
Abstract
Introduction
Data quality may impact the outcome of clinical trials; hence, there is a need to implement quality control strategies for the data collected. Traditional approaches to quality control have primarily used source data verification during on-site monitoring visits, but these approaches are hugely expensive as well as ineffective. There is growing interest in central statistical monitoring (CSM) as an effective way to ensure data quality and consistency in multicenter clinical trials.
Methods
CSM with SMART™ uses advanced statistical tools that help identify centers with atypical data patterns which might be the sign of an underlying quality issue. This approach was used to assess the quality and consistency of the data collected in the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial, involving 1495 patients across 232 centers in Japan.
Results
In the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial, very few atypical data patterns were found among the participating centers, and none of these patterns were deemed to be related to a quality issue that could significantly affect the outcome of the trial.
Discussion
CSM can be used to provide a check of the quality of the data from completed multicenter clinical trials before analysis, publication, and submission of the results to regulatory agencies. It can also form the basis of a risk-based monitoring strategy in ongoing multicenter trials. CSM aims at improving data quality in clinical trials while also reducing monitoring costs.
Similar content being viewed by others


Introduction
Data quality may impact the outcome of clinical trials; hence, there is a need to implement quality control strategies for the data collected. Until recently, data quality control was mainly implemented through extensive monitoring approaches, with on-site visits and exhaustive source data verification [1]. However, in a context where the complexity of clinical trials is continuously increasing, the limited efficiency and high cost of current practices have been questioned [2]. Instead of 100 % source data verification, the implementation of risk-based monitoring strategies is now recommended [3–6]. Risk-based monitoring suggests that monitoring should focus on “the most critical data elements and processes necessary to achieve the study objectives” [5, p. 5]. A diversification of monitoring methods is necessary to ensure trial validity [7]. In particular, central statistical monitoring (CSM) is specifically aimed at monitoring data quality over the course of the trial. CSM can also be used when a trial is completed, in order to provide a comprehensive assessment of data quality.
CSM methods have recently received increased attention in the clinical trial literature [8–12] as well as from regulatory agencies [5, 6]. The statistical concepts underlying CSM were proposed two decades ago to detect fraud [13–16], but there has since been a growing recognition that the same concepts can be extended to assess data quality in general [17].
In a nutshell, CSM is premised on the fact that in a well-conducted multicenter trial, the populations of patients, patterns of care, and data collection processes are similar across all centers; therefore, there should not be systematic center-to-center differences in the data. In such a situation, the differences observed in the values of any variable (such as clinical measurements, laboratory values, and reported adverse events) between centers should result only from systematic differences such as differences in patient populations, from the natural between-patient variability, and from the play of chance. The size and number of random differences between centers are expected to be reasonably small. If the data in a center differ significantly from those observed across the other centers, one may suspect that the difference is not simply due to the play of chance, and that there may be a cause for this difference. This cause has to be carefully investigated as it might be the sign of a quality issue (e.g., encoding problems, misunderstandings, inadequate trial conduct, errors, or fraud). It might also be the result of a population difference in the center, which is also an important aspect to document in the final analysis of the trial. To identify centers with unexpectedly different data, CSM relies on a number of statistical approaches. CSM uses the statistical distributions of all the variables in the clinical trial, so as to determine the “most critical data elements” on which risk-based monitoring suggests the monitoring should be focused.
CSM relies on three key ideas:
-
1.
All centers follow the same protocol and fill in the same case report form. Hence, the resulting data structure is the same for all centers, and the statistical distribution of the values collected for any given variable should not significantly differ from one center to another, in the absence of data issues.
-
2.
All variables are potentially indicative of the quality of the data collected in a center. Hence, CSM relies on powerful statistical methods that allow one to computationally investigate the database seen as a whole, without imposing any prior assumptions as to where risks may be present. A large number of generic statistical tests are performed so as to highlight issues in the values observed in the centers for all recorded variables. Of note, the analyses include all recorded variables, regardless of their meaning or importance.
-
3.
The data that have been collected in a center are compared with the data collected in all other centers. The amount of discrepancy is objectively and quantitatively evaluated through a large number of statistical tests.
In this article, we illustrate the use of CSM for a large phase III trial conducted in Japan, the Stomach Cancer Adjuvant Multi-institutional Trial Group (SAMIT) Trial, which included 1495 patients with locally advanced gastric cancer across 232 hospitals [18]. The main publication on this trial included the following statement [18, p. 889]: “Quality control was done with CluePoints (version 1.2). No atypical data patterns that would affect the analysis were found.” CluePoints is a company that provides a central monitoring platform aimed at identifying centers with atypical data patterns in a clinical trial, and at exploring the reasons for these atypical data [1]. This expertise relies on a patented CSM solution called SMART™ (for “statistical monitoring applied to research trials”). The purpose of this article is to describe the principles underlying SMART™ and to illustrate its use and outcome for the SAMIT trial.
