
Abstract
Here, we report brain white matter alterations in individuals clinically and genetically diagnosed with periodontal Ehlers–Danlos syndrome, a rare disease characterized by premature loss of teeth and connective tissue abnormalities. Eight individuals of two families clinically diagnosed with periodontal Ehlers–Danlos syndrome were included in the present study and underwent general physical, dental, and neurological examination. Whole exome sequencing was performed, and all patients included in the study underwent MRI of the brain. Whole exome sequencing revealed heterozygous C1R mutations c.926G>T (p.Cys309Phe, Family A) and c.149_150TC>AT (p.Val50Asp, Family B). All adult individuals (n = 7; age range 31 to 68 years) investigated by MRI had brain white matter abnormalities. The MRI of one investigated child aged 8 years was normal. The MRI pattern was suggestive of an underlying small vessel disease that is progressive with age. As observed in other leukoencephalopathies related to microangiopathies, the extent of the white matter changes was disproportionate to the neurologic features. Medical history revealed recurrent headaches or depression in some cases. Neurological examination was unremarkable in all individuals but one had mild cognitive decline and ataxia and experienced a seizure. The observation that periodontal Ehlers–Danlos syndrome caused by missense mutations in C1R is consistently associated with a leukoencephalopathy opens a new pathogenic link between the classical complement pathway, connective tissue, brain small vessels, and brain white matter abnormalities.
Similar content being viewed by others


Introduction
Periodontal Ehlers–Danlos syndrome (EDS) is a specific EDS subtype characterized by premature loss of teeth due to severe periodontitis, increased rate of infections, and connective tissue abnormalities like easy bruising, pretibial discolorations, joint hypermobility, and organ or vessel rupture. Periodontal EDS is caused by heterozygous mutations in C1R or C1S [1], which encode subunits C1r and C1s of the first component of the classical complement pathway. This pathway is central in microbial–host interactions. Its over-activation or deregulation may excessively amplify inflammation. Additionally, the C1q subunit—binding partner of the C1s-C1r tetramer—has a triple helical structure, which resembles collagen; some features of periodontal EDS may be due to abnormal C1r/C1s interaction with extracellular matrix components [1]. For example, interactions of C1q with bone morphogenetic protein-1 and tolloid-like proteinases, metalloproteinases that have major roles in extracellular matrix assembly, have recently been shown [2].
The different EDS subtypes are not usually associated with neurological symptomatology; brain MRI is typically unremarkable [3]. There are single reports on polymicrogyria [4,5,6,7], corpus callosum agenesis or hypoplasia [7, 8] periventricular heterotopia, ventricular dilatation or cerebral atrophy [7, 9] in hypermobile, classical, or unspecified EDS. Bi-allelic mutations in COL3A1, encoding type III (pro) collagen, cause cobblestone-like cortical malformation and white matter changes [10,11,12], but these are not observed in vascular EDS caused by heterozygous COL3A1 mutations. Leukoencephalopathy has been reported in a single individual with typical clinical features of periodontal EDS prior to identification of the causative gene defect [13]. Here, we report that leukoencephalopathy appears to be a general feature of periodontal EDS caused by C1R mutations.
Methods
Clinical investigations
Individuals of two families clinically diagnosed with periodontal EDS were included in the study. All patients included in the study underwent general physical and neurological examination. The clinical investigation list and the questionnaire for reporting on periodontal EDS were previously published in Kapferer-Seebacher et al. [14] as supplements. Briefly, clinical investigation included assessment of joint and skin features and photographs. Oral investigation included periodontal investigations, orthopantomogram, and intraoral photographs. The diagnosis of early severe periodontitis was based on radiologic evidence of severe alveolar bone loss (≥ 50%) at an age of ≤ 25 years or history of complete tooth loss due to tooth mobility at an age of ≤ 30 years. The questionnaire includes questions on previous dental treatments and smoking, joint and skin features, and other features like recurrent infections, hoarse voice, organ ruptures, and other systemic conditions. Family A was investigated at the VU University Medical Center, Amsterdam, in 2017. Clinical and genetic data of Family B have been previously reported by Kapferer-Seebacher et al. [1]. Neurologic investigation of family B was conducted in 2017 and 2018 at the Medical University of Innsbruck, Austria.
