
Abstract
Iguratimod (IGU) is a novel small molecule anti-rheumatic drug with the effect of non-steroidal anti-inflammatory drug and disease-modifying anti-rheumatic drug. IGU has various mechanisms of action, including inhibition of prostaglandin E2, tumor necrosis factor-α (TNF-α), interleukin-17 (IL-17) production, inhibition of macrophage migration inhibitory factor (MIF)-induced proinflammatory effects, inhibition of osteoclastogenesis, and promotion of osteoblastic differentiation. Ankylosing spondylitis (AS) is the major subtype of spondyloarthritis that affects the axial skeleton, causing inflammatory back pain, which can lead to impairments in structure and function and a decrease in quality of life. Theories on pathogenesis of AS include misfolding of human leukocyte antigen-B27 during its assembly leading to endoplasmic reticulum stress and unfolded protein response (UPR). Activation of UPR genes results in release of TNF-α and IL-17, which have been shown to be important in the development of AS. In addition, current evidence suggests the importance of cyclooxygenase-2/prostaglandin E2 pathway and MIF in the pathogenesis of AS. Current drugs for the treatment of AS are limited and exploration of effective drugs is needed. IGU may be effective for the treatment of AS given that its mechanisms of action are closely related to the pathogenesis of AS. In fact, several small-scale clinical trials have shown the efficacy of IGU for the treatment of AS. This article reviews the molecular mechanisms of IGU that are related to the pathogenesis of AS and clinical trials of IGU for the treatment of AS, providing a reference for future clinical application of IGU for AS.

Similar content being viewed by others
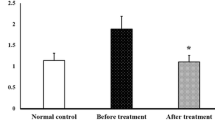

Data Availability
Not applicable.
Code availability
Not applicable.
References
Jiang H, Gao H, Wang Q, Wang M, Wu B (2020) Molecular mechanisms and clinical application of Iguratimod: a review. Biomed Pharmacother 122:109704
Tanaka K, Kawasaki H, Kurata K et al (1995) T-614, a novel antirheumatic drug, inhibits both the activity and induction of cyclooxygenase-2 (COX-2) in cultured fibroblasts. Jpn J Pharmacol 67(4):305–314
Aikawa Y, Yamamoto M, Yamamoto T et al (2002) An anti-rheumatic agent T-614 inhibits NF-kappaB activation in LPS- and TNF-alpha-stimulated THP-1 cells without interfering with IkappaBalpha degradation. Inflamm Res 51(4):188–194
Kohno M, Aikawa Y, Tsubouchi Y et al (2001) Inhibitory effect of T-614 on tumor necrosis factor-alpha induced cytokine production and nuclear factor-kappaB activation in cultured human synovial cells. J Rheumatol 28(12):2591–2596
Li G, Yamasaki R, Fang M et al (2018) Novel disease-modifying anti-rheumatic drug iguratimod suppresses chronic experimental autoimmune encephalomyelitis by down-regulating activation of macrophages/microglia through an NF-κB pathway. Sci Rep 8(1):1933
Wei Y, Sun X, Hua M et al (2015) Inhibitory effect of a novel antirheumatic drug T-614 on the IL-6-induced RANKL/OPG, IL-17, and MMP-3 expression in synovial fibroblasts from rheumatoid arthritis patients. Biomed Res Int 2015:214683
Luo Q, Sun Y, Liu W, Qian C, Jin B, Tao F, Gu Y, Wu X, Shen Y, Xu Q (2013) A novel disease-modifying antirheumatic drug, iguratimod, ameliorates murine arthritis by blocking IL-17 signaling, distinct from methotrexate and leflunomide. J Immunol 191(10):4969–4978
Bloom J, Metz C, Nalawade S et al (2016) Identification of iguratimod as an inhibitor of macrophage migration inhibitory factor (MIF) with steroid-sparing potential. J Biol Chem 291(51):26502–26514
Gan K, Yang L, Xu L et al (2016) Iguratimod (T-614) suppresses RANKL-induced osteoclast differentiation and migration in RAW264.7 cells via NF-κB and MAPK pathways. Int Immunopharmacol 35:294–300
Kuriyama K, Higuchi C, Tanaka K et al (2002) A novel anti-rheumatic drug, T-614, stimulates osteoblastic differentiation in vitro and bone morphogenetic protein-2-induced bone formation in vivo. Biochem Biophys Res Commun 299(5):903–909
Sieper J, Poddubnyy D (2017) Axial spondyloarthritis. Lancet 390(10089):73–84
Braun J, Sieper J (2007) Ankylosing spondylitis. Lancet 369(9570):1379–1390
Ranganathan V, Gracey E, Brown MA, Inman RD, Haroon N (2017) Pathogenesis of ankylosing spondylitis - recent advances and future directions. Nat Rev Rheumatol 13(6):359–367
Pedersen SJ, Maksymowych WP (2019) The pathogenesis of ankylosing spondylitis: an update. Curr Rheumatol Rep 21(10):58
