
Abstract
Iron–sulfur clusters are ubiquitous inorganic co-factors that contribute to a wide range of cell pathways including the maintenance of DNA integrity, regulation of gene expression and protein translation, energy production, and antiviral response. Specifically, the iron–sulfur cluster biogenesis pathways include several proteins dedicated to the maturation of apoproteins in different cell compartments. Given the complexity of the biogenesis process itself, the iron–sulfur research area constitutes a very challenging and interesting field with still many unaddressed questions. Mutations or malfunctions affecting the iron–sulfur biogenesis machinery have been linked with an increasing amount of disorders such as Friedreich’s ataxia and various cardiomyopathies. This review aims to recap the recent discoveries both in the yeast and human iron–sulfur cluster arena, covering recent discoveries from chemistry to disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron–sulfur clusters are metal prosthetic groups, synthesized and utilised in different cell compartments. They are present in nearly all organisms and required for a variety of protein biological functions, such as enzyme activity, protein regulation, and translation [1,2,3,4,5,6]. Fe–S clusters are considered to be among the most ancient catalysts, a concept that is supported by the elevated iron and sulfur levels in the environment and their unique characteristics, including the electron charge transfer activity and the formation into complexes [7, 8].
Despite their abundance, one of the many challenges which this field had to face early on was the detection of the clusters on proteins. The isolation of an intact complex of the protein with the cluster has been a difficult task, as the cluster can easily dissociate or change its redox state. These issues along with the lack of additional structural protein information were the cause for slow paced study of the clusters.
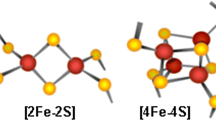
Consequently, although Fe–S clusters have been studied for decades, many aspects of their synthesis, transfer, incorporation on proteins, and role have still to be understood [9,10,11]. Even though Fe–S clusters can be found in different chemical structures, all of them derive from iron ions and sulfide (Fig. 1). The simplest form that can be found in nature is [2Fe–2S] and duplication of this form results to the cubic cluster [4Fe–4S]. In addition, loss of one Fe ions of that form leads to the non-symmetrical [3Fe–4S] structure [10]. Finally, another form is composed of a cubane [4Fe–4S] together with a [4Fe–3S] cluster resulting in the [8Fe–7S] cluster [3, 12].

Iron–sulfur clusters and proteins. a Example structures of known and well-characterised Fe/S clusters. Blue; Fe, black; sulfur. b Crystal structure of XPD helicase (PDB ID 3CRV) and c ferredoxin 2 (PDB ID 4ZHO) with a [4Fe–4S] and [2Fe–2S] clusters bound (black sticks)
These inorganic moieties pose an excellent example of co-factors that are highly reactive and thus suitable for many catalytic reactions. However, this feature is the main cause of their susceptibility as they are also prone to oxidation that leads to the Fe–S cluster inactivation. Many enzymes, such as DNA-binding proteins, have traded over iron for zinc or other metals that are less sensitive to oxidation and thus less likely to cause toxicity. Specifically, it was originally believed that glycosylases were the only DNA-binding enzymes that utilise Fe–S clusters [2, 13]. However, in 2006, another DNA enzyme, the helicase XfPD (Rad3 in Saccharomyces cerevisiae) was shown to bind to an iron–sulfur cluster within the catalytic domain of the protein. This finding indicated a role for iron–sulfur clusters in sensing DNA disruption (Fig. 1) and led to discovering more DNA-binding proteins that use Fe–S clusters, including all DNA polymerases [14,15,16]. The role of Fe–S clusters in processing nucleic acids is still not fully understood, although they are indispensable for enzyme activity. The main question focuses on why these metal moieties are preferred over others, when the risk of toxicity or DNA damage by iron is so great. This is a critical research area that can link Fe–S cluster chemistry with protein function, DNA damage, and subsequently diseases.
In this review, we will first describe the steps for the Fe–S biogenesis in the model organism Saccharomyces cerevisiae (budding yeast) and expand onto the relevant mechanisms in mammalian cells covering the recent information on the transport and delivery of the clusters into apoproteins. Given the importance that mitochondria have on Fe–S cluster formation, we will discuss any putative links between that pathway and the organelle’s biogenesis. Finally, some of the main human disorders caused by defects in proteins underpinning the Fe–S biogenesis machinery will be discussed.