Magnetic resonance imaging
All patients included in the study underwent MRI of the brain. We scored all MRIs according to a previously published standardized protocol [15].
Genomic analysis
Whole exome sequencing was performed on three affected individuals from family A. Genomic DNA was isolated from blood and 2.5 μg was sheared on a Covaris S2 instrument (Covaris, Woburn, MA). DNA libraries were prepared using Kapa Biosystems reagents (Kapa Biosystems, Wilmington, MA), and 1.0 μg of library was used for enrichment with Roche/NimbleGen SeqCap EZ MedExome (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) with 125 base pair paired-end reads. Over 92% of the capture region was covered ≥ 30× with a mean bait coverage of approximately 105× for each sample. Variant calling was performed using an in-house analysis pipeline. Alignment of sequence reads to the human genome (hg19) was performed with the Burrows–Wheeler Aligner tool (BWA-MEM v0.7.10) using default settings. Subsequently, Picard Tools (v1.111; http://picard.sourceforge.net/) was used for sorting and marking duplicates. For local realignment and base quality score recalibration, we used the Genome Analysis Toolkit (GATK; v3.3-0; Broad Institute, Cambridge, MA) and for variant calling, we used the GATK HaplotypeCaller. Variants were filter tagged using the GATK VariantFiltration and annotated by snpEff (v4.0). Variant prioritization was performed using Cartagenia Bench Lab NGS (Agilent Technologies, Santa Clara, USA). In short, a classification tree was used to select for variants present in all three affected individuals and virtually absent in control cohorts dbSNP build 142 (http://www.ncbi.nlm.nih.gov/projects/SNP), 1000 Genomes Phase 3 release v5.20130502, and ESP6500 (http://evs.gs.washington.edu/EVS/) as well as in-house controls. Prerequisite was that these variants had been genotyped in at least 200 alleles. Subsequently, the remaining variants were further prioritized based on literature, predicted (deleterious) effects on protein function by, e.g., truncating the protein, affecting splicing, amino acid change, and evolutionary conservation.
Results
Subjects, families and genetic results
Family A is a Caucasian Dutch four-generation family (Fig. 1); individuals A-II-1, A-III-1, and A-III-2 were available for clinical and genetic investigations. The C1R mutation c.926G>T (NM_001733.4) resulting in p.(Cys309Phe) was identified in all of them. This cysteine at position 309 is located in the Sushi/SCR/CCP domain and is involved in disulfide bond formation that stabilizes the C1r CCP1 module [1]. In addition, c.927C>G results in an amino acid change of the same amino acid p.(Cys309Trp) and was previously identified in other periodontal EDS patients [1]. No known or likely pathogenic variants were found in 154 genes related to leukoencephalopathies, including vascular leukoencephalopathies. All individuals were clinically diagnosed with periodontal EDS (for details see, Table 1; Figs. 1 and 2).

Pedigrees of the families described in this study. Above each pedigree, the C1R genotype in the affected individuals is specified on nucleotide and predicted protein levels. The whole pedigree of family B has been published elsewhere; identification codes are identical with Kapferer-Seebacher et al. [1]. The arrows indicate the individuals who were available for neurological examination. All indicated individuals were investigated by MRI and had brain white matter abnormalities, except individual A:III-1 at age 7 years

Pretibial discolorations in family A. Pretibial hemosiderotic plaques have been discribed in 83% of individuals affected with periodontal EDS (Kapferer-Seebacher et al. [1]). Photographs of shins (a) individual A-II-1, (b) individual A-III-2, and (c) individual A-III-1. No pretibial plaques were present in family B
At clinical investigation, the male individual A:II-1 was 58 years of age. In addition to signs of periodontal EDS, his medical history included diabetes type II, high blood pressure, hypercholesterolemia, and cardiac arrhythmias; he never smoked. From age 50 years he experienced slow and mild cognitive decline with memory, concentration and spatial orientation problems, dyspraxia, aggression, and depression. At age 57 years, he was admitted in a state of confusion during pneumonia, while in the hospital, he had a seizure lasting 20 min with a focal start in the right hand and secondary generalization. Neurological examination at age 57 years revealed mild tremor and slight cerebellar ataxia, no other abnormalities. Individual A:III-2 was 31 years of age at time of investigation. She never smoked, had a normal blood pressure, and had no complaints of cognitive decline. Neurological examination at age 31 years was normal. Individual A:III-1 was 8 years of age at time of investigation. He had mild learning problems, especially language problems, but followed normal level education. Neurological examination at age 8 years was normal.
