P-value ≤ 1 × 10− 7 or less. For χ2 period, there were 285,470 interactions for males and 38,411 for females. For MESA period, there were 100,701 interactions for males and 1,960,053 for females. The large differences in the number of interactions found for the two measures of circadian period are likely due to the differences in their distribution; χ2 period had a much lower range of values for period than MESA, particularly for the lower values of circadian period (Fig. 1). For RI, there were 29,465 interactions for males and 23,422 interactions for females. A total of 6401 unique epistatic interactions for χ2 and MESA period were within or within ± 1 kb of the core circadian clock genes: per, tim, Clk, cyc, Pdp1, vri, and sgg (Table S6). These previously unappreciated interactions suggested that circadian period could be modified by numerous interactions with the molecular circadian clock. Likewise, 189 epistatic interactions for RI contained canonical clock genes (Table S6). Despite pruning the data to a relatively high MAF and accounting for potential linkage disequilibrium, these large numbers of epistatic interactions were likely to have numerous false positives. We therefore followed up a subset of these interactions with further testing (see below).
Comparison with sleep genome-wide association
The circadian clock is thought to control the timing of sleep (Borbely 1982). We examined the degree of overlap between the variants we identified for circadian behavior with a previous genome-wide association study of sleep (Harbison et al. 2013). Only one variant overlapped between MESA period and a single sleep trait, average day bout length; two variants overlapped between χ2 period and waking activity CVE. However, if we considered the overlap among genes, 48 putative circadian genes overlapped with mean night sleep, night and day average bout length, and waking activity; 100 candidate circadian genes overlapped with measures of environmental sensitivity for night sleep, day sleep, day bout number, night average bout length, and waking activity (Table S7). Two of these genes were implicated in sleep and its regulation in other studies: Sh (Cirelli et al. 2005) and 5-HT1A (Yuan et al. 2006). Thus, while the overlap among polymorphic variants was low, the overlap between candidate genes in the sleep and circadian studies was moderate. Interestingly, of the 4440 genes predicted to interact epistatically with canonical clock genes, 991 genes were also implicated in sleep and waking activity (Harbison et al. 2013), 9.4% more than would be expected by random chance (Table S8).
Candidate genes potentially contributing to long circadian periods in DGRP_892
None of the significant polymorphisms we identified as associated with circadian period were in coding or known non-coding regulatory regions of the canonical clock genes. Furthermore, none of the DGRP variants predicted to alter coding sequence are found in canonical clock genes (per, tim, vri, Pdp1, Clk, sgg, cyc, and dbt) (Huang et al. 2014), nor did we find any naturally-occurring polymorphisms known to produce a long circadian period that were private to DGRP_892 (Konopka and Benzer 1971; Kloss et al. 1998). This suggests that the extreme circadian period seen in DGRP_892 may be due to the action of regulatory genes on core clock genes (Ray and Reddy 2016). Polymorphisms within or in close proximity to such regulatory genes may influence the extremely long Ʈ in DGRP_892 by changing transcript abundance (Albert and Kruglyak 2015), mRNA secondary structure (Nackley et al. 2006; Wang et al. 2007), or protein structure (Kimchi-Sarfaty et al. 2007). Thus, we examined transcript abundance over time of three canonical clock genes in DGRP_892: per, tim, and Pdp1, as well as 26 candidate genes from this study whose transcripts were previously reported to be either cycling and/or regulated by the circadian clock (Table S9). Six of these candidate genes came from the main effect analyses, while the remaining genes came from the epistatic analysis. Genes from the epistatic analysis interacted with either canonical clock genes or other genes identified in the GWA (Table S9). In addition, six candidate genes had intergenic variants proximal to predicted circadian regulatory motifs (i.e., E-boxes) that may influence promoter or terminator activity, messenger RNA (mRNA) conformation (stability), and subcellular localization of mRNAs and/or proteins (Komar et al. 1999; Komar 2007). Transcript levels for each gene were compared to Canton-S B, an isogenic line with a normal circadian period (23.81 ± 0.03 h in males; 24.05 ± 0.18 h in females). If these genes affect period in DGRP_892, we would expect to see differences in the magnitude of gene expression and/or cycling between the two lines.
Canonical circadian clock transcript levels
We examined transcript levels of per, tim, and Pdp1 at six circadian timepoints under both standard (LD) and constant dark (DD) lighting conditions. We first analyzed the changes in gene expression over time within each line (Table S10). As anticipated, transcripts of per were significantly different across time in Canton-S B, whether in DD or LD (FDR = 0.0001 and 0.0127, respectively), with a peak of expression around CT10 in DD and ZT10-14 in LD (post-hoc Tukey P ≤ 0.05). Likewise, tim expression was significantly different in Canton-S B across time for both lighting conditions (FDR = 0.0013 for DD and 0.0273 for LD); expression peaked at CT10 in DD and ZT10-14 in LD (post-hoc Tukey P ≤ 0.05). However, Pdp1 showed marginally non-significant differences in gene expression across time in Canton-S B under both lighting conditions (FDR = 0.0868 in DD and 0.0922 in LD), though there was a significant peak of expression in LD as ZT14 was higher than the other time points (post-hoc Tukey P ≤ 0.05). We used the JTK_CYCLE program to determine whether these genes were cycling rhythmically. Both per and tim cycled robustly in both LD and DD lighting conditions (FDR = 0.0002 and 0.0019 for per, respectively, and FDR = 0.0007 and 0.0488 for tim, respectively). Pdp1 exhibited significant cycling only in LD (FDR = 0.001). Thus, Canton-S B had the expected pattern of cycling gene expression for per and tim; Pdp1 appeared slightly damped.
In contrast to the canonical clock transcripts observed in Canton-S B, gene expression patterns differed in DGRP_892 (Table S10). per transcriptional cycling appeared damped in DGRP_892, particularly in DD (FDR = 0.0922 in DD and 0.0681 in LD). JTK_CYCLE analysis confirmed that per was not cycling in a 24-h period in LD or in DD. In LD, there was a peak of per expression at approximately ZT14 (post-hoc Tukey P ≤ 0.05). tim appeared to cycle robustly in DGRP_892 under both lighting conditions as expression changed across time (FDR = 0.0050 for DD and 0.0273 for LD) with a peak of expression at CT10 in constant darkness (post-hoc Tukey P ≤ 0.05). Analysis with JTK_CYCLE confirmed that tim had rhythmic expression in LD, while rhythmicity in DD was not formally significant (FDR = 0.0210 and 0.0735, respectively). Pdp1 transcripts were not differentially expressed across time in DGRP_892 under either lighting condition, but JTK_CYCLE analysis indicated that the transcripts were cycling in LD (P = 0.0430). Transcriptional cycling may therefore be impaired in both per and Pdp1 in DGRP_892.
We next asked whether there were significant differences in gene expression between Canton-S B and DGRP_892 in these canonical clock genes (Table S11). per expression was significantly different between the two lines (FDR = 0.0482), with DGRP_892 having higher levels in DD (FDR < 0.0001) (Fig. 3a). Gene expression between the two lines was not significantly different in LD (Fig. 3b). DGRP_892 had higher levels of tim than Canton-S B, whether gene expression was measured in LD or in DD (FDR < 0.0001) (Fig. 3c, d). tim expression levels in DGRP_892 were much higher than Canton-S B at CT14 in DD (FDR = 0.0387), and at ZT10 in LD (FDR = 0.0019) (Table S11). Pdp1 levels were higher in DGRP_892 than Canton-S B across time and lighting condition (FDR = 0.0388) (Fig. 3e, f), but the contrast between the two lines was only different in DD (FDR = 0.0360) when times or lighting conditions were considered separately. Given the wealth of reports of differences in transcript abundance in either sex (Jin et al. 2001; Arbeitman et al. 2002; Parisi et al. 2003; Ranz et al. 2003; Harbison et al. 2005; Wayne et al. 2007; Zhang et al. 2007; Ayroles et al. 2009; Huylmans and Parsch 2014; Huang et al. 2015), we expected to see differences in males and females. Only per and tim exhibited significant sex differences [PTreatment×Line×Sex = 0.0064 (FDR = 0.0482) and < 0.0001 (FDR = 0.0017), respectively]. Thus, transcriptional abundance differs in canonical clock genes in the DGRP_892 long period line relative to Canton-S B, but these differences are predominantly within genes controlling the negative feedback loop of the clock (i.e., per and tim), rather than the positive feedback loop (Pdp1).

Canonical clock gene expression profile of Canton-S B and DGRP_892. Normalized transcript levels are plotted against time for constant darkness (DD) and standard 12-h light:dark (LD) conditions. Canton-S B transcript levels are white; DGRP_892 transcript levels are gray. a, bper. c, dtim. e, fPdp1. CT circadian time, ZT zeitgeber time. Plots show the average of males and females. Significant differences between Canton-S B and DGRP_892 at a given time point are indicated by asterisks. *FDR < 0.05; ***FDR < 0.001. Source data is provided in Table S12
Candidate gene transcript levels
We assayed expression levels of 26 additional candidate genes predicted by the GWAS as described above. If some of these transcripts are changing in abundance over the 24-h day, that would suggest involvement in the circadian clock. As with the canonical clock genes, we examined reduced ANOVA models for each line separately to determine whether gene expression changed over time, which is one indication that the transcripts are potentially cycling. Rae1 and Cpr62Ba were differentially expressed across time in DGRP_892 in LD (FDR = 0.0433 and 0.0478, respectively), but the differences were confined to one sex only (Table S10). Post-hoc Tukey tests indicated that these genes may be fluctuating in females with a peak at ZT14 and in males with a peak at ZT10-14, respectively (Table S10). JTK_CYCLE analysis indicated that Rae1 may be rhythmic in females (P = 0.0269) but did not find rhythmicity in Cpr62Ba in males.