Schett G, Lories RJ, D’Agostino MA et al (2017) Enthesitis: from pathophysiology to treatment. Nat Rev Rheumatol 13(12):731–741
Ranganathan V, Ciccia F, Zeng F et al (2017) Macrophage migration inhibitory factor induces inflammation and predicts spinal progression in ankylosing spondylitis. Arthritis Rheum 69(9):1796–1806
Stojanović I, Cvjetićanin T, Lazaroski S, Stošić-Grujičić S, Miljković D (2009) Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology 126(1):74–83
Sieper J, Poddubnyy D (2016) New evidence on the management of spondyloarthritis. Nat Rev Rheumatol 12(5):282–295
Bhala N, Emberson J, Merhi A et al (2013) Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382(9894):769–779
van der Heijde D, Ramiro S, Landewe R et al (2017) 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis 76(6):978–991
Callhoff J, Sieper J, Weiß A, Zink A, Listing J (2015) Efficacy of TNFα blockers in patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis: a meta-analysis. Ann Rheum Dis 74(6):1241–1248
Boonen A, van der Heijde D, Severens JL et al (2006) Markov model into the cost-utility over five years of etanercept and infliximab compared with usual care in patients with active ankylosing spondylitis. Ann Rheum Dis 65(2):201–208
Baeten D, Sieper J, Braun J et al (2015) Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med 373(26):2534–2548
Tanaka K, Shimotori T, Makino S et al (1992) Pharmacological studies of the new antiinflammatory agent 3-formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-o ne. 1st communication: antiinflammatory, analgesic and other related properties. Arzneimittel-Forschung 42(7):935–944
Tanaka K, Makino S, Shimotori T et al (1992) Pharmacological studies of the new antiinflammatory agent 3-formylamino-7-methylsulfonylamino-6-phenoxy-4′-1-benzopyran-4-o ne. 2nd communication: effect on the arachidonic acid cascades. Arzneimittel-Forschung 42(7):945–950
Evans DM, Spencer CC, Pointon JJ et al (2011) Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet 43(8):761–767
Siegle I, Klein T, Backman JT et al (1998) Expression of cyclooxygenase 1 and cyclooxygenase 2 in human synovial tissue: differential elevation of cyclooxygenase 2 in inflammatory joint diseases. Arthritis Rheum 41(1):122–129
Paulissen SM, van Hamburg JP, Davelaar N et al (2013) Synovial fibroblasts directly induce Th17 pathogenicity via the cyclooxygenase/prostaglandin E2 pathway, independent of IL-23. J Immunol 191(3):1364–1372
Samuels JS, Holland L, Lopez M et al (2018) Prostaglandin E2 and IL-23 interconnects STAT3 and RoRγ pathways to initiate Th17 CD4 T-cell development during rheumatoid arthritis. Inflamm Res 67(7):589–596
Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15(6):362–374
Crew MD, Effros RB, Walford RL et al (1998) Transgenic mice expressing a truncated Peromyscus leucopus TNF-alpha gene manifest an arthritis resembling ankylosing spondylitis. J Interf Cytokine Res 18(4):219–225
Redlich K, Görtz B, Hayer S et al (2004) Overexpression of tumor necrosis factor causes bilateral sacroiliitis. Arthritis Rheum 50(3):1001–1005
Braun J, Bollow M, Neure L et al (1995) Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum 38(4):499–505
Iwakura Y, Ishigame H, Saijo S et al (2011) Functional specialization of interleukin-17 family members. Immunity 34(2):149–162
Miossec P, Korn T, Kuchroo VK (2009) Interleukin-17 and type 17 helper T cells. N Engl J Med 361(9):888–898
Xu Y, Zhu Q, Song J et al (2015) Regulatory effect of iguratimod on the balance of Th subsets and inhibition of inflammatory cytokines in patients with rheumatoid arthritis. Mediat Inflamm 2015:356040–356040
International Genetics of Ankylosing Spondylitis C, Cortes A, Hadler J et al (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet 45(7):730–738
Glatigny S, Fert I, Blaton MA et al (2012) Proinflammatory Th17 cells are expanded and induced by dendritic cells in spondylarthritis-prone HLA-B27-transgenic rats. Arthritis Rheum 64(1):110–120
Benham H, Rehaume LM, Hasnain SZ, Velasco J, Baillet AC, Ruutu M, Kikly K, Wang R, Tseng HW, Thomas GP, Brown MA, Strutton G, McGuckin MA, Thomas R (2014) Interleukin-23 mediates the intestinal response to microbial β-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheum 66(7):1755–1767
Mei Y, Pan F, Gao J et al (2011) Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol 30(2):269–273
Wendling D, Cedoz J-P, Racadot E, Dumoulin G (2007) Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine 74(3):304–305
Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, Thiel A, Radbruch A, Loddenkemper C, Sieper J (2011) Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther 13(3):R95–R95