Fe–S cluster pathways: from yeast to mammalian cells
For decades, it was presumed that Fe–S clusters are incorporated on apoproteins in a spontaneous fashion, a theory that was proved insufficient, since in vitro successful integration required toxic Fe levels. A more plausible theory was that there are specific pathways responsible for the biosynthesis of Fe–S for the final delivery to apoproteins. Later, studies on the fields of iron chemistry, metabolism, and oxidative stress provided the evidence that changed the perception of Fe–S biosynthesis [17,18,19]. One of the main breakthroughs was the investigation of the enzyme nitrogenase [20], that catalyses the reduction of dinitrogen, known as ‘nitrogen fixation’. Dennis Dean and colleagues performed genetic and biochemical studies on the Gram-negative facultative anaerobe Klebsiella pneumoniae bacterium identifying genes involved in the nitrogen fixation process. The comparison between the sequences of the nifUSV gene clusters from K. pneumoniae and Azotobacter vinelandii revealed identical organization of the genomes as well as a high degree of sequence homology [21]. The studies made on A. vinelandii were crucial to the discovery of Fe–S biogenesis mechanism as three operons dedicated to the Fe–S biogenesis were found, one of them being the nif operon, involved in the biogenesis of nitrogenase [22]. Since then, the iron–sulfur cluster synthesis and assembly pathways have begun to be described, in addition to the sensing iron-level mechanisms, providing a better understanding of the complicated chemistry of iron [23, 24]. Bacteria Fe–S cluster biogenesis machineries have been reviewed elsewhere [25, 26], and thus, are not the focus of this review. The data summarised below have been acquired from studies mainly in yeast and mammalian cell lines. Since the pathways are conserved, we only shortly refer to the yeast system and analyse, in more detail, the mammalian as a way to introduce the relevance and implication of this co-factor in a wide range of diseases.
Fe–S clusters in Saccharomyces cerevisiae
Iron–sulfur cluster proteins can be found in most cell compartments from mitochondria to the nucleus. Due to their role complexity, the need for a simple model system to study these molecules was apparent. The yeast S. cerevisiae has been used in the past thoroughly to study complex biological processes as a simple, easy to grow eukaryotic model organism and provided the necessary platform for the investigation of the highly conserved Fe–S pathways.
Free iron (as well as sulfides) can be toxic to the cell [27, 28], rendering its regulation, uptake, and assembly essential processes that should be controlled constantly. Three biosynthesis pathways have been described so far that contribute to that end; two at the mitochondrial level [iron–sulfur cluster (ISC)] and one at the cytosolic compartments [cytosolic iron–sulfur cluster assembly (CIA)] (Fig. 2) [2, 11, 29, 30]. In the mitochondrial matrix, the first stage of the ISC pathway comprises of the iron–sulfur cluster synthesis, with the extraction of sulfur from cysteine. This reaction is catalysed by the enzyme cysteine desulfurase, Nfs1. Next is the cluster assembly, a step that is accomplished on the scaffold protein Isu1 by a group of enzymes that utilise the available iron and sulfur, as shown in Fig. 2. The Fe–S cluster is subsequently released from the scaffold protein and transferred by chaperones to glutaredoxin 5 (Grx5). The cooperation between Grx5 and co-chaperones, Jac1 and Ssq1, leads to the final incorporation of the clusters onto the target apoproteins [31]. Apart from the simple cluster, the mitochondrial ISC machinery can also produce the more complex forms that are required for the function of several proteins in the organelle. The synthesis of a cubic cluster from the initially formed [2Fe–2S] requires a conversion reaction occurring later in the pathway. Alternatively, the [2Fe–2S] clusters follow the putative mitochondrial export route to be used by the CIA system.

Fe/S cluster biogenesis. The process includes three different stages, two in mitochondria (A and B), and one in the cytosol (C). In the mitochondrial matrix, the ISC machinery (A) is responsible for the formation of the clusters and the maturation of the apoproteins. An unknown compound (X-S) is exported from the matrix (B) through the ISC export pathway. In the cytosol (C), the CIA machinery takes over for the incorporation of the clusters in the proteins
The exported form of the cluster has been the subject of many studies over the years; and despite substantial progress in the field, the identity of the exported species remains speculative. Originally, it was shown that the export pathway involves three components, one membrane channel protein (Atm1), one sulfhydryl oxidase (Erv1), and one reducing factor [glutathione (GSH)]. The equivalent human homologues will be reviewed later. The most recent data show that the exported cluster could be stabilised by glutathione molecules [32, 33]. Specifically, crystal structures revealed that reduced glutathione (GSH) was associated with both the yeast and bacterial pore Atm1, whereas bacterial Atm1 could also bind to oxidised glutathione (GSSG). However, there are still no concrete data of transported conjugates of glutathione and iron–sulfur cluster, rendering the real nature compound elusive. In addition, there are studies that contradict the presence of an export pathway, based on the inconclusive evidence of an exported cluster from mitochondria and the cytosolic presence of the initial proteins involved in the cluster formation [34].
In the cytoplasm, a [2Fe–2S] cluster that is in turn bound by the two monothiol glutaredoxins, Grx3 and Grx4 [35]. The two proteins act as a homodimer coordinating the cluster with the assistance of two glutathione molecules [35, 36]. The Fe–S cluster can be further used for the biogenesis of cytosolic and nuclear Fe–S proteins. Specifically, the two proteins Tah18–Dre2 provide the necessary electron donors to deliver the assembly of the cluster on the first scaffold protein complex of the CIA machinery, Cfd1–Nbp35 [37, 38]. Next, the cluster is transferred through the pathway molecules to the cytosolic apoproteins for their maturation (Fig. 2).
Fe–S clusters in mammalian cells
As noted by the fact that the assembly involved in the biogenesis of Fe–S clusters is highly conserved throughout evolution, mammalian Fe–S cluster biogenesis follows a path that resembles both the yeast and bacterial pathways. The bacterial classical system is mainly regulated by the Isc gene [39] and homologues of the proteins encoded by this gene can be found in mammals. In this section, we will focus on the mammalian biogenesis machinery as a basis to show its involvement in several diseases such as neurodegenerative disorders.