Family B is a five-generation Caucasian Austrian family, whose clinical and genetic data were previously reported (Family 1 in Kapferer-Seebacher et al.) [1]. The identified C1R mutation c.149_150TC>AT (p.Val50Asp) segregated with 15 individuals clinically affected by periodontal EDS in this family; in Fig. 1, only individuals relevant for the present study are depicted, with the same numbers as used in Kapferer-Seebacher et al. [1]. Individuals B:III-2, B:III-10, B:IV-1, B:IV-2, and B:IV-10 were available for the present neurologic analysis. All investigated individuals of family B had never smoked or were light smokers (≤ 5 cigarettes per day), and had no hypertension or hypercholesterinemia. All affected individuals in this family showed normal intellectual performance without evidence of cognitive decline (Fig. 3).

Oral features. a Intraoral radiograph of individual B:IV-10 at age 32 years. Notice severe periodontal bone loss in the lower jaw, especially teeth no. 44, 45, and 34, 35. b Intraoral photographs at age 34 years. Teeth no. 34 to 36 have been lost. Severe gingival recession (exposed tooth roots) and lack of attached gingiva with mucosa extending to the gingival margins is specific for periodontal EDS
In male individual B:III-10, neurological examination at age 68 years was normal. The female individual B:III-2 reported on frequent headaches; neurological examination at age 56 years was normal. In female individual B:IV-10, neurological examination at age 43 years was normal. The female individual B:IV-1 suffered from depression. Neurological examination at age 34 years was normal. The female individual B:IV-2 was not available for clinical neurologic investigation, but reported frequent fainting spells and dizziness, and a history of frequent headaches in childhood.
Magnetic resonance imaging
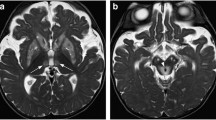
In family A, individual A:II-1 underwent MRI at age 57 years (Fig. 4a (a–f)). It showed no cerebral or cerebellar atrophy. Almost diffuse, homogeneous cerebral white matter signal abnormalities were present, with an anterior-posterior gradient, in which the posterior cerebral white matter was somewhat better preserved. A thin rim of directly subcortical white matter was relatively, but not entirely spared. The anterior part of the temporal lobe and external and extreme capsules were affected. The corpus callosum and internal capsule were spared. Within the basal nuclei and thalami numerous small foci of high T2 signal were seen, which in part had a low signal on FLAIR, indicative of enlarged perivascular spaces. Two foci in the putamen and head of the caudate nucleus on the left were larger and cystic, indicative of lacunar infarctions. Within the brain stem, a lacunar infarction was present in the pons. Inhomogeneous T2 hyperintensities were present in the pons, the lower part of the midbrain and the cerebellar white matter. Gradient echo images showed evidence of a few microbleeds in the basal nuclei on the left and the dentate nucleus on both sides. No areas of diffusion restriction or abnormal contrast enhancement were seen. Individual A:III-2 underwent MRI at 31 years (Fig. 4a (g and h)). It showed signal abnormalities in the deep parietal and periventricular frontal and occipital white matter of limited extent. No signal abnormalities elsewhere, enlarged perivascular spaces, lacunar infarcts, areas of diffusion restriction, or microbleeds were visible. Individual A:III-1 underwent MRI at 7 years. It was normal.