Methods
Data from the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial
The key elements of the SAMIT trial are presented below (from Tsuburaya et al. [18]):
-
Trial short identifier: SAMIT.
-
Trial registration: University Hospital Medical Information Network Clinical Trials Registry number C000000082.
-
Trial funding: Epidemiological and Clinical Research Information Network.
-
Number of randomized patients: 1495.
-
Number of centers: 232.
-
Country: Japan.
-
Timeline: Randomization between August 3, 2004 and September 29, 2009; follow-up for 72 months.
-
Patient population: Patients aged 20–80 years with T4a or T4b gastric cancer, who had had D2 dissection and an Eastern Cooperative Oncology Group performance score of 0–1.
-
Main findings: Sequential treatment did not improve disease-free survival, and tegafur–uracil was not inferior to S-1 (in fact S-1 was superior to tegafur–uracil) in terms of disease-free survival.
-
Trial conclusion: S-1 monotherapy should remain the standard treatment for locally advanced gastric cancer in Japan.
The database of the SAMIT trial was transferred to CluePoints for analysis by SMART™ in May 2013. The database contained all clinical data as well as some metadata, including unique identifiers for patients, centers, and visits. All non-free-text variables were included in the analysis, as all of them were considered potentially indicative of data quality issues. In the remainder of this article, actual center identifiers have been replaced by three random letters to preserve anonymity.
Statistical tests
The core of SMART™ is a battery of purpose-designed statistical tests that are applied to all variables in the clinical database [8]. These tests have been designed and improved during the last decade so as to efficiently detect a large number of anomalous patterns in clinical trials. For each statistical test, the underlying principle is to compare the distribution of a variable in a center with the distribution of that variable across all other centers so as to assess whether an atypical pattern is found with respect to the variable in this center. For most variables in the database, several tests are actually applied, and these investigate different aspects of the distribution: the mean, the variability around the mean, etc. The actual set of tests performed on a variable is automatically selected according to the statistical properties of the variable: for instance, date values are not analyzed in the same way as a sequence of measurements over time. The tests are applied for all centers that have entered at least two patients in the trial.
This multiple testing procedure aims to detect signals reflecting atypical data patterns in some of the centers involved in the clinical trial. The signals fall into four broad families:
-
1.
Data inconsistencies related to reporting. These signals detect atypical numbers of missing values or atypical numbers of records. For instance, an atypically low number of records for adverse events might be an indication of a misunderstanding of the protocol, leading to the non-reporting of non-serious adverse events. Alternatively, an atypically high number of missing laboratory values might indicate a delay in data transfer from the laboratory to the database.
-
2.
Data inconsistencies related to data tendency. These signals detect inconsistencies in the global distribution of the values of a variable in a center, across all patients and all visits. For instance, one might find a center where patients have atypically low body temperatures, which might be an indication of miscalibrated thermometers. On the other hand, an atypically high weight for the patients in a center might be an indication of an atypical population—for instance, if the center is specialized in the treatment of obesity.
-
3.
Data inconsistencies related to visit-to-visit evolution. These signals detect anomalies in the evolution of a variable over time. For instance, the blood pressure might be atypically stable for the patients in a center, which might be an indication that blood pressure measurements are not taken at each visit, but are simply copied (“propagated”) from previous visits. Or, an atypically high within-patient variability for most laboratory results might indicate that some blood samples were erroneously or intentionally taken from another subject.
-
4.
Data inconsistencies related to days. These signals detect anomalies in the proportion of measurements recorded on Saturdays or Sundays. In most countries, Saturdays and/or Sundays are not typical working days, so the number of analyses performed on those days is usually lower than on weekdays. An atypically high proportion of Saturdays and Sundays might be a sign of fabricated data, or might simply be related to atypical working days in the center.
It is important to keep in mind that data inconsistencies are statistical signals indicating that the data from a center differ from data across all other centers; whether these statistical signals are related to quality issues in the trial cannot be determined automatically and requires further scrutiny by individuals familiar with the trial.