We were particularly interested in those genes with differential gene expression between Canton-S B and DGRP_892 (Table S11 and S12). Nine genes had differences in gene expression between Canton-S B and DGRP_892 across sexes, times, and lighting conditions: AGO2 (FDR = 0.0705; marginally significant, but see below), CG42321 (FDR < 0.0001), Dop1R2 (FDR = 0.0442), GlcT-1 (FDR = 0.0450), GluRIIA (FDR = 0.0141), Mdr65 (FDR = 0.0263), Rae1 (FDR = 0.0206), Tep4 (FDR = 0.0029), and tnc (FDR = 0.0198) (Fig. 4a–h; Fig. S6). Three genes had significant differences in expression between Canton-S B and DGRP_892 that were specific to lighting conditions: AGO2 (FDR = 0.0016), GlcT-1 (FDR = 0.0055), and tnc (FDR = 0.0353) (Table S11). The expression of CG42321 tended to be higher in DGRP_892 than Canton-S B under all conditions; CT06, CT14, and CT18 were significantly different in DD (FDR = 0.0269, 0.0193, and 0.0374, respectively) (Table S13). When gene expression for Dop1R2 and GluRIIA was averaged across all timepoints, there were significant differences in gene expression between Canton-S B and DGRP_892. For Dop1R2, DGRP_892 had higher average expression (Fig. 4e, f), while for GluRIIA, Canton-S B had higher average expression (Fig. S6A and B). Canton-S B had higher GlcT-1 expression than DGRP_892 at CT14 in DD (PTime = 0.0003; FDR = 0.0348) (Fig. 4g, h). tnc transcript levels were higher in Canton-S B at all timepoints in LD. The remaining genes showed more complex patterns of expression across time. These line-specific differences in gene expression potentially contribute to the long period in DGRP_892.

Candidate gene expression profiles of Canton-S B and DGRP_892. Normalized transcript levels are plotted against time as in Fig. 3. a, bAGO2. c, dCG42321. e, fDop1R2. g, hGlcT-1. The remaining genes are plotted in Figure S6
Thus, nine candidate genes from the GWAS had differential expression between Canton-S B and DGRP_892, seven of which were predicted to have epistatic effects with other candidate genes or the canonical circadian clock genes (Table S9). Whether these genes are causal in the long period of DGRP_892 remains to be elucidated, but differences in gene expression indicate that they may have a role.
Behavioral tests of candidate genes with gene expression differences
We tested candidate genes with differences in gene expression for their role in circadian period and rhythmicity index using available mutations and RNAi knockdowns. We tested CG42321, Cpr62Ba, and tnc using Minos {ET1} element lines (Bellen et al. 2011).The Minos lines have an control line with an identical genetic background, which makes them ideal for candidate gene tests. However, the Cpr62Ba and tnc stocks had been maintained via crosses to a different strain, which altered the genetic background. Therefore, for these two mutants, we tested flies that were heterozygous as well as homozygous for the Minos element. Minos allele CG42321MB05800 had a significant effect on χ2 period (Table 1 and Table S15). tncMB04464 mutants had significantly altered rhythmicity index (Table 1).
Furthermore, we tested several genes having Minos {y[+mDint2]=MIC} insertions available in CG42321, Cpr62Ba, Dop1R2, GlcT-1, Mdr65, Tep4, and tnc. (Venken et al. 2011). In addition, we tested available deletions in AGO2 and GluRIIA, a P{w[+mC]=EP1} insertion in GlcT-1, P{w[+mC] y[+mDint2]=EPgy2} insertions in Tep4 and tnc, and a P{y[+mDint2] w[BR.E.BR]=SUPor-P} insertion in Mdr65. One mutant allele of CG42321, CG42321MI08838, affected χ2 period, but a second allele, CG42321MI06777, did not (Table 2). Additionally, the CG42321MI06777 allele affected RI, but only as a heterozygote (Table 1). Both of the Cpr62BaMI12486 and Cpr62BaMI03734 alleles tested affected χ2 when homozygous. The Cpr62BaM12486 allele only targets the first intron of the -RB isoform of Cpr62Ba, while the Cpr62BaMI03734 and Cpr62BaMB12091 alleles putatively affect both isoforms (Table S14). The Dop1R2MI08664 allele affected RI in males only, with a difference of – 0.1297 from the control; no other effects of this mutation were observed (Table 1). The GlcT-1MI06082 allele had no effect on circadian traits; however, the GlcT-1G5974 allele did, impacting both χ2 and RI. Both GlcT-1 insertions target the transcribed portion of the gene; however, the GlcT-1G5974 is upstream of the coding region of the gene, while the GlcT-1MI06082 allele is just downstream of it. Two alleles of Tep4, Tep4MI13472 and Tep4EY04656, affected circadian period and the Tep4MI13472 allele affected RI as well. In addition, male-specific effects on χ2 were observed in the tncEY16369 allele.
Finally, we tested AGO2, Dop1R2, GlcT-1, GluRIIA, Mdr65, Rae1, Tep4, and tnc via RNAi knockdown (Table 1 and S16). These genes are expressed in adult male and female heads (Brown et al. 2014); thus, it is possible that gene expression occurs in clock neural cells. To test the potential effect of these genes on the circadian clock, we drove expression in circadian clock neural cells by crossing these RNAi constructs to Pdf-GAL4 and tim-GAL4. Pdf-GAL4 expression is confined to 4 of 5 small ventral lateral neurons and the large ventral lateral neurons (Renn et al. 1999) while tim-GAL4 is expressed more broadly, targeting the ventral lateral neurons as well as the dorsal neurons (groups 1–3) and the dorsal lateral neurons (Kaneko and Hall 2000). We first tested whether RNAi knockdown using both GAL4 drivers would affect circadian phenotypes. χ2 period was affected in AGO2, GlcT-1, Tep4, and tnc knockdown animals, while RI was affected in Dop1R2 and GluRIIA knockdowns (Table 1). Closer inspection revealed that the effect on χ2 period was due to differences between the RNAi knockdowns and the y[1] v[1]; P{y[+t7.7]=CaryP}attP40 control line crossed to tim-GAL4; the χ2 period of crosses to Pdf-GAL4 were in the same direction, but not statistically significant. This raised the interesting possibility that the effects of these genes on circadian period require the larger group of clock-related neurons. Alternatively, the tim-GAL4 driver might have been more efficient at reducing gene expression than the Pdf-GAL4 driver.
In summary, tests of mutants and RNAi knockdown flies further implicated our candidate genes as affecting circadian rhythms. The strongest evidence was for CG42321, Cpr62Ba, GlcT-1, and Tep4 affecting χ2 period. Multiple constructs of these genes in varied genetic backgrounds affected circadian behavior. Effects on RI were more fleeting, but evidence suggests that Dop1R2, Tep4, and tnc alter RI.
Behavioral tests of candidate genes predicted to have large effect sizes
We also conducted tests on candidate genes predicted to have the largest effect sizes on χ2 period and rhythmicity index. We tested available Minos insertions in CG11073, CG13243, CG34355, CG42672, Mp, Prosap, Ptp99A, sano, and SKIP, which were predicted to have large effects on χ2 period (Table S5); bru1, CG32052, CG6123, flw, and Sh were predicted to have large effects on rhythmicity index. Although they were predicted to affect rhythmicity index, CG32052 and CG6123 had effects on χ2 period, with an increase of 3.52 and 1.63 h over the control period, respectively (Table 1 and Table S17). An attempt to excise the Minos element from CG32052 revealed that the long-period phenotype did not map to this gene. bru1 had a significant effect on rhythmicity index, differing − 0.0831 from the control. Prosap and SKIP altered rhythmicity index, though they were predicted to affect circadian period. Thus, mutations in four additional candidate genes impacted circadian period and rhythmicity index.
Discussion
Here we assessed circadian period (Ʈ) and rhythmicity index (RI) in a natural population of flies. The Ʈ estimates calculated by MESA were derived from the highest peak in the power spectral density for each fly, and the low range of Ʈ estimated with MESA did not agree with the low range of χ2 period calculations (see Fig. 1). Low periods of ~ 15 h are rare but have been observed in an ultrafast allele of period in flies (Konopka et al. 1994). It has been suggested the low period values calculated using MESA may reflect ultradian rhythms (Dowse and Ringo 1987, 1989); alternatively, recent work proposes that high peaks in the power spectral density corresponding to low circadian periods may actually be harmonics of the true circadian period (Lazopulo and Syed 2016). However, both of these methods enabled us to identify candidate genes that could be verified with further testing.
If we considered the more narrowly distributed χ2 to represent the true extent of the variation in the DGRP, it would still be far more variable than estimates of circadian period in humans using a forced desynchrony protocol, which have coefficients of variation in the range of 0.54–0.58% (Czeisler et al. 1999). However, chronotype, a related measure, has a heritability of 0.12–0.47 in humans [reviewed in (Kalmbach et al. 2016)] (Toomey et al. 2015; Jones et al. 2016; Lane et al. 2016); bed time, a similar measure, has a heritability of 0.22 (Gottlieb et al. 2007). Furthermore, heritability for circadian period in mice ranges from 0.21 to 0.55 (Hofstetter et al. 1995, 1999). These studies indicate a strong influence of both genetic and environmental factors on circadian period. Similarly, we found relatively low heritability estimates for circadian period in this study of flies, as observed previously (Emery et al. 1995).