MRI. a Family A, (a–f) individual A:II-1 at age 57 years; (g, h) individual A:III-2 at age 31 years. The FLAIR (a, d) and T2-weighted (b, c) images in the older patient show virtually diffuse cerebral white matter signal abnormalities, including also the external and extreme capsules (c) and the anterior temporal white matter (d). Lacunar infarcts are seen in the head of the caudate nucleus on the left (arrow in b), left putamen (arrow in c), and pons (arrow in d). Numerous small areas of abnormal signal are seen in the basal ganglia and thalami (c) and the pons (d). Gradient echo images show microbleeds in the basal nuclei and dentate nucleus (arrows in e and f). The FLAIR (g, h) images in the younger patient show a thin periventricular rim as well as a few larger areas of abnormal signal in the deep cerebral white matter. b Family B, (a–d) individual B:III-10 at age 68 years (e, f); individual B:III-2 at age 56 years and (g, h) individual B:IV-10 at 43 years. In the oldest patient, the T2-weighted images show extensive and confluent periventricular and deep cerebral white matter abnormalities (a–d), as well as numerous enlarged perivascular spaces in the white matter (a–c) and basal nuclei (d). There is a mild generalized atrophy. The FLAIR images in the younger patients (e–h) show a thin periventricular rim as well as multifocal small and lager larger areas of abnormal signal in the deep cerebral white matter
In family B, individual B:III-10 underwent MRI at age 68 years (Fig. 4b (a–d)). It showed slight generalized cerebral atrophy with enlarged lateral ventricles and subarachnoid spaces. Extensive, homogeneous cerebral white matter signal abnormalities were present, without anterior–posterior gradient. The subcortical white matter was preserved in all areas and the anterior part of the temporal lobe and external and extreme capsules were not affected. The corpus callosum, internal capsule, brain stem, and cerebellum were spared. Numerous enlarged perivascular spaces were present spread over the cerebral hemispheres, involving white matter, basal nuclei and thalami. No lacunar infarcts, areas of diffusion restriction or abnormal enhancement after contrast were present. MR angiography and venography did not reveal vessel abnormalities. Individual B:III-2 underwent MRI at age 56 years. It showed numerous enlarged perivascular spaces. FLAIR showed a thin, continuous periventricular rim of abnormal signal and numerous small and larger spots of abnormal signal spread over the deep cerebral white matter. No lacunar infarcts, areas of diffusion restriction, or abnormal contrast enhancement were present. MR angiography and venography did not reveal vessel abnormalities. Individual B:IV-10 underwent MRI at age 43 years. The abnormalities were similar to those of individual B:III-2, but less extensive. Individual B:IV-1 underwent MRI at age 35 years. It only showed a very thin, continuous periventricular rim and a few small spots of abnormal signal spread over the deep cerebral white matter on FLAIR. MRI of individual B:IV-2 was obtained at age 33 years because of the neurological complaints described above. It revealed numerous enlarged perivascular spaces in the cerebral white matter. Additionally, a thin, continuous periventricular rim of abnormal signal and multiple small focal lesions were present in the deep cerebral white matter. No areas of diffusion restriction, microbleeds or abnormal contrast enhancement were observed.
Discussion
We report two families with eight individuals clinically and genetically diagnosed with autosomal-dominant periodontal EDS who underwent MRI of the brain. In both families, periodontal EDS is associated with an adult-onset leukoencephalopathy. In young-adult patients, MRI shows multifocal signal changes in the deep cerebral white matter and enlarged perivascular spaces. In older patients, the leukoencephalopathy has become extensive to virtually diffuse and additional lesions and lacunar infarctions are present in the basal nuclei, thalami, pons, and cerebellum. One older patient has microbleeds. The latter MRI pattern is suggestive of an underlying small vessel disease [16]. Most common risk factors for leukoencephalopathies caused by small vessel disease are hypertension, hypercholesterolemia, diabetes mellitus, and age [17, 18]. Genetic vascular leukoencephalopathies, including CADASIL [19], CARASIL [20], CARASAL [21], and COL4A1- or COL4A2-related disorders in adults [22, 23], cause a similar MRI pattern. With the anterior temporal white matter abnormalities and involvement of the external and extreme capsules, the MRI pattern of our EDS patients is in fact very similar to that of CADASIL [20].