Statistical test results: P values
Each statistical test performed by SMART™ returns a P value; hence each P value is associated with a triplet (center × variable × test). The smaller the P value of a test for a given center, the higher the amount of discrepancy between this center and all other centers with respect to the corresponding variable. The application of SMART™ to a clinical database returns a large number of P values. For instance, the analysis of the SAMIT trial, with 232 centers, of which 168 had included at least two patients, generated 61,488 P values. An automated quality control procedure was applied to remove the P values deemed unreliable or uninformative. For the SAMIT trial, 47,389 P values successfully passed the quality control procedure.
It is obviously neither possible nor useful to consider each of the thousands of P values individually. Only the most striking statistical signals are identified, and the related triplets (center × variable × test) are flagged for further scrutiny. Two complementary approaches are used to make use of the large set of P values. First, “extreme” P values are individually examined. Second, an overall data inconsistency score (DIS) is computed for each center to provide a summary of the data inconsistencies observed in the center across all tests and all variables.
Extreme P values
The P value of a test is interpreted as the probability of observing solely by chance a result at least as extreme as the one actually observed for the test. Observing a very low P value for a given test in a center points to an atypical data pattern in the values observed for the corresponding variable in this center.
A threshold must be chosen to consider a P value as being “extreme.” Given that about 5 × 104 P values were generated by SMART™ in the SAMIT trial, a threshold of 10−6 would seem appropriate using a Bonferroni correction, to ensure that the extreme P values were significant at the 0.05 level after adjustment for multiplicity. However, the tests were highly correlated; hence, the Bonferroni correction would be overly conservative and a less extreme threshold of 10−5 was arbitrarily chosen.
To graphically support the analysis of extreme P values, tests were grouped by the type of data inconsistency detected (reporting, data tendency, dates or visit-to-visit evolution, as described earlier).
Data inconsistency scores
The DIS is a numeric value ranging from 0 to 10 or greater. It is calculated so as to reflect the overall number of atypical data patterns in a center, across all variables, taking into account all of the aspects of the data distribution that are investigated by the statistical tests of SMART™. As such, the DIS is a summary of all P values associated with a center (technically, the DIS is equal to minus the logarithm to base 10 of an overall P value). The higher the DIS, the higher the confidence that the data collected in the center exhibit atypical patterns as compared with the other centers in the trial. A gain of one unit on the DIS scale reflects a tenfold decrease in the probability that data inconsistency was detected in the center by the random play of chance.
The DIS serves as a basis for highlighting centers with major atypical data patterns, those data patterns being either very strong statistical signals located in a few variables or, alternatively, medium statistical signals found across a larger set of variables. Thereby, the DIS is a necessary complement to the extreme P values, as it provides a comprehensive view of the overall degree of data inconsistency in a given center.
Centers with a DIS higher than a given threshold are identified as outliers. The threshold is chosen in regard of the risk of identifying a center as presenting data inconsistencies when in fact it does not. This risk is measured by the false discovery rate, which is defined as the proportion of centers that might be falsely identified as being outliers with that threshold. In general, a threshold of 3 for the DIS has been found adequate for reasonable identification of centers as outliers. This is the case for the SAMIT trial. Once the outliers have been identified, the reasons for their discrepancy with respect to the other centers are identified by a review of the individual statistical tests, and ultimately of the raw clinical data.
Results
The SAMIT trial data were analyzed with SMART™ (version 1.7). A total of 61,488 tests were performed, which generated 47,389 quality control-checked P values. The underlying data inconsistencies were investigated.
Centers with extreme P values
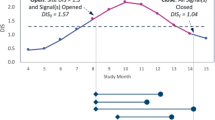
Four centers, identified as ZXB, UFE, HDM, and NBQ, had at least one extreme P value (Fig. 1). Center ZXB has the most extreme P value, related to an atypical pattern in dates, which indicates that a given variable is recorded atypically often during weekends in the center. Center NBQ has three extreme P values related to the visit-to-visit evolutions of three variables, which indicate an atypical behavior of the measurements over time. Center UFE also has an extreme P value in the visit-to-visit evolution of a variable. Finally, center HDM has an extreme P value related to an atypical rate of reporting for a variable. Those four centers presenting at least one extreme P value require further investigation so as to determine whether their statistically atypical character might result from a quality issue.

Assessment of the quality and consistency of the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial: individual statistical test results by test category. Four centers had a total of six tests with a P value <10−5
In center ZXB (six patients), all dates for a variable encoding the start date of concomitant treatments were on a Saturday. These start dates were considered plausible, as this center actually opened on Saturdays, whereas most other centers in the trial did not.