Surveys of natural populations of Drosophila have identified latitudinal clines (Kyriacou et al. 2008; Rand et al. 2010; Sawyer et al. 1997) in polymorphic variants of canonical clock genes. These variants are associated with other traits that have a clear link to environmental conditions, particularly temperature: diapause, thermal tolerance, and eclosion (Sawyer et al. 1997; Rand et al. 2010; Kyriacou et al. 2008; Pegoraro et al. 2014; Tauber et al. 2007). Thus, polymorphisms influencing circadian period may be maintained as the result of acclimatization (Kyriacou et al. 2008).
In addition to circadian period, RI was highly variable, ranging from 0.05 to 0.55 in the DGRP. Interestingly, seven lines were classified as arrhythmic in one sex: one line had arrhythmic males, while six lines had arrhythmic females. For the most part, the RI in the opposite sex of these lines was not high (≤ 0.17), indicating that both sexes were at best weakly rhythmic. Given the importance of the circadian clock in regulating biological processes, it is puzzling why arrhythmicity would be maintained in nature. It may be that less robust (i.e., more variable) circadian behavior gives these animals an adaptive advantage. Such an advantage has been observed explicitly in cyanobacteria, where arrhythmic strains had higher growth in constant conditions compared to strains with a functional clock (Woelfle et al. 2004). However, strains with functional clocks were more fit in changing circadian conditions than arrhythmic strains (Woelfle et al. 2004; Ouyang et al. 1998). Wildtype flies produced more eggs in constant light conditions, which produce arrhythmic activity patterns; but constant light also reduced lifespan (Sheeba et al. 2000). Thus, the fitness advantage of variable circadian behavior depends heavily on the environmental conditions encountered. Future work will focus on the discovery of trade-offs between these circadian behaviors and other traits as pleiotropic gene action may be the basis for the maintenance of these differences (Carbone et al. 2006).
Our GWAS identified new candidate genes for circadian behavior. Interestingly, no canonical clock genes were identified for Ʈ, while only one polymorphism in Pdp1 was associated with RI. Only ten of the genes we identified here had been associated with circadian rhythms previously. This is analogous to linkage and genome-wide association studies in chronotype and sleep in humans (Pagani et al. 2016; Jones et al. 2016; Lane et al. 2016; Hu et al. 2016) and in circadian behavior in mice (Shimomura et al. 2001), where few canonical clock genes were implicated. Further, 24.7% of the associated polymorphisms were intergenic, suggesting that circadian rhythms may be modified via transcriptional regulation, if the polymorphisms fall in enhancer or promoter regions (Massouras et al. 2012; Heap et al. 2009). Comparison of this study to a previous study of sleep in the same population showed some overlap between genes associated with both suites of traits, and these comparisons revealed genes largely involved in developmental processes, including neuronal development. Interestingly, a single candidate gene, raw, overlaps with a homolog (RNASEL) identified in human GWAS studies of chronotype (Hu et al. 2016; Lane et al. 2016; Jones et al. 2016), and 16 additional genes have homologs identified in human GWAS studies of sleep (Spada et al. 2016; Cade et al. 2016) (Table 2). The homology results should be interpreted with caution but suggest a conserved architecture between flies and humans. Thus, some of the genes we identified have been implicated in mammalian studies of related circadian traits and of sleep.
In this study, we identified a line with a very long circadian period, DGRP_892. Both males and females of this line have circadian periods that average 31 h. Not many other observations of long circadian periods in this range in flies have been made. Konopka and Benzer described an allele of per that resulted in a 28.5-h circadian period (Konopka and Benzer 1971); Smith et al. (2008) found 33-h periods resulting from a dominant negative allele of the casein kinase α subunit; Rothenfluh et al. (2000) identified long-period mutations in timeless created via EMS mutagenesis; and Blau and Young noted a 28-h period when vrille expression was altered (Blau and Young 1999). We measured gene expression in this line for several candidate genes on the hypothesis that polymorphic variants in and around the candidate gene might alter gene expression (Heap et al. 2009; Massouras et al. 2012). per transcript levels are known to be delayed in perL mutants (Hardin et al. 1990), which have a long circadian period. We did not find the amino acid substitution of per reported to cause the long period (Baylies et al. 1987) in DGRP_892. Nor were there any obvious variants private to DGRP_892 that could affect the clock; yet gene expression in per was altered in these animals. Pdp1 also had altered transcriptional oscillations in DGRP_892, though the effects were smaller. Delays in Pdp1 expression are positively correlated with altered circadian period (Hardin et al. 1990; Muskus et al. 2007; Chen et al. 2013). These observations suggest differential regulation of both the positive and the negative feedback loop of the circadian clock in DGRP_892, though the effect on the negative feedback loop was more pronounced.
We examined gene expression in 26 candidate genes identified in this study as possible mediators of canonical clock gene expression differences in DGRP_892. We selected genes with candidate polymorphisms in coding as well as non-coding regions. We observed altered gene expression in nine candidate genes (AGO2, CG42321, Dop1R2, GlcT-1, GluRIIA, Mdr65, Rae1, Tep4, and tnc). With the exception of Rae1 and Mdr65, we also observed changes in circadian behavior in mutants and RNAi knockdowns of these genes. We describe the functions of these genes briefly here. A role for AGO2 in mammalian circadian rhythms has been reported (Lee et al. 2013). CG42321 is a predicted gene with an unknown function but is a strong candidate for circadian period in this study. Previous work identified CG42321 as a target of Clk (Abruzzi et al. 2011). A Translating Ribosome Affinity Purification study in fly clock cells identified Cpr62Ba as cycling (Huang et al. 2013). Dop1R2 has a role in neuronal inhibition of sleep-promoting neurons in the dorsal fan-shaped body (Pimentel et al. 2016); here we found that Dop1R2 affected rhythmicity index. GlcT-1 has a role in developmental apoptosis (Kohyama-Koganeya et al. 2004), and is predicted to be cycling in mammals (Hughes et al. 2010). Four of the candidate genes—GluRIIA, Mdr65, Rae1 and Tep4—have mammalian homologs which have been reported to be cycling in the SCN and other specific mammalian organs (Hughes et al. 2010). GluRIIA encodes a glutamate receptor capable of calcium ion transfer (Han et al. 2015) predicted to cycle in peripheral mouse organs (Hughes et al. 2010). Mdr65 is a gene involved in the transport of xenobiotic substances across the fly’s neural protective barrier (Mayer et al. 2009). Rae1 functions to limit synaptic growth (Tian et al. 2011). In addition to cycling in mammals, Tep4 is a Drosophila immune response gene that has been previously shown to be regulated in a circadian manner (Ceriani et al. 2002). Though a direct role of tenectin (tnc) in circadian rhythms is unknown in Drosophila, its mammalian homolog RTN3 (reticulon 3) is reported to cycle in the mouse liver (Hughes et al. 2010) and SCN (Panda et al. 2002). tnc expression is regulated by ecdysone and it encodes an integrin ligand involved in wing morphogenesis; it is also expressed in Drosophila CNS (Fraichard et al. 2006, 2010).
Nine genes had differential gene expression in DGRP_892 relative to Canton-S B, and several of these (GlcT-1, Mdr65, and Tep4; see Table S9) were predicted to interact with canonical circadian clock genes. Yet few of the genes we tested had significant differences in transcript abundance in a 24-h period. Rae1 was the only gene with significant differences in gene expression at different times. One potential reason for this is that we only assayed six timepoints over the circadian day; additional timepoints may help resolve these differences. In addition, more replication would improve these measures, as the statistical power for measurements of gene expression under different environmental conditions is low relative to differences detectable by genotype (Lin et al. 2016b). Alternatively, transcriptional cycling of molecules may not be required to modify circadian behavior (Ray and Reddy 2016); the genotype-specific differences in expression would then be relevant to the phenotypic differences we observed. Finally, studies have reported that synonymous SNPs may lead to altered protein translation kinetics and thereby altered protein conformation rather than affecting transcription (Kimchi-Sarfaty et al. 2007; Komar et al. 1999). It would be worth exploring whether these unique synonymous variants affect protein functions which in turn result in aberrant circadian phenotypes in DGRP_892.
We also tested Minos mutants in 14 genes predicted to have large effect sizes on χ2 period and rhythmicity index. CG6123, a gene with unknown function, were predicted to affect rhythmicity index but instead increased period. Likewise, Prosap, which affects the number of synaptic boutons in the neuro-muscular junction (Harris et al. 2016) and SKIP, a potassium-channel interacting protein with a role in olfaction (Tunstall et al. 2012), affected rhythmicity index; however, they were predicted to have effects on period. Only bru1 affected rhythmicity index as predicted. These pleiotropic mutational effects have been previously observed for sleep as well (Wu et al. 2018; Harbison et al. 2013). This may indicate that polymorphic variants have strong context-dependent effects on sleep and circadian rhythms (Chandler et al. 2017). Alternatively, an improvement in phenotypic modeling may yield more accurate predictions.