The process of the de novo synthesis of Fe–S clusters takes place in mitochondria and is driven by the ISC machinery. Furthermore, it has been suggested that Fe–S biogenesis can also take place in the cytosol and the nucleus [40,41,42]. The synthesis starts with the delivery of both the sulfur and iron ions onto the scaffold protein ISCU [43]. The sulfur is provided by a cysteine desulfurase (NFS1) which removes and delivers it from l-cysteine onto ISCU. This delivery process is driven by the formation of transient persulfide on the active cysteine of NFS1, which forms a complex with ISD11 (also known as LYRM4) and the mitochondrial acyl carrier protein (ACP) [44]. The importance of the NFS1 and ISD11 has been shown by the inefficient maturation of Fe–S proteins in HeLa cells with depleted NFS1 and the accumulation of ferric iron and inactivation of aconitase in cells with downregulated ISD11, respectively [45, 46]. An allosteric regulation role for the sulfur transfer onto the ISCU–NFS1–ISD11–ACP complex has been suggested for the mammalian frataxin (FXN) [47, 48]. The iron source for the cluster assembly is not known yet; however, FXN has also been proposed as one of the iron entry regulators during the [4Fe–4S] cluster formation [49]. It is interesting to note that unlike the mammalian FXN, its bacterial homologue CyaY exerts a negative regulatory effect on the generation of Fe–S instead of its activation role present in mammals [50]. The mechanisms and players for the insertion of Fe into the complex need to be elucidated, but it has been suggested that Fe is required for the sulfur to be delivered form NFS1 [51]. Electrons are required for the generation of the [2Fe–2S] cluster and they are likely transferred from NAD(P)H to ferredoxin reductase (FDXR) and onto ferredoxin (FDX) [52].
Following its formation by the ISC machinery, the Fe–S cluster needs to be transferred to recipient apoproteins. Although little is known about this process in mammals, evidence based on yeast and bacteria suggests that the chaperone HSPA9 (HscA in bacteria) binds to the co-chaperone HSC20 (HscB in bacteria) and targets the ISCU complex for the release of the [2Fe–2S] cluster, likely mediated by a conformational change in ISCU that favours the release [53]. It has been proposed that the cluster can be released to glutaredoxin 5 (GLRX5), as the yeast Ssq1 has been shown to interact with GLRX5 to facilitate the transfer of the cluster [54]. This concept is supported by the discovery of [2Fe–2S] clusters buried in human GLRX5 revealed by its crystal structure [55]. The complex involved in late maturation of [4Fe–4S] clusters is composed of the human proteins ISCA1, ISCA2 and IBA57. This complex does not interact directly with the initial ISC machinery. The importance of each component was shown by decreased activity of aconitase, lipoic acid synthase, and the complex I of the respiratory chain in HeLa cells, where the three proteins were downregulated [56]. Finally, the trafficking from the ISCA1–ISCA2–IBA57 complex to apoproteins is facilitated by NFU1. The bacterial homologue Nfu1 interacts with the [4Fe–4S] cluster machinery and target proteins, suggesting a similar role for the human protein [57]. Furthermore, the Bol1 and Bol3 targeting factors have been involved in the maturation of a specific set of [4Fe–4S] proteins [57, 58].
The mechanism of the assembly, trafficking, and maturation of Fe–S clusters has been mainly described by the work made with the homologous proteins in both bacteria and yeast, but the relevance of most of these components in human physiology is shown by their relation to various diseases. In the following sections, we will discuss several diseases that have been linked to alterations in components involved in Fe–S cluster biogenesis.
Is there a link between mitochondria biogenesis components and Fe–S biogenesis?
The mitochondria import pathways are critical for mitochondria biogenesis, whilst the Fe–S cluster biogenesis is considered as one of the most essential functions of mitochondria. There are some fragmented studies that suggested a link between these two important processes, but whether this is true remains unclear. Specifically, the IMS oxidoreductase Mia40, one of the two key components for the MIA pathway responsible for targeting many intermembrane space proteins, has been shown (both in vivo and in vitro) to bind to iron/sulfur clusters, in addition to its well-established role in mitochondria biogenesis [59, 60]. Two monomers can coordinate the cluster through the catalytic CPC motif. However, it remains unclear whether the presence of Fe–S on Mia40 has any relevance for its import function. In mammalian cells deletion of Mia40 is associated with increased iron levels in mitochondria, but again whether this is a direct effect on Fe–S cluster availability/transport or an indirect effect of reduced import of some other protein(s) that are Mia40-dependent is not known.
Another link of the two processes is through the CIA component, Dre2, that has been shown to localize both in the cytosol and in mitochondria [61,62,63]. Dre2 interacts with Mia40 independently of the presence of its Fe–S clusters. This interaction results in the introduction of two disulfide bonds in the Dre2 structure. However, it was later demonstrated that the localization of Dre2 is difficult to prove unambiguously as this protein associates tightly with the outer membrane of mitochondria and resists proteolytic hydrolysis [64]. Even though the role of Dre2 in the cytosol as part of the CIA machinery is understood, a putative role for a mitochondria-associated sub-population has not been determined yet. One could speculate that the mitochondrial association may be triggered by specific determinants potentially assisting the delivery of the iron–sulfur clusters from the matrix into the cytosol.