Ebihara S, Date F, Dong Y, Ono M (2015) Interleukin-17 is a critical target for the treatment of ankylosing enthesitis and psoriasis-like dermatitis in mice. Autoimmunity 48(4):259–266
Raychaudhuri SK, Saxena A, Raychaudhuri SP (2015) Role of IL-17 in the pathogenesis of psoriatic arthritis and axial spondyloarthritis. Clin Rheumatol 34(6):1019–1023
Gravallese EM, Schett G (2018) Effects of the IL-23-IL-17 pathway on bone in spondyloarthritis. Nat Rev Rheumatol 14(11):631–640
Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, McInnes I, van Laar JM, Landewé R, Wordsworth P, Wollenhaupt J, Kellner H, Paramarta J, Wei J, Brachat A, Bek S, Laurent D, Li Y, Wang YA, Bertolino AP, Gsteiger S, Wright AM, Hueber W (2013) Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet 382(9906):1705–1713
Calandra T, Roger T (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3(10):791–800
Kozaci LD, Sari I, Alacacioglu A et al (2010) Evaluation of inflammation and oxidative stress in ankylosing spondylitis: a role for macrophage migration inhibitory factor. Mod Rheumatol 20(1):34–39
Baerlecken NT, Nothdorft S, Stummvoll GH, Sieper J, Rudwaleit M, Reuter S, Matthias T, Schmidt RE, Witte T (2014) Autoantibodies against CD74 in spondyloarthritis. Ann Rheum Dis 73(6):1211–1214
Riechers E, Baerlecken N, Baraliakos X et al (2019) Sensitivity and specificity of autoantibodies against CD74 in nonradiographic axial Spondyloarthritis. Arthritis Rheum 71(5):729–735
Wang X, Ma C, Li P et al (2017) Effects of iguratimod on the levels of circulating regulators of bone remodeling and bone remodeling markers in patients with rheumatoid arthritis. Clin Rheumatol 36(6):1369–1377
Wang XT, Li P, Xu TS et al (2016) Effect of iguratimod and methotrexate on RANKL and OPG expression in serum and IL-1β-induced fibroblast-like synoviocytes from patients with rheumatoid arthritis. Cell Mol Biol 62(12):44–50
Taurog JD, Chhabra A, Colbert RA (2016) Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med 374(26):2563–2574
Sambrook PN, Geusens P (2012) The epidemiology of osteoporosis and fractures in ankylosing spondylitis. Ther Adv Musculoskelet Dis 4(4):287–292
Im CH, Kang EH, Ki JY, Shin DW, Choi HJ, Chang EJ, Lee EY, Lee YJ, Lee EB, Kim HH, Song YW (2009) Receptor activator of nuclear factor kappa B ligand-mediated osteoclastogenesis is elevated in ankylosing spondylitis. Clin Exp Rheumatol 27(4):620–625
Sieper J, Poddubnyy D, Miossec P (2019) The IL-23-IL-17 pathway as a therapeutic target in axial spondyloarthritis. Nat Rev Rheumatol 15(12):747–757
Neerinckx B, Lories R (2017) Mechanisms, impact and prevention of pathological bone regeneration in spondyloarthritis. Curr Opin Rheumatol 29(4):287–292
Watad A, Bridgewood C, Russell T et al (2018) The early phases of ankylosing spondylitis: emerging insights from clinical and basic science. Front Immunol 9:2668
Luo Y, Zheng N, Wu R (2018) Is iguratimod effective in refractory axial spondyloarthritis? Scand J Rheumatol 47(6):518–520
Qiu YY, Tang Y, Rui JB, Li J (2016) Clinical observation on treatment of refractory ankylosing spondylitis by Iguratimod. J Jiangsu Univ (Medicine Edition) 26(3):235–239
Huang BJ, Ma JX (2018) Observation of short-term efficacy of iguratimod in the treatment of ankylosing spondylitis. Chin Commun Doctors 34(13):92–93
Lin YP, Liu H, Gao JT (2019) Initial clinical observation on treatment of ankylosing spondylitis by Iguratimod. J Clin Rational Drug Use 12(14):9–13
Xu BJ, Mo XQ, Xue XQ, Wu Y (2019) Study of the efficacy and safety of iguratimod in the treatment of ankylosing spondylitis. J New Med 50(12):915–918
Zeng HQ, Kong WH, Zhuang P, Dong HJ, Yi ZH, Chen XP, Fu CZ, Ye ZH (2016) Observation of efficacy of iguratimod in the treatment of ankylosing spondylitis. Hainan Med J 27(1):118–120
Funding
Science and technology general project of Guangzhou, China. Funding number: 201804010028.
Author information
Authors and Affiliations
Contributions
Yang Cui had the original idea for the article, Suling Liu performed the literature search, data analysis and drafted the manuscript. Yan Cui and Xiao Zhang critically revised the manuscript.
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, S., Cui, Y. & Zhang, X. Molecular mechanisms and clinical studies of iguratimod for the treatment of ankylosing spondylitis. Clin Rheumatol 40, 25–32 (2021). https://doi.org/10.1007/s10067-020-05207-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05207-z