In this study, we have identified candidate polymorphisms associated with circadian behavior. Although we have not yet established that these variants are causal, our future goal is to elucidate mechanisms leading to phenotypic variance. This work is part of a systems genetics approach (Ray and Reddy 2016) and has identified previously unappreciated polymorphisms for circadian behavior.
Materials and methods
Quantitative circadian rhythm phenotypes
We measured circadian phenotypes on 167 lines of the Drosophila Genetic Reference Panel (DGRP), a collection of inbred lines derived from wild-caught flies (Mackay et al. 2012; Huang et al. 2014). Flies were maintained on standard culture medium (http://flystocks.bio.indiana.edu/Fly_Work/media-recipes/bloomfood) at 25 °C and 60–65% relative humidity, under a 12-h light: dark (LD) cycle until circadian measurements were made. We recorded fourteen continuous days of activity in constant darkness (DD) using the Drosophila Activity Monitoring System (Trikinetics, Waltham, MA). Activity counts were sampled in 1-min intervals. Flies were placed on 5% sucrose, 1.5% agar food for the duration of the circadian measurements as standard culture medium becomes too dry for the flies to eat after 14 days. The DGRP lines were randomly divided into four equal blocks. We replicated circadian measurements three times for each block of lines. Replicate measurements were conducted at 2-week intervals. Eight flies of each sex per line were measured in each replicate, resulting in circadian measurements for 24 flies per sex per line, or 48 flies per line. We assumed that we would be able to detect differences of 1 h (for MESA and χ2 period) and 0.1 for RI at 80% statistical power (in practice, we were able to detect differences in period as low as 0.765 and 0.359 h for MESA and χ2 period, and differences as low as 0.045 for RI at 80% power) (Sokal and Rohlf 1995). Eight male and eight female w1118; Canton-S B flies were measured in each replicate as a control. Although the effects of social exposure on circadian rhythms are not known, both social exposure and mating status affect sleep in flies (Ganguly-Fitzgerald et al. 2006; Isaac et al. 2010); we therefore collected virgin males and females from each line and housed them at 30 flies per same-sex vial for 4 days prior to making the activity measurements. We discarded the measures from flies that did not live through the recording period; we also discarded the first day of recording as the flies were recovering from CO2 exposure. All other measurements, including any outliers, were retained in the analysis.
We calculated the rhythmicity index (RI) and period length (Ʈ) for the DGRP population. To make these calculations, we first summed the activity counts into 30-min bins, which reduces the potential for spurious patterns due solely to the choice of bin size (Dowse and Ringo 1994). To determine RI, we calculated the autocorrelelogram for each fly using the equation
where \({r_k}\) is the autocorrelation coefficient at lag \(k\), \({x_t}\) is the activity level at time \(t\), \({x_{t+k}}\) is the activity level at time \(t+k\), and \(\overline {x}\) is the mean activity level for the fly (Chatfield 2003). We defined RI as the correlation coefficient \({r_k}\) of the third highest peak in the autocorrelelogram (Levine et al. 2002). In addition to a sinusoidal component, the autocorrelelograms of some flies exhibit a linear trend, which can skew the rhythmicity index (Levine et al. 2002). To account for this linear trend, we first fitted a regression line to each autocorrelelogram. If the autocorrelelogram had a nominally significant linear component (P < 0.05), we adjusted the RI for that trend by subtracting the calculated RI of the predicted regression line. We classified flies as rhythmic or arrhythmic using the 95% confidence interval \(\frac{2}{{\sqrt N }}\), where \(N\) is the number of activity measurements (Levine et al. 2002; Chatfield 2003). Since we summed our activity measures into 30-min bins, N = 672 and the 95% confidence interval for the autocorrelation coefficient was \({r_k}\) = 0.0772. Flies with an RI below this value were classified as arrhythmic.
We used two methods to calculate Ʈ: Maximum Entropy Spectral Analysis (MESA) (Burg 1967), and the chi-squared periodogram (Enright 1965). MESA analysis assumes that the time series data is composed of an autoregressive function \(X\left( t \right)\) and a stochastic or noise function \({Z_t}\) (Levine et al. 2002; Burg 1967). At a given time \(t\), \(X(t)\) is
where the \(a\) coefficients are estimated from the data, \(n\) is number of terms, \(t\) is time, and \(Z\) is the stochastic function. A frequency-based spectrum \(S(\omega )\) can then be calculated via Fourier transform from the equation
where \(P\) is the output power or spectral density, \(p\) is the number of coefficients, and \(i=\sqrt { - 1}\) (Levine et al. 2002; Burg 1967; Dowse 2007, 2009; Dowse and Ringo 1989). Circadian period can then be estimated as the time at which the maximum spectral density peak occurs (Dowse and Ringo 1989). We used BATCHMES, a freely available program, to implement MESA [H. Dowse, personal communication, and (Dowse 2007)].
Furthermore, power spectral density graphs were examined for each rhythmic fly; for most flies, the spectral density contained a single dominant peak. In the graphs for some flies, we did not observe an interpretable peak. These calculations for these flies were removed from further analysis.
We also calculated period using the chi-squared periodogram. This method first assumes a period \(P\) for the data. If the activity data are broken up into units of period \(P\), then the differences between these units can be used to calculate a χ2 statistic to determine how well the assumed period fits the actual data. We used an in-house C# program (R. Sean Barnes) to estimate the \({Q_p}\) statistic using the equation
where \({X_i}~\) is the activity counts for a given unit of time in a data set of \(N\) values, \(M\) is the mean of all \(N\) values, and \({M_h}\) is the mean activity counts for each time unit for \(K\) values (Refinetti 2004; Enright 1965). We calculated \({Q_p}\) for a range of possible periods\(~P\), from 12 h to 32 h (Refinetti 2004; Enright 1965). \({Q_p}\) has a χ2 probability distribution with \(P - 1\) degrees of freedom (Refinetti 2004). If \({Q_p}~\)was not significant according to the χ2 distribution, we did not estimate the period; if it was, we used the highest \({Q_p}\) value to estimate circadian period (Refinetti 2004). If we judged the fly to be arrhythmic using the rhythmicity index criteria, we did not estimate circadian period.
Quantitative genetic analyses
We partitioned the variance in each circadian parameter using the ANOVA model: Y = µ + B + S + L(B) + S × L(B) + R(B) + S × R(B) + R × L(B) + S × R × L(B) + ε, where L (line), B (block) and R (replicate) are random effects, S (sex) is a fixed effect, and ε is the error variance. We used reduced models to partition the variance for each sex separately. We estimated variance components using the restricted maximum likelihood (REML) method. We calculated broad sense heritability as H2 = (σ2L + σ2SL)/(σ2L + σ2SL + σ2E) for sexes combined, where σ2L is the variance component among lines, σ2SL is the line-by-sex variance component, and σ2E is the sum of all other sources of variation. We used H2 = σ2L/(σ2L + σ2E) to estimate broad-sense heritability for sexes separately. We calculated the cross-sex genetic correlation rMF as σ2L/√(σ2LM × σ2LF), where σ2L is the variance component among lines for sexes combined, σ2LM is the variance component among lines for males separately, and σ2LF is the variance component among lines for females separately. We calculated the genetic correlation rG between circadian and sleep traits as rG = cov12/√(σL12 × σL22) (Falconer and Mackay 1996), where cov12 is the covariance between traits 1 and 2, and σL12 and σL22 are the among-line variances for traits 1 and 2, respectively.
Genotype-phenotype associations
We associated the line mean of each circadian rhythm parameter with all 1,920,276 segregating sites in the DGRP having a minor allele frequency of 0.05 or greater using the DGRP2 web-based analysis (http://dgrp2.gnets.ncsu.edu) (Huang et al. 2014), which implements the FaST-LMM algorithm (Lippert et al. 2011). This analysis first adjusts the phenotypic line means for the effects of Wolbachia pipientis infection and major chromosomal inversions [In(2L)t, In(2R)NS, In(3R)P, In(3R)K, and In(3R)Mo] where present. The DGRP2 web-based analysis fits a linear mixed model that incorporates any cryptic genetic relatedness present in the lines. The linear mixed model is \(y={\mathbf{X}}b+{\mathbf{Z}}u+e\), where \(y\) is the phenotypic line mean adjusted for effects of Wolbachia infection and segregating polymorphic inversions, \({\mathbf{X}}\) represents the covariance relationship matrix for the fixed variant effect \(b\), \({\mathbf{Z}}\) is the incidence matrix for the random polygenic effect \(u\), and \(e\) is the residual effect. False discovery rate (FDR) corrections for multiple tests were computed using the method of Benjamini and Hochberg (Benjamini and Hochberg 1995). As line DGRP_892 had a very long period, we removed DGRP_892 from the GWA of circadian period, as well as the epistatic analysis of circadian period described below since leaving it in would violate assumptions of normality (Mackay et al. 2012). The power to detect significant associations varies with normalized effect size (a/σG) and minor allele frequency. We expected to have the power to detect normalized effect sizes of 0.5 or greater where minor alleles were common, i.e., the frequency was close to 0.5 (Mackay et al. 2009). We used DIOPT to identify human homologs of candidate genes identified in the genome-wide analysis (Hu et al. 2011).