Leukoencephalopathy has been previously reported in a single individual diagnosed with periodontal EDS [13]. Unfortunately, the patient is not available for clinical follow-up and genetic confirmation of the diagnosis, but the reported clinical manifestations including pretibial skin discoloration are pathognomonic for periodontal EDS. Considering the consistent association of a vascular leukoencephalopathy with periodontal EDS in three families, it is likely that it is causally related to periodontal EDS. Neurologic features in the patient reported by Spranger et al. [13] were limited to recurrent headaches and several episodes of drop-attacks without loss of consciousness or other neurologic deficits; intellectual performance was reported normal. Also in our patients, neurological deficits are mild or absent. As in other small vessel diseases, the leukoencephalopathy in periodontal EDS is disproportionate to the severity of the clinical disease [21, 24]. Individual A:II-1 is the only patient, who presented with neurological complaints. He also is the only one with multiple vascular risk factors, including diabetes type II, hypertension, and hypercholesterolemia, which might have contributed to the disease.
The pathogenesis of the leukoencephalopathy caused by C1R mutations in periodontal EDS is unclear. All mutations identified so far appear to cause periodontal EDS through a gain of function effect; none of the mutations identified to date alter the enzymatic region of the molecules. There is evidence that the mutations cause intracellular retention of C1r/C1s protein and expansion of the endoplasmatic reticulum which may interfere with processing of collagen and/or other components of the exctracellular matrix (ECM) [1]. In addition, abnormal activation of the complement 1 subunits causing intracellular and extracellular presence of serine protease may alter ECM matrix proteins and trigger an immune response that could lead to arteriolar damage and white matte changes as observed in other microangiopathies. No brain autopsy data are available from affected individuals, but histopathologic investigation of urticaria1 lesion in one patient revealed small-vessel vasculitis consisting of neutrophils, mononuclear cells, and endothelial disruption (leukocytoclastic vasculitis) [25]. Capillary fragility plays a major role in periodontal EDS, and easy bruising is a major criterion of periodontal EDS [26]. However, this is also found in other forms of EDS without MRI abnormalities, and there is no evidence that vascular connective tissue fragility is the primary cause of the apparently insidious development of leukoencephalopathy in pEDS.
In conclusion, leukoencephalopathy appears to be part of the clinical spectrum in periodontal EDS with C1R mutations, as it was evident in all adult individuals studied by MRI known to us. The MRI pattern is suggestive of an underlying small vessel disease that progresses with age. The extent of MRI changes is disproportionate to the presence of neurologic disease features. Longitudinal studies and histopathological analyses in patients with periodontal EDS will have to elucidate the exact cause and neurological consequences of these findings.