Most of the mitochondrial biogenesis pathways are well conserved. One exception is the trypanosomatids, a family of protozoan parasites. It has been shown that oxidoreductase Mia40 is absent from this organism, while the Erv1 homologue seems to take over the entire function of the Mia/Erv1 pathway [65, 66]. It is not clear how this system in T. brucei can operate mechanistically. The investigation of the Fe–S cluster export pathway in this organism will be very exciting.
Intriguingly, the initial results that linked lower levels of Erv1 in a yeast temperature mutant to a role of this protein in Fe–S cluster were later disputed by another study. This showed that glutathione levels in this strain were surprisingly lower and it is the effect of the levels of glutathione, the apparent cause for the observed defect in Fe–S cluster biogenesis [67]. These authors went to a great extent to investigate different strains and conditions relevant to both Erv1 and Mia40, and concluded that there is no link of the levels of iron to Erv1 or Mia40.
Another case of an iron–sulfur cluster protein that has been shown to have mitochondrial association is mitoNEET [68]. Specifically, this protein is anchored to the outer membrane facing the cytosol; and even though it has a zinc finger domain, it binds to iron, specifically [2Fe–2S] clusters. Many studies focus on this protein, as its overexpression is linked with various effects, such as enhanced lipid uptake, reduced membrane potential, less ROS production, and inhibition of iron transport from the cytosol in mitochondria [69]. All the above point to a role of mitoNEET as an important mitochondrial regulator and an interesting potential target for therapies. MitoNEET has already been used as a target of the insulin-sensitizing thiazolidinedione diabetes drugs [70].
The case of oxymonad Monocercomonoides sp.
Mitochondria are considered in general indispensable for viability. Supporting this concept, in the past, there have been reports of organisms surviving in low oxygen environments with a reduced form of this organelle. Recently, however, a very interesting study reported the first case of eukaryotic organism completely devoid of mitochondria, the oxymonad Monocercomonoides sp. [71]. It appears that the absence of mitochondria in this case is not an ancestral characteristic, but instead a secondary loss incident. The data are based mainly in phylogenetic analysis and genome searches for known genes, normally selected as markers in mitochondrial studies, for example membrane translocases. The searches proved unsuccessful as no such sequences are present in the Monocercomonoides sp. genome. Regarding the Fe–S cluster biosynthesis pathways, however, this organism contains a cytosolic sulfur mobilization system (SUF), that has been described in bacteria, instead of the mitochondrial iron–sulfur cluster assembly pathways (ISC). In addition to the SUF, the group identified other essential genes for glycolytic proteins including enzymes for anaerobic glycolysis, indicating that the mitochondrial absence is solid. Thus, the existence of Monocercomonoides sp. highlights a unique exception to the concept that mitochondria are essential for viability in all eukaryotes.
Iron sensing and regulation
Well-studied and known reactions that utilise Fe–S clusters include the sulfur donors in biosynthesis, the mitochondrial electron transport chain reactions, catalysis by aconitase, etc. These reactions have been linked with human diseases via elevated iron levels, mutations in enzymes, or DNA damage.
As mentioned before, even though iron is vital for the cell processes, excess amounts can lead to oxidative damage and toxicity. Cells have adapted to address to such a challenge, by developing specific mechanisms to strategically regulate iron intake, storage, and utilization according to the variations in cell environment.
Iron regulatory proteins (IRPs) are responsible for controlling the translation of genes involved in those specific mechanisms by binding to non-coding sequences of the corresponding mRNAs, known as iron-responsive elements (IREs). This mechanism depends on the cell iron conditions, in addition to structural features of the enzyme [72]. Thus, in the presence of high iron levels, IRP1 binds a cubic iron–sulfur cluster and function as a cytosolic aconitase. There are many dedicated studies on IRP1, their structural features, interaction with hypoxia inducing factors that are covered in other reviews [73, 74].
Another example is in Saccharomyces cerevisiae, where the iron metabolism is regulated by the transcription factors Aft1/Aft2 and Yap5, in accordance to the available iron levels [24, 36]. The first two are implicated with the activation of genes when the iron levels are low, while Yap5 is involved when the iron levels are extremely high. In particular, these proteins receive a signal from the mitochondrial ISC, in the form of a cluster, and with the help of the monothiol glutaredoxins Grx3 and Grx4, they regulate their transcriptional function [35, 75,76,77]. All genes associated with iron sensing and regulation include a specific DNA sequence in their promoter, the iron regulatory element (known as IRE), that is recognised by the transcription factors.
There are also many indications that tripeptide glutathione (GSH) plays an essential role in cellular iron metabolism [78, 79]. Defects in glutathione biosynthesis can lead to accumulation of iron in mitochondria [80, 81].
Diseases associated with iron–sulfur clusters
The involvement of iron–sulfur clusters in the vast majority of the cell processes makes it clear that many diseases are linked with malfunctions or mutations in pathways that include them. Below, we mention some of the many known and studied diseases associated with Fe–S pathways, also summarised in Table 1.