Pairwise epistatic associations
All possible variant pairs having minor allele frequencies of 0.15 or greater were tested for epistatic interactions. Pairs were pruned for high linkage disequilibrium (r2 ≥ 0.8) in 100-bp windows shifted in 10-bp increments, leaving 666,202 variants for the analysis. We applied the model Y = µ + M1 + M2 + M1 × M2 + ε, where M1 is the first marker considered, M2 is the second marker considered, and ε is the error term. Phenotypes were adjusted for the presence of Wolbachia and inversions as well as the top 11 genotypic principal components. We ran the model using FastEpistasis 2.03 (Schupbach et al. 2010), refined the results and obtained P-values using the F distribution with appropriate degrees of freedom, and retained those interactions (i.e., M1 × M2 terms) having a P-value of 1 × 10− 7 or less. These analyses were performed separately for males and females.
Assessment of selected candidate genes in a long-period line
One of the lines, DGRP_892, had an unusually long circadian period. We characterized gene expression over time in the heads of males and females (Hardin et al. 1990) from this line for 29 candidate genes implicated by the GWA, and under two lighting conditions. We also examined gene expression changes over time in this line for three canonical clock genes: per, tim, and Pdp1. w1118;Canton-S B (hereafter referred to as Canton-S B) was used as a control for comparison to DGRP_892 as it had normal circadian behavior (Fig. S7).
Sample preparation
DGRP_892 and Canton-S B control flies were maintained under standard culture medium, 25 °C, 60–75% relative humidity and lighting conditions (LD) as outlined above. After eclosion, 30 virgin males and 30 virgin females were collected and maintained in sex-separate vials for 3 days. Gene expression was measured in fly heads for two separate lighting conditions: constant darkness (DD), and LD. For the DD condition, flies were first entrained in an incubator in LD for 3 days and then transferred to DD. On the 3rd day of DD, flies were flash-frozen on dry ice every 4 h starting at circadian time (CT) CT02 and stored at -80 °C until further processing. For the LD assay flies were collected and flash-frozen in − 80 °C every 4 h starting at Zeitgeber time (ZT) ZT02 after 6 days in LD. Frozen flies were then transferred to dry ice to remove the heads from the bodies. We replicated this procedure three times to produce 144 samples (2 conditions, LD and DD; 3 biological replicates, 2 sexes, 2 genotypes, and 6 time points).
RNA extraction
Thirty flies per sample were decapitated on dry ice by gently tapping on the micro centrifuge tubes; heads were transferred to Omni-ruptor tubes (Omni International, Kennesaw, GA) with 4 RNase/DNase-free metal beads (Omni International, Kennesaw, GA) and homogenized in 125 µl of Trizol reagent (Thermo Fisher Scientific, Grand Island, NY) using the Omni-bead Ruptor (Omni International, Kennesaw, GA). The tubes were centrifuged briefly after homogenization and an additional 375 µl of Trizol was added to make the total volume of 500 µl; the mixture was incubated at room temperature for 5 min. Next, 100 µl of chloroform (Mallinckrodt Baker, Center Valley, PA) was added to each tube and mixed for 15 s by shaking. The tubes were incubated at room temperature for 3 min and then centrifuged at 4 °C, 13,000 rpm for 15 min. The aqueous phase was transferred to new Eppendorf tubes and 20 µl of 3M sodium acetate (Quality Biological, Gaithersburg, MD) was added to each sample. Afterward, to eliminate proteins from nucleic acid samples, 250 µl of phenol–chloroform (Sigma, St. Louis, MO) was added and the samples were centrifuged at 4 °C, 13,000 rpm for 15 min. The aqueous phase was transferred to new tubes and 3 µl glycogen (Qiagen, Valencia, CA) and 300 µl of isopropanol (Mallinckrodt Baker, Center Valley, PA) were added followed by centrifugation at 4 °C, 13,000 rpm for 30 min. The supernatants were discarded while avoiding the pellet. The pellets were washed twice in 75% ethanol, centrifuging at 4 °C, 7500 rpm for 5 min. Pellets were air dried for 10–15 min and dissolved in 87.5 µl of RNase-free water (Quality Biological, Gaithersburg, MD). DNase treatment was performed on RNA samples to eliminate any genomic DNA contamination using 10 µl Buffer RDD (Qiagen, Valencia, CA) and 2.5 µl DNase-I stock solution (Qiagen, Valencia, CA) followed by incubation at room temperature for 10 min. The RNA samples were stored at − 80 °C until further processing. RNA was purified using the RNAeasy Kit (Qiagen, Valencia, CA), per the manufacturer’s instructions.
Single-stranded cDNA was made from total RNA using the High-Capacity cDNA Reverse Transcription Kit per the manufacturer’s instructions (Thermo Fisher Scientific, Grand Island, NY). A total of 250 ng RNA was used for each 20 µl reaction. The reverse transcription reactions were performed on a PTC-250 thermocycler (MJ Research, Foster City, CA).
Real-time quantitative PCR
The cDNA samples were diluted by 20 × for all unknown samples. To create a standard curve, equal portions of each sample were combined and diluted by 5 ×, 20 ×, 100 ×, and 500 ×. PCR reactions were carried out in 384-well plates (Thermo Fisher Scientific, Grand Island, NY) using SYBR Green PCR Master Mix (Thermo Fisher Scientific, Grand Island, NY) according to the manufacturer’s instructions. The PCR reactions were performed on an Applied Biosystems QuantStudio™ 12K Flex Real-Time PCR System using the manufacturer’s protocol (Thermo-Fisher Scientific, Grand Island, NY). All primers were designed using Oligo Analyzer Software 3.1 (Integrated DNA Technologies, Coralville, Iowa). Actin-5c was used as an internal control for the total RNA content in each sample. Primer sequence lengths ranged from 19 to 22 bases and were synthesized by IDTDNA (Integrated DNA technologies, Coralville, Iowa). All primer sequences assayed are provided in Table S9.
Quality control and normalization
Gene expression was estimated from the raw data obtained from QuantStudio™ 12K Flex Real-Time PCR System using standard curves. We first checked the r2 values of the standard curve by fitting the linear regression of the log transformation of the quantity and the cycle threshold (CT) values. Four different amounts of total RNA (1000, 200, 40 and 8 ng) were used as the known quantities in the standard curve. We found that r2 value was low for some genes and could be improved after removing the smallest quantity of RNA. To be consistent, therefore, we used the highest three values for quantity in the standard curve for all genes. None of the samples were estimated to have quantities below 40 ng.
Each plate had two technical replicates for each RNA sample under each light/sex/genotype/candidate gene condition. After fitting the standard curve, we averaged the two technical replicates for use in subsequent data analyses. As the samples were necessarily distributed across several plates, we used Quantile (Q) normalization (Bolstad et al. 2003) to correct the bias caused by potential plate effects (Mar et al. 2009). In addition, we also normalized the data using the quantity of Actin-5c in each genotype/sex/rep/condition as a reference (Ling and Salvaterra 2011; Ponton et al. 2011) by dividing the Q-normalized quantities of the target genes by the Q-normalized quantities of Actin-5c, which did not significantly vary over time or genotype (Fig. S8). The resulting ratios were used in the data analysis detailed below.
Candidate gene expression analysis
We examined the differences in gene expression using the ANOVA model Y = µ + L + S + Tr + T + L × S + L × Tr + L × T + S × Tr + S × T + Tr × T + L × S × Tr + L × S × T + L × Tr × T + S × Tr × T + L × Tr × S × T + ε, where Y is gene expression, L and S are as defined previously, Tr is lighting condition (LD or DD), T is time, and ε is the error term. We used reduced models to examine gene expression differences in Canton-S B and DGRP_892 by lighting condition, sex, and time. To determine whether the transcripts within a given line and lighting condition might be cycling over the circadian day, we applied the reduced model Y = µ + S + T + S + T × S + ε and used post-hoc Tukey analysis to determine which time points were significantly different from one another. To control for multiple tests, we calculated false-discovery rates using the method of Benjamini and Hochberg (Benjamini and Hochberg 1995). Genes having significant differential expression over time were then analyzed using JTK_CYCLE (Hughes et al. 2010). JTK_CYCLE applies the Jonckheere-Terpstra-Kendall algorithm to a range of potential circadian period lengths assuming a symmetrical cosine distribution and reports the period of the data, if rhythmic, and a P-value (Hughes et al. 2010).
Verification of candidate genes
We tested circadian behavior in P-element mutants and RNAi knockdown flies in 10 candidate genes identified by the GWAS which either had significant differences in gene expression between DGRP_892 and Canton-S B or exhibited evidence of transcriptional cycling. We tested AGO2, CG42321, Cpr62Ba, Dop1R2, GlcT-1, GluRIIA, Mdr65, Rae1, Tep4, and tnc (Table S14). We tested available Minos elements (Mi{ET1}, Mi{y[+mDint2]=MIC}) (Bellen et al. 2011; Venken et al. 2011), P-element insertion lines (Bellen et al. 2011, 2004), imprecise excisions (Hain et al. 2010), null alleles (Petersen et al. 1997), and RNAi knockdown lines (Ni et al. 2011; Perkins et al. 2015). Lines with Mi{ET1} insertions were created in an isogenic w1118 background (w1118[5905]) (Bellen et al. 2011) ; hence we used this background as a control. We used a y[1] w[1] [1495] control with Mi{y[+mDint2]=MIC} and all other P-element insertions. We used w1118 [3605] as a control for the AGO2 and GluRIIA mutants; these deletions were not viable as homozygotes and were tested as heterozygotes. The TRiP RNAi constructs were derived from two lines: y[1] v[1]; P{y[+t7.7]=CaryP}attP40 and y[1] v[1]; P{y[+t7.7]=CaryP}attP2. If the RNAi transgene was inserted on the 2nd chromosome attP40 site, we used y[1] v[1]; P{y[+t7.7]=CaryP}attP40 (36304) as a control. Likewise, if the RNAi transgene was inserted on the 3rd chromosome attP2 site, we used P{y[+t7.7]=CaryP}attP2 (36303) as a control (Perkins et al. 2015). All RNAi lines and their respective controls were crossed with Pdf-GAL4 and tim-GAL4 driver lines (Kaneko and Hall 2000). The Pdf-GAL4 line drives gene expression in the ventrolateral neurons of the brain (Renn et al. 1999). The tim-GAL4 driver drives tim expression in the ventrolateral neurons as well as additional neuronal subsets (DN1, DN2, and DN3 neurons as well as dorsolateral neurons) (Kaneko and Hall 2000). Note that the cross between tim-GAL4 and UAS-Rae1-RNAi was lethal.