In center UFE (two patients), an atypically low variability was observed for the drug volume. The latter was either 400 mg or missing in this center, whereas values observed in other centers ranged between 0 and 4100 mg. After investigation, this atypical pattern was attributed to the play of chance, especially since only two patients were randomized in this center.
In center HDM (two patients), the last confirmation date for which the patient did not relapse was not reported for the two patients in the center. Further investigation showed that these last confirmation dates had not been entered in the database, but were in fact available.
In center NBQ (17 patients), several time sequences of blood-test-related measurements had an atypical first value. The reason for this atypical data pattern was that the center recorded zero in the case report form when a measurement had not been made.
Centers with extreme data inconsistency scores
Figure 2 shows the DIS assigned to the 168 centers participating in the SAMIT trial with more than two patients, plotted as a function of the center size (i.e., the number of randomized patients in the center). Each center is represented by a bubble, with the bubble size proportional to the center size. Centers labeled JZD, BMY, and VSF were identified as outliers (DIS higher than 3). For each of these centers, the probability of false detection as an outlier was lower than 5.6 %, as indicated by the false discovery rate scale.

Assessment of the quality and consistency of the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial: data inconsistency score (DIS) as a function of the center size. Three centers had a DIS greater than 3 (false discovery rate, FDR, <5.6 %)
In center JZD (two patients), the two patients had atypically similar initial values for several laboratory results and symptom grades. Moreover, they tended to have a similar trend of evolution over time for some laboratory results and symptom grades. Nothing was found in the data that would suggest that this finding could not simply have arisen from the play of chance, given the small size of the center.
In center BMY (two patients), there was an atypically low number of records and an atypically high number of missing values. In particular, one of the patients had no records for laboratory values or symptom grades. This patient had withdrawn his consent. The other patient had stopped the treatment in the first course of the trial.
In center VSF (24 patients), several recorded dates fell on a Saturday. These dates were considered plausible, as the center actually opened on Saturdays, whereas most other centers in the trial did not.
Discussion
We have described the method used by SMART™ to assess the quality and consistency of multicenter clinical trial data, and we have discussed the results of this approach applied to the SAMIT trial. Remarkably few atypical data patterns were found in the database of the SAMIT trial. These atypical patterns were further investigated so as to determine whether they were related to quality issues for the trial. As a result, some were easily explained by atypical center openings hours or patient features; some were found to be data recording problems; and some were deemed to be due to the play of chance, given the small size of the center. None of these data patterns were suspected to have had any impact on the outcome of the trial, nor was there any evidence of misconduct that could have affected the safety of the patients. Although it can never be concluded for certain that a trial is free of any errors that affect its quality, statistical monitoring of the data provides a quantitative and qualitative assessment of the data quality [17]. In the case of the SAMIT trial, this assessment suggested that the protocol was consistently followed across the participating centers [18].
The SAMIT trial had been completed when the SMART™ analyses were performed; hence, these analyses served as a last quality check before the database was locked for final analyses. CSM can thus prove useful to provide a check of the quality of the data of completed multicenter clinical trials before analysis, publication, and submission of the results to regulatory agencies. CSM can also be more generally used for ongoing trials to detect issues as early as possible, and to take remedial actions as required. Atypical data patterns that are uncovered by CSM range from atypical patient population or center characteristics, unintentional errors, sloppiness, and tampering to ultimately fraud [1]. Possible actions to confirm the issues identified include simply querying of some of the data in some centers, sending on-site monitors to perform source data verification, controlling the adherence to the protocol, or verifying the calibration of some instruments. If serious issues are confirmed in some centers, actions can range from additional training to improving the understanding of the protocol to closure of the center in the case of extreme problems. CSM can therefore contribute to a risk-based monitoring strategy in accordance with the Food and Drug Administration and European Medicines Agency guidelines [5, 6]. CSM is highly efficient and cost-saving compared with traditional approaches to data quality control, and as such, it can be expected to become an integral part of clinical trial monitoring in the future [8].
References
Buyse M. Centralized statistical monitoring as a way to improve the quality of clinical trials. Appl Clin Trials. 2014. http://www.appliedclinicaltrialsonline.com/print/199784. Accessed 20 Feb 2015.
Morrison BW, Cochran CJ, White JG, Harley J, Kleppinger CF, Liu A, et al. Monitoring the quality of conduct of clinical trials: a survey of current practices. Clin Trials. 2011;8:342–9.
Smith CT, Stocken DD, Dunn J, Cox T, Ghaneh P, Cunningham D, et al. The value of source data verification in a cancer clinical trial. PLoS One. 2012;7:e51623.