Friedreich’s ataxia
Friedreich’s ataxia (FRDA) is the most common disease associated with dysfunction of Fe–S biogenesis. It is an autosomal recessive neurodegenerative disease with an incidence of 1/50,000 in Caucasian population [82]. FRDA is caused by a homozygous guanine–adenine–adenine (GAA) repeat expansion within the first intron of the frataxin gene (FXN) located on the long arm of chromosome 9 [83]. The main symptoms have been associated with FRDA dysarthria (a motor speech disorder), scoliosis, muscle weakness, and loss of position sense. In addition, FRDA can lead to diabetes and cardiomyopathy.
As mentioned before, frataxin is involved in Fe–S clusters biogenesis. Thus, alterations linked to iron metabolism are present in FRDA. The pathophysiology of FRDA comprises deficit of aconitase and respiratory chain complexes, presence of oxidative damage markers in blood and urine, and intracellular iron accumulation [82, 84,85,86]. Currently, no successful treatment is available for FRDA. One main reason for this is the lack of understood detailed understanding of mechanisms of Fe–S cluster biogenesis and appropriate disease models. However, some treatments involving iron chelators like deferiprone are already in clinical trials and are listed in the Friedreich’s Ataxia Research Alliance; and others are still being developed and tested in animals (i.e., gene therapy targeting the FXN gene) [87, 88].
ISCU and FDX2 myopathies
Myopathy is a disorder of skeletal muscles with the presence of impairment of muscle fibers. The ISCU myopathy is an autosomal recessive disorder characterised by exercise intolerance that leads to increased lactate and pyruvate concentrations and it was first identified in Sweden [89]. This disorder is caused by severely reduced levels of ISCU protein in individuals that share a point mutation in the fourth intron of the ISCU gene that causes a premature stop codon. This mutation amplifies a polypyrimidine tract (tctttg to tctttc), which is normally a weak splice acceptor. As a consequence of this, the strengthened site allows the inclusion of an aberrant exon into the transcript [90]. The reduced levels of a functional ISCU protein lead to Fe–S clusters biogenesis and Fe homeostasis alterations in samples from patients [91]. A mutation of a conserved glycine residue into glutamate has been shown to interfere with the interaction of the scaffold protein with NFS1 and HSC20 [92]. The use of specific anti-sense oligonucleotide targeting the ISCU gene to correct the reading frame has shown promising results in fibroblast derived from patients [93].
Similar to the ISCU myopathy, mutations in the FDX1L gene which disrupts the initiation translation site of the FDX2 protein caused proximal muscle myopathy associated with myoglobinuria and lactic acidosis together with reduced activity of aconitase and affected complex II [94].
Infantile complex II/III deficiency
A lethal autosomal recessive disease caused by a mutation that leads to substitution of arginine into glutamine in the cysteine desulfurase, NFS1, has been identified. It is characterised by lactic acidosis, muscle deficiencies in respiratory complex II and III that have as consequence multisystem organ failure [95]. Little is known about this disorder as just three patients have been reported so far, but its lethality as well as the strong impairment of both cytosolic and mitochondrial Fe–S proteins in NFS1 knocked down HeLa cells stress the importance of the activity of this protein.
Respiratory chain complexes’ deficiency
A homozygous mutation in LYRM4, the gene encoding for ISD11, was found in two patients with decreased oxidative phosphorylation. The mutation was identified in a patient with deficiency of complex I, II, and III in muscle and liver by massive parallel sequencing (MitoExome sequencing) [96]. The same mutation was identified in another affected patient who presented additional complex IV deficiency and who died while neonate. The differences between the outcomes of both patients are suggested to be due to the availability of cysteine in the new-born period. Furthermore, in vitro experiments showed no desulfurase activity of NFS1 when expressed together with mutant ISD11, reflecting the importance role of this protein for the stabilisation of the desulfurase [96].
X-linked sideroblastic anemia and ataxia
The ATP-binding cassette, sub-family B, member 7 (ABCB7) is a membrane associated protein located in mitochondria. ABCB7 is a transporter present in the inner membrane of mitochondria and its depletion in HeLa cells showed Fe accumulation within mitochondria [97]. Mutations in the ABCB7 gene cause a recessive disorder, namely X-linked sideroblastic anemia and ataxia (XLSA/A) [98]. XLSA/A is an early onset disease characterised by a blood disorder in which erythrocytes do not produce enough haemoglobin (sideroblastic anemia) and slow-progressive movement problems (slow-progressive ataxia) [99]. The mutations on XLSA/A patients are in the transmembrane domain, but still, complete understanding of ABCB7 function is needed to fully characterise this disorder and can develop a possible cure.
Sideroblastic anemia or variant non-ketonic hyperglycinemia and iron overload
Sideroblastic anemias are a group of heterogeneous disorders that share common features like mitochondrial iron overload, high numbers of ringed sideroblasts, and affected erythropoiesis. Among the different types of sideroblastic anemia, one version is caused by a large deletion on the GLRX5 gene. This mutation in intron 1 leads to complete loss of GLRX5 protein function and the subsequent impairment of Fe–S biogenesis together with the activation of the iron-responsive element (IRE)-binding activity of iron regulatory protein 1 (IRP1) [100].