We also tested available Minos elements Mi{ET1} for candidate genes having large predicted combined-sex effect sizes for χ2 period and RI: bru1, CG11073, CG13243, CG17839, CG32052, CG34355, CG42672, CG6123, flw, Mp, Prosap, Ptp99A, sano, Sh, and SKIP (Table S14). The w1118[5905] line was used as the control for all mutations. Note that the sano insertion was tested as a heterozygote balanced over CyO as homozygous mutants were lethal.
Flies were maintained under standard culture (cornmeal-molasses-agar medium, 25 °C, 60–75% relative humidity) and lighting conditions (12-h light: dark cycle) (LD). Five virgin male and five females were used for setting up cultures in an LD incubator. For RNAi knockdowns, five male flies containing UAS-constructs were crossed with five GAL4 females. Twenty virgin males and twenty virgin females were collected and maintained in sex-separate vials for 3 days at LD. Individual flies were then transferred to DAM2 monitors (Trikinetics, Waltham, MA) for for 4 days in LD followed by 7 days in DD to assess the entrainment and free-running behavior. Flies were set up on 5% sucrose, 2% agar food. Sixteen flies per sex per line were tested, for a total of 32 flies per line. We then calculated χ2 and RI as indicated above. For all Minos element, P-element, and deletion mutants, we analyzed the data using the ANOVA model Y = µ + G + S + G × S + ε, where G is genotype and S is sex. For the RNAi knockdown flies, we used the model Y = µ + GAL4 + UAS + S + GAL4 × UAS + GAL4 × S + UAS × S + GAL4 × UAS × S + ε, where GAL4 is the effect of the Pdf or tim GAL4 driver, UAS is the effect of the RNAi knockdown line, and S is sex. Mutant and RNAi knockdowns were compared to controls in a pairwise fashion using a post-hoc Tukey test.
References
Abruzzi KC, Rodriguez J, Menet JS et al (2011) Drosophila CLOCK target gene characterization: implications for circadian tissue-specific gene expression. Genes Dev 25:2374–2386
Albert FW, Kruglyak L (2015) The role of regulatory variation in complex traits and disease. Nat Rev Genet 16:197–212
Arbeitman MN, Furlong EE, Imam F et al (2002) Gene expression during the life cycle of Drosophila melanogaster. Science 297:2270–2275
Arya GH, Magwire MM, Huang W et al (2015) The genetic basis for variation in olfactory behavior in Drosophila melanogaster. Chem Senses 40:233–243
Ayroles JF, Carbone MA, Stone EA et al (2009) Systems genetics of complex traits in Drosophila melanogaster. Nat Genet 41:299–307
Baylies MK, Bargiello TA, Jackson FR et al (1987) Changes in abundance or structure of the per gene product can alter periodicity of the Drosophila clock. Nature 326:390–392
Bellen HJ, Levis RW, Liao G et al (2004) The BDGP gene disruption project: Single transposon insertions associated with 40% of Drosophila genes. Genetics 167:761–781
Bellen HJ, Levis RW, He Y et al (2011) The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics 188:731–743
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Blau J, Young MW (1999) Cycling vrille expression is required for a functional Drosophila clock. Cell 99:661–671
Boivin DB (2000) Influence of sleep-wake and circadian rhythm disturbances in psychiatric disorders. J Psychiatry Neurosci 25:446–458
Bolstad BM, Irizarry RA, Astrand M et al (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193
Borbely AA (1982) A two process model of sleep regulation. Hum Neurobiol 1:195–204
Brown JB, Boley N et al (2014) Diversity and dynamics of the Drosophila transcriptome. Nature 512:393–399
Bunney WE, Bunney BG (2000) Molecular clock genes in man and lower animals: Possible implications for circadian abnormalities in depression. Neuropsychopharmacology 22:335–345
Burg JP (1967) Maximum entropy spectral analysis. In: 37th annual international meeting: society of exploration geophysics
Cade BE, Chen H, Stilp AM et al (2016) Genetic associations with obstructive sleep apnea traits in Hispanic/Latino Americans. Am J Respir Crit Care Med 194:886–897
Carbone MA, Jordan KW, Lyman RF et al (2006) Phenotypic variation and natural selection at Catsup, a pleiotropic quantitative trait gene in Drosophila. Curr Biol 16:912–919
Ceriani MF, Hogenesch JB, Yanovsky M et al (2002) Genome-wide expression analysis in Drosophila reveals genes controlling circadian behavior. J Neurosci 22:9305–9319
Chandler CH, Chari S, Kowalski A et al (2017) How well do you know your mutation? Complex effects of genetic background on expressivity, complementation, and ordering of allelic effects. PLoS Genet 13:e1007075
Chatfield C (2003) The analysis of time series. CRC Press, Boca Raton
Chen Z, Yoo SH, Takahashi JS (2013) Small molecule modifiers of circadian clocks. Cell Mol Life Sci 70:2985–2998
Cirelli C, Bushey D, Hill S et al (2005) Reduced sleep in Drosophila Shaker mutants. Nature 434:1087–1092
Czeisler CA, Duffy JF, Shanahan TL et al (1999) Stability, precision, and near-24-hour period of the human circadian pacemaker. Science 284:2177–2181
Dembeck LM, Boroczky K, Huang W et al (2015a) Genetic architecture of natural variation in cuticular hydrocarbon composition in Drosophila melanogaster. Elife 4:4
Dembeck LM, Huang W, Magwire MM et al (2015b) Genetic architecture of abdominal pigmentation in Drosophila melanogaster. PLoS Genet 11:e1005163
Dowse HB (2007) Statistical analysis of biological rhythm data. Methods Mol Biol 362:29–45
Dowse HB (2009) Analyses for physiological and behavioral rhythmicity. Methods Enzymol 454(Pt A 454):141–174
Dowse HB, Ringo JM (1987) Further evidence that the circadian clock in Drosophila is a population of coupled ultradian oscillators. J Biol Rhythms 2:65–76
Dowse HB, Ringo JM (1989) The search for hidden periodicities in biological time-series revisited. J Theor Biol 139:487–515
Dowse HB, Ringo JM (1994) Summing locomotor activity data into ‘bins’: how to avoid artifact in spectral analysis. Biol Rhythm Res 25:2–14
Dunlap JC, Loros JJ, Colot HV et al (2007) A circadian clock in Neurospora: how genes and proteins cooperate to produce a sustained, entrainable, and compensated biological oscillator with a period of about a day. Cold Spring Harb Symp Quant Biol 72:57–68
Emery PT, Morgan E, Birley AJ (1994) An investigation of natural genetic variation in the circadian system of Drosophila melanogaster: rhythm characteristics and methods of quantification. Chronobiol Int 11:72–84
Emery PT, Birley AJ, Morgan E (1995) Investigation of natural genetic variation in the circadian system of Drosophila melanogaster. II. Biometrical analyses of locomotor activity rhythms recorded in constant darkness. Chronobiol Int 12:77–86
Enright JT (1965) The search for rhythmicity in biological time-series. J Theor Biol 8:426–468
Falconer DS, Mackay TF (1996) Introduction to quantitative genetics. Longman, Essex
Fernandez MP, Chu J, Villella A et al (2007) Impaired clock output by altered connectivity in the circadian network. Proc Natl Acad Sci USA 104:5650–5655
Fraichard S, Bouge AL, Chauvel I et al (2006) Tenectin, a novel extracellular matrix protein expressed during Drosophila melanogaster embryonic development. Gene Expr Patterns 6:772–776
Fraichard S, Bouge AL, Kendall T et al (2010) Tenectin is a novel alphaPS2betaPS integrin ligand required for wing morphogenesis and male genital looping in Drosophila. Dev Biol 340:504–517
Ganguly-Fitzgerald I, Donlea J, Shaw PJ (2006) Waking experience affects sleep need in Drosophila. Science 313:1775–1781
Garlapow ME, Huang W, Yarboro MT et al (2015) Quantitative genetics of food intake in Drosophila melanogaster. PLoS ONE 10:e0138129
Gottlieb DJ, O’Connor GT, Wilk JB (2007) Genome-wide association of sleep and circadian phenotypes. BMC Med Genet 8(Suppl):S1–S9
Grandin LD, Alloy LB, Abramson LY (2006) The social zeitgeber theory, circadian rhythms, and mood disorders: review and evaluation. Clin Psychol Rev 26:679–694
Grenier JK, Arguello JR, Moreira MC et al (2015) Global diversity lines-a five-continent reference panel of sequenced. G3 5:593–603
Grierson AB, Hickie IB, Naismith SL et al (2016) Circadian rhythmicity in emerging mood disorders: state or trait marker? Int J Bipolar Disord 4:3
Hain D, Bettencourt BR, Okamura K et al (2010) Natural variation of the amino-terminal glutamine-rich domain in Drosophila argonaute2 is not associated with developmental defects. PLoS ONE 5:e15264
Han TH, Dharkar P, Mayer ML et al (2015) Functional reconstitution of Drosophila melanogaster NMJ glutamate receptors. Proc Natl Acad Sci USA 112:6182–6187
Harbison ST, Chang S, Kamdar KP et al (2005) Quantitative genomics of starvation stress resistance in Drosophila. Genome Biol 6:R36