References
Kapferer-Seebacher I, Pepin M, Werner R, Aitman TJ, Nordgren A, Stoiber H, Thielens N, Gaboriaud C, Amberger A, Schossig A, Gruber R, Giunta C, Bamshad M, Bjorck E, Chen C, Chitayat D, Dorschner M, Schmitt-Egenolf M, Hale CJ, Hanna D, Hennies HC, Heiss-Kisielewsky I, Lindstrand A, Lundberg P, Mitchell AL, Nickerson DA, Reinstein E, Rohrbach M, Romani N, Schmuth M, Silver R, Taylan F, Vandersteen A, Vandrovcova J, Weerakkody R, Yang M, Pope FM, Molecular Basis of Periodontal EDSC, Byers PH, Zschocke J (2016) Periodontal Ehlers-Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Genet 99(5):1005–1014. https://doi.org/10.1016/j.ajhg.2016.08.019
Lacroix M, Tessier A, Dumestre-Perard C, Vadon-Le Goff S, Gout E, Bruckner-Tuderman L, Kiritsi D, Nystrom A, Ricard-Blum S, Moali C, Hulmes DJS, Thielens NM (2017) Interaction of complement defence collagens C1q and mannose-binding lectin with BMP-1/tolloid-like proteinases. Sci Rep 7(1):16958. https://doi.org/10.1038/s41598-017-17318-w
Castori M, Voermans NC (2014) Neurological manifestations of Ehlers-Danlos syndrome(s): a review. Iran J Neurol 13(4):190–208
Echaniz-Laguna A, de Saint-Martin A, Lafontaine AL, Tasch E, Thomas P, Hirsh E, Marescaux C, Andermann F (2000) Bilateral focal polymicrogyria in Ehlers-Danlos syndrome. Arch Neurol 57(1):123–127
Ezzeddine H, Sabouraud P, Eschard C, El Tourjuman O, Bednarek N, Motte J (2005) Bilateral frontal polymicrogyria and Ehlers-Danlos syndrome. Arch Pediatr 12(2):173–175. https://doi.org/10.1016/j.arcped.2004.11.021
Mathew T, Sinha S, Taly AB, Arunodaya GR, Srikanth SG (2005) Neurological manifestations of Ehlers-Danlos syndrome. Neurol India 53(3):339–341
Verrotti A, Sparta MV, Monacelli D, Porto R, Castagnino M, Russo Raucci A, Compagno F, Viglio S, Foiadelli T, Nicita F, Grosso S, Spalice A, Chiarelli F, Marseglia G, Savasta S (2014) Long-term prognosis of patients with Ehlers-Danlos syndrome and epilepsy. Epilepsia 55(8):1213–1219. https://doi.org/10.1111/epi.12699
Dotti MT, De Stefano N, Mondillo S, Agricola E, Federico A (1999) Neurological involvement and quadricuspid aortic valve in a patient with Ehlers-Danlos syndrome. J Neurol 246(7):612–613
Hagino H, Eda I, Takashima S, Takeshita K, Sugitani A (1985) Computed tomography in patients with Ehlers-Danlos syndrome. Neuroradiology 27(5):443–445
Vandervore L, Stouffs K, Tanyalcin I, Vanderhasselt T, Roelens F, Holder-Espinasse M, Jorgensen A, Pepin MG, Petit F, Khau Van Kien P, Bahi-Buisson N, Lissens W, Gheldof A, Byers PH, Jansen AC (2017) Bi-allelic variants in COL3A1 encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts. J Med Genet 54(6):432–440. https://doi.org/10.1136/jmedgenet-2016-104421
Horn D, Siebert E, Seidel U, Rost I, Mayer K, Abou Jamra R, Mitter D, Kornak U (2017) Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities. Am J Med Genet Part A 173(9):2534–2538. https://doi.org/10.1002/ajmg.a.38345
Jorgensen A, Fagerheim T, Rand-Hendriksen S, Lunde PI, Vorren TO, Pepin MG, Leistritz DF, Byers PH (2015) Vascular Ehlers-Danlos syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur J Hum Genet EJHG 23(6):796–802. https://doi.org/10.1038/ejhg.2014.181
Spranger S, Spranger M, Kirchhof K, Steinmann B (1996) Ehlers-Danlos syndrome type VIII and leukodystrophy. Am J Med Genet 66(2):239–240. https://doi.org/10.1002/(SICI)1096-8628(19961211)66:2<239::AID-AJMG23>3.0.CO;2-T
Kapferer-Seebacher I, Lundberg P, Malfait F, Zschocke J (2017) Periodontal manifestations of Ehlers-Danlos syndromes: a systematic review. J Clin Periodontol 44(11):1088–1100. https://doi.org/10.1111/jcpe.12807
van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J (1999) Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 213(1):121–133. https://doi.org/10.1148/radiology.213.1.r99se01121
Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O'Brien JT, Barkhof F, Benavente OR, Black SE, Brayne C, Breteler M, Chabriat H, Decarli C, de Leeuw FE, Doubal F, Duering M, Fox NC, Greenberg S, Hachinski V, Kilimann I, Mok V, Oostenbrugge R, Pantoni L, Speck O, Stephan BC, Teipel S, Viswanathan A, Werring D, Chen C, Smith C, van Buchem M, Norrving B, Gorelick PB, Dichgans M, STfRVco nE (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12(8):822–838. https://doi.org/10.1016/S1474-4422(13)70124-8
Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC (2002) Subcortical ischaemic vascular dementia. Lancet Neurol 1(7):426–436
Sahathevan R, Brodtmann A, Donnan GA (2012) Dementia, stroke, and vascular risk factors; a review. Int J Stroke 7(1):61–73. https://doi.org/10.1111/j.1747-4949.2011.00731.x
Pantoni L, Pescini F, Nannucci S, Sarti C, Bianchi S, Dotti MT, Federico A, Inzitari D (2010) Comparison of clinical, familial, and MRI features of CADASIL and NOTCH3-negative patients. Neurology 74(1):57–63. https://doi.org/10.1212/WNL.0b013e3181c7da7c
Nozaki H, Nishizawa M, Onodera O (2014) Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 45(11):3447–3453. https://doi.org/10.1161/STROKEAHA.114.004236
Bugiani M, Kevelam SH, Bakels HS, Waisfisz Q, Ceuterick-de Groote C, Niessen HW, Abbink TE, Lesnik Oberstein SA, van der Knaap MS (2016) Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology 87(17):1777–1786. https://doi.org/10.1212/WNL.0000000000003251
van der Knaap MS, Smit LM, Barkhof F, Pijnenburg YA, Zweegman S, Niessen HW, Imhof S, Heutink P (2006) Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol 59(3):504–511. https://doi.org/10.1002/ana.20715
Gunda B, Mine M, Kovacs T, Hornyak C, Bereczki D, Varallyay G, Rudas G, Audrezet MP, Tournier-Lasserve E (2014) COL4A2 mutation causing adult onset recurrent intracerebral hemorrhage and leukoencephalopathy. J Neurol 261(3):500–503. https://doi.org/10.1007/s00415-013-7224-4
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG (2009) Cadasil. Lancet Neurol 8(7):643–653. https://doi.org/10.1016/S1474-4422(09)70127-9
Hoffman GS, Filie JD, Schumacher HR Jr, Ortiz-Bravo E, Tsokos MG, Marini JC, Kerr GS, Ling QH, Trentham DE (1991) Intractable vasculitis, resorptive osteolysis, and immunity to type I collagen in type VIII Ehlers-Danlos syndrome. Arthritis Rheum 34(11):1466–1475
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B (2017) The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C: Semin Med Genet 175(1):8–26. https://doi.org/10.1002/ajmg.c.31552
Acknowledgements
We thank Dagmar Slot, Academic Centre for Dentistry Amsterdam (ACTA), University of Amsterdam and VU University, for organization of patients’ clinical oral investigation (family A).
Funding
Open access funding provided by Austrian Science Fund (FWF). The study was supported by the Dutch ZonMw TOP grant 91211005 and Austrian Science Fund FWF (I 2909-B30).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The study was conducted with the probands’ understanding and full written consent as part of the Biobank for Rare Diseases and the pilot study MRI in rare neurologic diseases, both approved by the Ethics Committee of the Medical University Innsbruck, Austria (study nos. UN4501 and 1074/2017); the study was also approved by the Ethics Committee for gene identification research on patients with unclassified leukoencephalopathies at the VU University Medical Center Amsterdam (2010/267), in accordance with the Declaration of Helsinki of 1975 as revised in 2013.
Disclosures
The authors report no disclosures relevant to the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kapferer-Seebacher, I., Waisfisz, Q., Boesch, S. et al. Periodontal Ehlers–Danlos syndrome is associated with leukoencephalopathy. Neurogenetics 20, 1–8 (2019). https://doi.org/10.1007/s10048-018-0560-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-018-0560-x