Multiple mitochondrial dysfunctions syndromes
Multiple mitochondrial dysfunctions syndromes (MMDS) are due to mutations in proteins involved in biogenesis of Fe–S clusters and are characterised by mitochondria affected at multiple levels. MMDS 1 is caused by mutations in affecting NFU1 and was first identified in three siblings of Mexican origins, while MMDS 2 is the result of a mutation that leads to premature stop codon in the BOLA3 gene causing severe epileptic encephalopathy and elevated lactate and glycine levels. Finally, MMDS 3 is caused by a homozygous mutation that diminishes the activity of IBA57 which leads to defects in mitochondrial respiratory complexes I and II [102–105].
Conclusion
Despite the rapid progress in the Fe–S field, there are still many unanswered questions regarding the interactions between the individual proteins, the formation of complexes, the identity or even the presence of an X-S exported material from the mitochondria, and the delivery of the clusters into the recipient apoproteins. Research in this field will help to understand the determinants of many diseases associated with iron cluster protein pathways. Furthermore, the development of appropriate cell and mouse models for these diseases is crucial to further investigate potential therapeutic strategies.
Abbreviations
- Aft:
-
Activator of ferrous transport
- Atm1:
-
ATP-binding cassette (ABC) transporter
- Cfd1:
-
Complement factor D
- CIA:
-
Cytosolic iron–sulfur cluster assembly
- Erv1:
-
Essential for respiration and vegetative growth 1
- GFER:
-
Growth factor, augmenter of liver regeneration
- Grx/GLRX:
-
Glutaredoxin
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidised glutathione
- HSP9:
-
Heat shock protein 9
- HSC20:
-
Heat shock cognate protein 20
- IRP:
-
Iron regulatory proteins
- IRES:
-
Iron-responsive elements
- ISC:
-
Iron–sulfur cluster
- Isu1:
-
Iron–sulfur cluster assembly scaffold protein
- Jac1:
-
J-type accessory chaperone 1
- Nbp35/NUBP1:
-
Nucleotide-binding protein
- Rad3:
-
TFIIH/NER complex ATP-dependent 5′–3′ DNA helicase subunit RAD3
- Ssq1:
-
Stress-seventy sub-family Q 1
- SUF:
-
Sulfur mobilization system
- XPD:
-
Xeroderma pigmentosum group D helicase
- Yap5:
-
Yeast AP-5
References
Andreini C, Rosato A, Banci L (2017) The relationship between environmental dioxygen and iron–sulfur proteins explored at the genome level. PLoS ONE 12:e0171279. https://doi.org/10.1371/journal.pone.0171279
Fuss JO, Tsai C-L, Ishida JP, Tainer JA (2015) Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim Biophys Acta 1853:1253–1271. https://doi.org/10.1016/j.bbamcr.2015.01.018
Beinert H (2000) Iron–sulfur proteins: ancient structures, still full of surprises. J Biol Inorg Chem 5:2–15
Snyder CH, Merbitz-Zahradnik T, Link TA, Trumpower BL (1999) Role of the Rieske iron–sulfur protein midpoint potential in the protonmotive Q-cycle mechanism of the cytochrome bc1 complex. J Bioenerg Biomembr 31:235–242
Hunsicker-Wang LM, Heine A, Chen Y et al (2003) High-resolution structure of the soluble, respiratory-type Rieske protein from Thermus thermophilus: analysis and comparison. Biochemistry 42:7303–7317. https://doi.org/10.1021/bi0342719
Peters JW, Stowell MH, Soltis SM et al (1997) Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 36:1181–1187. https://doi.org/10.1021/bi9626665
Fontecave M (2006) Iron–sulfur clusters: ever-expanding roles. Nat Chem Biol 2:171–174. https://doi.org/10.1038/nchembio0406-171
Imlay JA (2006) Iron–sulphur clusters and the problem with oxygen. Mol Microbiol 59:1073–1082. https://doi.org/10.1111/j.1365-2958.2006.05028.x
Rouault TA (2014) Mammalian iron–sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol 16:45–55. https://doi.org/10.1038/nrm3909
Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Structure, function, and formation of biological iron–sulfur clusters. Annu Rev Biochem 74:247–281. https://doi.org/10.1146/annurev.biochem.74.082803.133518
Lill R, Dutkiewicz R, Freibert SA et al (2015) The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron–sulfur proteins. Eur J Cell Biol 94:280–291. https://doi.org/10.1016/j.ejcb.2015.05.002
Beinert H, Holm RH, Münck E (1997) Iron–sulfur clusters: nature’s modular, multipurpose structures. Science 277:653–659
Boal AK, Yavin E, Lukianova OA et al (2005) DNA-bound redox activity of DNA repair glycosylases containing [4Fe–4S] clusters†. Biochemistry 44:8397–8407. https://doi.org/10.1021/bi047494n
Rudolf J, Makrantoni V, Ingledew WJ et al (2006) The DNA repair helicases XPD and FancJ have essential iron–sulfur domains. Mol Cell 23:801–808. https://doi.org/10.1016/j.molcel.2006.07.019