Harbison ST, McCoy LJ, Mackay TF (2013) Genome-wide association study of sleep in Drosophila melanogaster. BMC Genom 14:281
Hardin PE (2011) Molecular genetic analysis of circadian timekeeping in Drosophila. Adv Genet 74:141–173
Hardin PE, Hall JC, Rosbash M (1990) Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343:536–540
Harris KP, Akbergenova Y, Cho RW et al (2016) Shank modulates postsynaptic Wnt signaling to regulate synaptic development. J Neurosci 36:5820–5832
Heap GA, Trynka G, Jansen RC et al (2009) Complex nature of SNP genotype effects on gene expression in primary human leucocytes. BMC Med Genomics 2:1
Helfrich-Forster C, Nitabach MN, Holmes TC (2011) Insect circadian clock outputs. Essays Biochem 49:87–101
Hofstetter JR, Mayeda AR, Possidente B et al (1995) Quantitative trait loci (QTL) for circadian rhythms of locomotor activity in mice. Behav Genet 25:545–556
Hofstetter JR, Possidente B, Mayeda AR (1999) Provisional QTL for circadian period of wheel running in laboratory mice: quantitative genetics of period in RI mice. Chronobiol Int 16:269–279
Hu Y, Flockhart I, Vinayagam A et al (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform 12:357
Hu Y, Shmygelska A, Tran D et al (2016) GWAS of 89,283 individuals identifies genetic variants associated with self-reporting of being a morning person. Nat Commun 7:10448
Huang Y, Ainsley JA, Reijmers LG et al (2013) Translational profiling of clock cells reveals circadianly synchronized protein synthesis. PLoS Biol 11:e1001703
Huang W, Massouras A, Inoue Y et al (2014) Natural variation in genome architecture among 205 Drosophila melanogaster genetic reference panel lines. Genome Res 24:1193–1208
Huang W, Carbone MA, Magwire MM et al (2015) Genetic basis of transcriptome diversity in Drosophila melanogaster. Proc Natl Acad Sci USA 112:E6010–E6019
Hughes ME, Hogenesch JB, Kornacker K (2010) JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms 25:372–380
Hunter CM, Huang W, Mackay TF et al (2016) The genetic architecture of natural variation in recombination rate in Drosophila melanogaster. PLoS Genet 12:e1005951
Huylmans AK, Parsch J (2014) Population- and sex-biased gene expression in the excretion organs of Drosophila melanogaster. G3 4:2307–2315
Isaac RE, Li C, Leedale AE et al (2010) Drosophila male sex peptide inhibits siesta sleep and promotes locomotor activity in the post-mated female. Proc Biol Sci 277:65–70
Jin W, Riley RM, Wolfinger RD et al (2001) The contributions of sex, genotype, and age to transcriptional variance in Drosophila melanogaster. Nat Genet 29:389–395
Jones SE, Tyrrell J, Wood AR et al (2016) Genome-wide association analyses in 128,266 individuals identifies new morningness and sleep duration loci. PLoS Genet 12:e1006125
Jordan KW, Craver KL, Magwire MM et al (2012) Genome-wide association for sensitivity to chronic oxidative stress in Drosophila melanogaster. PLoS ONE 7:e38722
Kalmbach DA, Schneider LD, Cheung J et al (2016) Genetic basis of chronotype in humans: Insights from three landmark GWAS. Sleep. https://doi.org/10.1093/sleep/zsw1048
Kaneko M, Hall JC (2000) Neuroanatomy of cells expressing clock genes in Drosophila: transgenic manipulation of the period and timeless genes to mark the perikarya of circadian pacemaker neurons and their projections. J Comp Neurol 422:66–94
Kimchi-Sarfaty C, Oh JM, Kim IW et al (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315:525–528
King E, Merkes CM, McNeil CL et al (2012a) Genetic dissection of a model complex trait using the Drosophila synthetic population resource. Genome Res 22:1558–1566
King EG, Macdonald SJ, Long AD (2012b) Properties and power of the Drosophila synthetic population resource for the routine dissection of complex traits. Genetics 191:935–949
Kloss B, Price JL, Saez L et al (1998) The Drosophila clock gene double-time encodes a protein closely related to human casein kinase 1e. Cell 94:97–107
Kohyama-Koganeya A, Sasamura T, Oshima E et al (2004) Drosophila glucosylceramide synthase: a negative regulator of cell death mediated by proapoptotic factors. J Biol Chem 279:35995–36002
Komar AA (2007) Genetics. SNPs, silent but not invisible. Science 315:466–467
Komar AA, Lesnik T, Reiss C (1999) Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett 462:387–391
Konopka RJ, Benzer S (1971) Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci USA 68:2112–2116
Konopka RJ, Hamblen-Coyle MJ, Jamison CF et al (1994) An ultrashort clock mutation at the period locus of Drosophila melanogaster that reveals some new features of the fly’s circadian system. J Biol Rhythms 9:189–216
Kumar S, Mohan A, Sharma VK (2005) Circadian dysfunction reduces lifespan in Drosophila melanogaster. Chronobiol Int 22:641–653
Kyriacou CP, Peixoto AA, Sandrelli F et al (2008) Clines in clock genes: fine-tuning circadian rhythms to the environment. Trends Genet 24:124–132
Lane JM, Vlasac I, Anderson SG et al (2016) Genome-wide association analysis identifies novel loci for chronotype in 100,420 individuals from the UK Biobank. Nat Commun 7:10889
Lazopulo A, Syed S (2016) A mathmatical model provides mechanistic links to temporal patterns in Drosophila daily activity. BMC Neurosci 17:14
Lee KH, Kim SH, Lee HR et al (2013) MicroRNA-185 oscillation controls circadian amplitude of mouse Cryptochrome 1 via translational regulation. Mol Biol Cell 24:2248–2255
Levine JD, Funes P, Dowse HB et al (2002) Signal analysis of behavioral and molecular cycles. BMC Neurosci 3:1
Lin Y, Chen ZX, Oliver B et al (2016a) Microenvironmental gene expression plasticity among individual Drosophila melanogaster. G3 (Bethesda) 6:4197–4210
Lin Y, Golovnina K, Chen Z-X et al (2016b) Comparison of normalization and differential expression analyses using RNA-Seq data from 726 individual Drosophila melanogaster. BMC Genomics 17:28
Ling D, Salvaterra PM (2011) Robust RT-qPCR data normalization: validation and selection of internal reference genes during post-experimental data analysis. PLoS ONE 6:e17762
Lippert C, Listgarten J, Liu Y et al (2011) FaST linear mixed models for genome-wide association studies. Nat Methods 8:833–835
Mackay TF, Lyman RF (2005) Drosophila bristles and the nature of quantitative genetic variation. Philos Trans R Soc Lond B Biol Sci 360:1513–1527
Mackay TF, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10:565–577
Mackay TF, Richards S, Stone EA et al (2012) The Drosophila melanogaster genetic reference panel. Nature 482:173–178
Magnusson A, Boivin D (2003) Seasonal affective disorder: an overview. Chronobiol Int 20:189–207
Mar JC, Kimura Y, Schroder K et al (2009) Data-driven normalization strategies for high-throughput quantitative RT-PCR. BMC Bioinform 10:110
Massouras A, Waszak SM, Albarca-Aguilera M et al (2012) Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet 8:e1003055
Mattis J, Sehgal A (2016) Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol Metab 27:192–203
Mayer F, Mayer N, Chinn L et al (2009) Evolutionary conservation of vertebrate blood-brain barrier chemoprotective mechanisms in Drosophila. J Neurosci 29:3538–3550
McHill AW, Wright KP Jr (2017) Role of sleep and circadian disruption on energy expenditure and in metabolic predisposition to human obesity and metabolic disease. Obes Rev 18(Suppl 1):15–24
McHill AW, Melanson EL, Higgins J et al (2014) Impact of circadian misalignment on energy metabolism during simulated nightshift work. Proc Natl Acad Sci USA 111:17302–17307
Mizrak D, Ruben M, Myers GN et al (2012) Electrical activity can impose time of day on the circadian transcriptome of pacemaker neurons. Curr Biol 22:1871–1880
Morgante F, Sorensen P, Sorensen DA et al (2015) Genetic architecture of micro-environmental plasticity in Drosophila melanogaster. Sci Rep 5:09785
Muskus MJ, Preuss F, Fan JY et al (2007) Drosophila DBT lacking protein kinase activity produces long-period and arrhythmic circadian behavioral and molecular rhythms. Mol Cell Biol 27:8049–8064
Nackley AG, Shabalina SA, Tchivileva IE et al (2006) Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314:1930–1933
Ni JQ, Zhou R, Czech B et al (2011) A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8:405–407
Nitta KR, Jolma A, Yin Y et al (2015) Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. Elife 4:e04837
Ouyang Y, Andersson CR, Kondo T et al (1998) Resonating circadian clocks enhance fitness in cyanobacteria. Proc Natl Acad Sci USA 95:8660–8664
Pagani L, St Clair PA, Teshiba TM et al (2016) Genetic contributions to circadian activity rhythm and sleep pattern phenotypes in pedigrees segregating for severe bipolar disorder. Proc Natl Acad Sci USA 113:E754–E761