Bakobaki JM, Rauchenberger M, Joffe N, McCormack S, Stenning S, Meredith S. The potential for central monitoring techniques to replace on-site monitoring: findings from an international multi-centre clinical trial. Clin Trials. 2012;9:257–64.
US Department of Health and Human Services, Food and Drug Administration. Guidance for industry: oversight of clinical investigations—a risk-based approach to monitoring. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM269919.pdf. Accessed 20 Feb 2015.
European Medicines Agency. Reflection paper on risk based quality management in clinical trials. EMA/INS/GCP/394194/2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500110059.pdf. Accessed 20 Feb 2015.
Baigent C, Harrel F, Buyse M, Emberson JR, Altman DG. Ensuring trial validity by data quality assurance and diversification of monitoring methods. Clin Trials. 2008;5:49–55.
Venet D, Doffagne E, Burzykowski T, Beckers F, Tellier Y, Genevois-Marlin E, et al. A statistical approach to central monitoring of data quality in clinical trials. Clin Trials. 2012;9:705–7013.
Kirkwood A, Cox T, Hackshaw A. Application of methods for central statistical monitoring in clinical trials. Clin Trials. 2013;10:783–806.
Edwards P, Shakur H, Barnetson L, Prieto D, Evans S, Robert I. Central and statistical monitoring in the clinical randomization of an antifibrinolytic in significant haemorrhage (CRASH_2) trial. Clin Trials. 2014;11:336–43.
Lindblad AS, Manukyan Z, Purohit-Sheth T, Gensler G, Okwesili P, Meeker-O’Connel A, et al. Central site monitoring: results from a test of accuracy in identifying trials and sites failing food and drug administration inspection. Clin Trials. 2014;11(2):205–17.
Pogue JM, Devereaux PJ, Thorlund K, Yusuf S. Central statistical monitoring: detecting fraud in clinical trials. Clin Trials. 2013;10:225–35.
Buyse M, George S, Evans S, Geller NL, Ranstan J, Scherrer B, et al. The role of biostatistics in the prevention, detection and treatment of fraud in clinical trials. Stat Med. 1999;18:3435–51.
Evans S. Statistical aspects of the detection of fraud. In: Lock S, Wells F, Farthing M, editors. Fraud and misconduct in medical research. 3rd ed. London: BMJ Publishing Group; 2001. p. 186–204.
Buyse M, Evans S. Fraud in clinical trials. In: Armitage P, Colton T, editors. Encyclopaedia of biostatistics. New York: Wiley; 2005. p. 2023–31.
Al-Marzouki S, Evans S, Marshall T, Roberts I. Are these data real? Statistical methods for the detection of data fabrication in clinical trials. BMJ. 2005;331:267–70.
George SL, Buyse M. Data fraud in clinical trials. Clin Investig. 2015;15:161–73.
Tsuburaya A, Yoshida K, Kobayashi M, Yoshino S, Takahashi M, et al. Sequential paclitaxel followed by tegafur and uracil (UFT) or S-1 versus UFT or S-1 monotherapy as adjuvant chemotherapy for T4a/b gastric cancer (SAMIT): a phase 3 factorial randomised controlled trial. Lancet Oncol. 2014;15:886–93.
Acknowledgments
The development of SMART™ received financial support from the Walloon Government under the BioWin framework (consortium agreement no. 6741). The authors gratefully acknowledge the help of Koji Oba with the interpretation of the statistical signals found in the SAMIT trial. The authors thank the Epidemiological and Clinical Research Information Network at Nagoya University School of Medicine for the preparation of the database of the SAMIT trial.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Catherine Timmermans and Erik Doffagne are employees of CluePoints (Mont-Saint-Guibert, Belgium). Tomasz Burzykowski is an employee and stockholder of International Drug Development Institute (Louvain-la-Neuve, Belgium). Marc Buyse is an employee of International Drug Development Institute (Cambridge, MA, USA) and a stockholder of International Drug Development Institute and CluePoints. The other authors declare that they have no conflict of interest. Marc Buyse, Tomasz Burzykowski, Erik Doffagne and David Venet are named inventors on US patent application no. 13/452,338 for SMART™.
Ethical standards
See the main publication on the SAMIT trial [18].
Rights and permissions
About this article

Cite this article
Timmermans, C., Doffagne, E., Venet, D. et al. Statistical monitoring of data quality and consistency in the Stomach Cancer Adjuvant Multi-institutional Trial Group Trial. Gastric Cancer 19, 24–30 (2016). https://doi.org/10.1007/s10120-015-0533-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-015-0533-9