Pokharel S, Campbell JL (2012) Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron–sulfur cluster domain. Nucleic Acids Res 40:7821–7830. https://doi.org/10.1093/nar/gks534
Netz DJA, Stith CM, Stümpfig M et al (2011) Eukaryotic DNA polymerases require an iron–sulfur cluster for the formation of active complexes. Nat Chem Biol 8:125–132. https://doi.org/10.1038/nchembio.721
Kispal G, Csere P, Prohl C, Lill R (1999) The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J 18:3981–3989. https://doi.org/10.1093/emboj/18.14.3981
Strain J, Lorenz CR, Bode J et al (1998) Suppressors of superoxide dismutase (SOD1) deficiency in Saccharomyces cerevisiae. Identification of proteins predicted to mediate iron–sulfur cluster assembly. J Biol Chem 273:31138–31144
Schilke B, Voisine C, Beinert H, Craig E (1999) Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 96:10206–10211
Zheng L, White RH, Cash VL et al (1993) Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc Natl Acad Sci USA 90:2754–2758
Beynon J, Ally A, Cannon M, Cannon F, Jacobson M, Cash V, Dean D (1987) Comparative organization of nitrogen fixation-specific genes from Azotobacter vinelandii and Klebsiella pneumoniae: DNA sequence of the nifUSV genes. J Bacteriol 169(9):4024–4029
Rouault TA, Maio N (2017) Biogenesis and functions of mammalian iron–sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J Biol Chem 292(31):12744–12753. https://doi.org/10.1074/jbc.R117.789537
Bird AJ (2015) Cellular sensing and transport of metal ions: implications in micronutrient homeostasis. J Nutr Biochem 26:1103–1115. https://doi.org/10.1016/j.jnutbio.2015.08.002
Martínez-Pastor MT, Perea-García A, Puig S (2017) Mechanisms of iron sensing and regulation in the yeast Saccharomyces cerevisiae. World J Microbiol Biotechnol 33:75. https://doi.org/10.1007/s11274-017-2215-8
Py B, Barras F (2010) Building Fe–S proteins: bacterial strategies. Nat Rev Microbiol 8:436–446. https://doi.org/10.1038/nrmicro2356
Blanc B, Gerez C, Ollagnier de Choudens S (2015) Assembly of Fe/S proteins in bacterial systems. Biochim Biophys Acta Mol Cell Res 1853:1436–1447. https://doi.org/10.1016/j.bbamcr.2014.12.009
Lane DJR, Merlot AM, Huang ML-H et al (2015) Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta 1853:1130–1144. https://doi.org/10.1016/j.bbamcr.2015.01.021
Kabil O, Motl N, Banerjee R (2014) H2S and its role in redox signaling. Biochim Biophys Acta 1844:1355–1366. https://doi.org/10.1016/j.bbapap.2014.01.002
Lill R, Hoffmann B, Molik S et al (2012) The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta 1823:1491–1508. https://doi.org/10.1016/j.bbamcr.2012.05.009
Stehling O, Smith PM, Biederbick A et al (2007) Investigation of iron–sulfur protein maturation in eukaryotes. Methods Mol Biol 372:325–342. https://doi.org/10.1007/978-1-59745-365-3_24
Uzarska MA, Dutkiewicz R, Freibert SA, Lill R, Mühlenhoff U (2013) The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation. Mol Biol Cell 12:1830–1841. https://doi.org/10.1091/mbc.E12-09-0644 PMID: 23615440
Srinivasan V, Pierik AJ, Lill R (2014) Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 343:1137–1140. https://doi.org/10.1126/science.124672933
Lee JY, Yang JG, Zhitnitsky D et al (2014) Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science 343:1133–1136. https://doi.org/10.1126/science.1246489
Rouault TA (2015) Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol 16(1):45–55. https://doi.org/10.1038/nrm3909
Mühlenhoff U, Molik S, Godoy JR et al (2010) Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron–sulfur cluster. Cell Metab 12:373–385. https://doi.org/10.1016/j.cmet.2010.08.001
Ueta R, Fujiwara N, Iwai K, Yamaguchi-Iwai Y (2012) Iron-induced dissociation of the Aft1p transcriptional regulator from target gene promoters is an initial event in iron-dependent gene suppression. Mol Cell Biol 32:4998–5008. https://doi.org/10.1128/MCB.00726-12
Netz DJA, Stümpfig M, Doré C et al (2010) Tah18 transfers electrons to Dre2 in cytosolic iron–sulfur protein biogenesis. Nat Chem Biol 6:758–765. https://doi.org/10.1038/nchembio.432
Lill R, Srinivasan V, Mühlenhoff U (2014) The role of mitochondria in cytosolic-nuclear iron–sulfur protein biogenesis and in cellular iron regulation. Curr Opin Microbiol 22:111–119. https://doi.org/10.1016/j.mib.2014.09.015
Zheng L, Cash VL, Flint DH, Dean DR (1998) Assembly of iron–sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem 273:13264–13272. https://doi.org/10.1074/JBC.273.21.13264
Tong WH, Rouault T (2000) Distinct iron–sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J 19(21):5692–5700