Panda S, Hogenesch JB, Kay SA (2002) Circadian rhythms from flies to human. Nature 417:329–335
Parisi M, Nuttall R, Naiman D et al (2003) Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299:697–700
Patke A, Murphy PJ, Onat OE et al (2017) Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell 169:203–215, e213
Pegoraro M, Noreen S, Bhutani S et al (2014) Molecular evolution of a pervasive natural amino-acid substitution in Drosophila cryptochrome. PLoS ONE 9:e86483
Perkins LA, Holderbaum L, Tao R et al (2015) The transgenic RNAi project at Harvard Medical School: resources and validation. Genetics 201:843–852
Petersen SA, Fetter RD, Noordermeer JN et al (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19:1237–1248
Pimentel D, Donlea JM, Talbot CB et al (2016) Operation of a homeostatic sleep switch. Nature 536:333–337
Ponton F, Chapuis MP, Pernice M et al (2011) Evaluation of potential reference genes for reverse transcription-qPCR studies of physiological responses in Drosophila melanogaster. J Insect Physiol 57:840–850
Rand DM, Weinreich DM, Lerman D et al (2010) Three selections are better than one: clinal variation of thermal QTL from independent selection experiments in Drosophila. Evolution 64:2921–2934
Ranz JM, Castillo-Davis CI, Meiklejohn CD et al (2003) Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300:1742–1745
Ray S, Reddy AB (2016) Cross-talk between circadian clocks, sleep-wake cycles, and metabolic networks: dispelling the darkness. BioEssays 38:394–405
Refinetti R (2004) Non-stationary time series and the robustness of circadian rhythms. J Theor Biol 227:571–581
Renn SCP, Park JH, Rosbash M et al (1999) A pdf neuropeptide gene mutation and ablation of PDF neurons each cause severe abnormalities of behavioral circadian rhythms in Drosophila. Cell 99:791–802
Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418:935–941
Reutrakul S, Knutson KL (2015) Consequences of circadian disruption on cardiometabolic health. Sleep Med Clin 10:455–468
Reutrakul S, Van Cauter E (2014) Interactions between sleep, circadian function, and glucose metabolism: implications for risk and severity of diabetes. Ann N Y Acad Sci 1311:151–173
Rothenfluh A, Abodeely M, Price JL et al (2000) Isolation and analysis of six timeless alleles that cause short- or long-period circadian rhythms in Drosophila. Genetics 156:665–675
Sawyer LA, Hennessy JM, Peixoto AA et al (1997) Natural variation in a Drosophila clock gene and temperature compensation. Science 278:2117–2120
Scheer FA, Wright KP Jr, Kronauer RE et al (2007) Plasticity of the intrinsic period of the human circadian timing system. PLoS ONE 2:e721
Schupbach T, Xenarios I, Bergmann S et al (2010) FastEpistasis: a high performance computing solution for quantitative trait epistasis. Bioinformatics 26:1468–1469
Sehgal A, Rothenfluh-Hilfiker A, Hunter-Ensor M et al (1995) Rhythmic expression of timeless: a basis for promoting circadian cycles in period gene autoregulation. Science 270:808–810
Sheeba V, Sharma VK, Shubha K et al (2000) The effect of different light regimes on adult lifespan in Drosophila melanogaster is partly mediated through reproductive output. J Biol Rhythms 15:380–392
Shi M, Zheng X (2013) Interactions between the circadian clock and metabolism: there are good times and bad times. Acta Biochim Biophys Sin (Shanghai) 45:61–69
Shimomura K, Low-Zeddies SS, King DP et al (2001) Genome-wide epistatic interaction analysis reveals complex genetic determinants of circadian behavior in mice. Genome Res 11:959–980
Shorter J, Couch C, Huang W et al (2015) Genetic architecture of natural variation in Drosophila melanogaster aggressive behavior. Proc Natl Acad Sci USA 112:E3555–E3563
Smith EM, Lin J-M, Meissner R-A et al (2008) Dominant-negative CK2alpha induces potent effects on circadian rhythmicity. PLoS Genet 4:e12
Sokal RR, Rohlf J (1995) Biometry. W. H. Freeman and Company, New York
Spada J, Scholz M, Kirsten H et al (2016) Genome-wide association analysis of actigraphic sleep phenotypes in the LIFE Adult Study. J Sleep Res 25:690–701
Szular J, Sehadova H, Gentile C et al (2012) Rhodopsin 5- and Rhodopsin 6-mediated clock synchronization in Drosophila melanogaster is independent of retinal phospholipase C-beta signaling. J Biol Rhythms 27:25–36
Takahashi JS (1993) Circadian-clock regulation of gene expression. Curr Opin Genet Dev 3:301–309
Tauber E, Zordan M, Sandrelli F et al (2007) Natural selection favors a newly derived timeless allele in Drosophila melanogaster. Science 316:1895–1898
Tian X, Li J, Valakh V et al (2011) Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat Neurosci 14:1267–1275
Toomey R, Panizzon MS, Kremen WS et al (2015) A twin-study of genetic contributions to morningness-eveningness and depression. Chronobiol Int 32:303–309
Tunstall NE, Herr A, de Bruyne M et al (2012) A screen for genes expressed in the olfactory organs of Drosophila melanogaster identifies genes involved in olfactory behaviour. PLoS ONE 7:e35641
Ueda HR, Matsumoto A, Kawamura M et al (2002) Genome-wide transcriptional orchestration of circadian rhythms in Drosophila. J Biol Chem 277:14048–14052
Unkless RL, Rottschaefer SM, Lazzaro BP (2015) A genome-wide association study for nutritional indices in Drosophila. G3 5:417–425
Vaisnav M, Xing C, Ku H-C et al (2014) Genome-wide association analysis of radiation resistance in Drosophila melanogaster. PLoS ONE 9:e104858
Van Dongen HPA, Kerkhof GA, Dinges DF (2004) Human circadian rhythms. In: Sehgal A (ed) Molecular biology of circadian rhythms. Wiley, Hoboken
Venken KJ, Schulze KL, Haelterman NA et al (2011) MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods 8:737–743
Videnovic A, Zee PC (2015) Consequences of circadian disruption on neurologic health. Sleep Med Clin 10:469–480
Wang P, Lyman RF, Shabalina SA et al (2007) Association of polymorphisms in odorant-binding protein genes with variation in olfactory response to benzaldehyde in Drosophila. Genetics 177:1655–1665
Wayne ML, Telonis-Scott M, Bono LM et al (2007) Simpler mode of inheritance of transcriptional variation in male Drosophila melanogaster. Proc Natl Acad Sci USA 104:18577–18582
Weber AL, Khan GF, Magwire MM et al (2012) Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLoS ONE 7:e34745
Woelfle MA, Ouyang Y, Phanvijhitsiri K et al (2004) The adaptive value of circadian clocks: an experimental assessment in cyanobacteria. Curr Biol 14:1481–1486
Wu JF, Wang Y, Wu SH (2008) Two new clock proteins, LWD1 and LWD2, regulate Arabidopsis photoperiodic flowering. Plant Physiol 148:948–959
Wu KJ, Kumar S, Serrano Negron YL et al (2018) Genotype influences day-to-day variability in sleep in Drosophila melanogaster. Sleep. https://doi.org/10.1093/sleep/zs1205
Yu W, Hardin PE (2006) Circadian oscillators of Drosophila and mammals. J Cell Sci 119:4793–4795
Yuan Q, Joiner WJ, Sehgal A (2006) A sleep-promoting role for the Drosophila serotonin receptor 1A. Curr Biol 16:1051–1062
Zee PC, Badr MS, Kushida C et al (2014) Strategic opportunities in sleep and circadian research: report of the Joint Task Force of the Sleep Research Society and American Academy of Sleep Medicine. Sleep 37:219–227
Zhang Y, Sturgill D, Parisi M et al (2007) Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450:233–237
Zwarts L, Vanden Broeck L, Cappuyns E et al (2015) The genetic basis of natural variation in mushroom body size in Drosophila melanogaster. Nat Commun 6:10115
Acknowledgements
This work was supported by the Intramural Research Program of the NIH, National Heart Lung and Blood Institute to S. T. H. and NIH GM 45146 to T. F. C. M. The authors would like to thank Y. Lin for technical assistance.
Author information
Authors and Affiliations
Contributions
Conception and design: STH, TFCM. Acquisition of data: SK, LJM, KRS. Analysis and interpretation of data: STH, SK, WH, KRS. Drafting or revising the article: STH, SK, WH, KRS, TFCM.
Corresponding author
Ethics declarations
Conflict of interest
Susan T. Harbison, Shailesh Kumar, Wen Huang, Lenovia J. McCoy, Kirklin R. Smith, Trudy F. C. Mackay declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Additional information
Edited by Charalambos Kyriacou.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Harbison, S.T., Kumar, S., Huang, W. et al. Genome-Wide Association Study of Circadian Behavior in Drosophila melanogaster. Behav Genet 49, 60–82 (2019). https://doi.org/10.1007/s10519-018-9932-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-018-9932-0