Tong WH, Jameson GN, Huynh BH, Rouault TA (2003) Subcellular compartmentalization of human Nfu, an iron–sulfur cluster scaffold protein, and its ability to assemble a [4Fe–4S] cluster. Proc Natl Acad Sci USA 100(17):9762–9767
Tong WH, Rouault TA (2006) Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron–sulfur cluster biogenesis and iron homeostasis. Cell Metab 3:199–210
Muhlenhoff U, Gerber J, Richhardt N, Lill R (2003) Components involved in assembly and dislocation of iron–sulfur clusters on the scaffold protein Isu1p. EMBO J 22:4815–4825. https://doi.org/10.1093/emboj/cdg446
Van Vranken JG, Jeong M-Y, Wei P et al (2016) The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron–sulfur cluster biogenesis. Elife 5. https://doi.org/10.7554/elife.17828
Biederbick A, Stehling O, Rosser R et al (2006) Role of human mitochondrial Nfs1 in cytosolic iron–sulfur protein biogenesis and iron regulation. Mol Cell Biol 26:5675–5687. https://doi.org/10.1128/MCB.00112-06
Shi Y, Ghosh MC, Tong W-H, Rouault TA (2009) Human ISD11 is essential for both iron–sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum Mol Genet 18:3014–3025. https://doi.org/10.1093/hmg/ddp239
Parent A, Elduque X, Cornu D et al (2015) Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat Commun 6:5686. https://doi.org/10.1038/ncomms6686
Tsai C-L, Barondeau DP (2010) Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry 49:9132–9139. https://doi.org/10.1021/bi1013062
Colin F, Martelli A, Clémancey M et al (2013) Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J Am Chem Soc 135:733–740. https://doi.org/10.1021/ja308736e
Bridwell-Rabb J, Iannuzzi C, Pastore A, Barondeau DP (2012) Effector role reversal during evolution: the case of frataxin in Fe–S cluster biosynthesis. Biochemistry 51:2506–2514. https://doi.org/10.1021/bi201628j
Pandey A, Pain J, Ghosh AK et al (2015) Fe–S cluster biogenesis in isolated mammalian mitochondria. J Biol Chem 290:640–657. https://doi.org/10.1074/jbc.M114.610402
Shi Y, Ghosh M, Kovtunovych G et al (2012) Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron–sulfur cluster biogenesis. Biochim Biophys Acta Mol Cell Res 1823:484–492. https://doi.org/10.1016/j.bbamcr.2011.11.002
Cai K, Frederick RO, Kim JH et al (2013) Human mitochondrial chaperone (mtHSP70) and cysteine desulfurase (NFS1) bind preferentially to the disordered conformation, whereas co-chaperone (HSC20) binds to the structured conformation of the iron–sulfur cluster scaffold protein (ISCU). J Biol Chem 288:28755–28770. https://doi.org/10.1074/jbc.M113.482042
Uzarska MA, Dutkiewicz R, Freibert S-A et al (2013) The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation. Mol Biol Cell 24:1830–1841. https://doi.org/10.1091/mbc.E12-09-0644
Johansson C, Roos AK, Montano SJ et al (2011) The crystal structure of human GLRX5: iron–sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem J 433:303–311. https://doi.org/10.1042/BJ20101286
Sheftel AD, Wilbrecht C, Stehling O et al (2012) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe–4S] protein maturation. Mol Biol Cell 23:1157–1166. https://doi.org/10.1091/mbc.E11-09-0772
Melber A, Na U, Vashisht A et al (2016) Role of Nfu1 and Bol3 in iron–sulfur cluster transfer to mitochondrial clients. Elife 5:e15991. https://doi.org/10.7554/eLife.15991
Uzarska MA, Nasta V, Weiler BD et al (2016) Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron–sulfur proteins. Elife 5. https://doi.org/10.7554/elife.16673
Spiller MP, Ang SK, Ceh-Pavia E et al (2013) Identification and characterization of mitochondrial Mia40 as an iron–sulfur protein. Biochem J 455:27–35. https://doi.org/10.1042/BJ20130442
Murari A, Thiriveedi VR, Mohammad F et al (2015) Human mitochondrial MIA40 (CHCHD4) is a component of the Fe–S cluster export machinery. Biochem J 471:231–241. https://doi.org/10.1042/BJ20150012
Vernis L, Facca C, Delagoutte E et al (2009) A newly identified essential complex, Dre2–Tah18, controls mitochondria integrity and cell death after oxidative stress in yeast. PLoS ONE 4:e4376. https://doi.org/10.1371/journal.pone.0004376
Banci L, Bertini I, Ciofi-Baffoni S et al (2011) Anamorsin is a [2Fe–2S] cluster-containing substrate of the Mia40-dependent mitochondrial protein trapping machinery. Chem Biol 18:794–804
Zhang Y, Lyver ER, Nakamaru-Ogiso E et al (2008) Dre2, a conserved eukaryotic Fe/S cluster protein, functions in cytosolic Fe/S protein biogenesis. Mol Cell Biol 28:5569–5582. https://doi.org/10.1128/MCB.00642-08
Peleh V, Riemer J, Dancis A, Herrmann J (2014) Protein oxidation in the intermembrane space of mitochondria is substrate-specific rather than general. Microb Cell 1:81–93. https://doi.org/10.15698/mic2014.01.130
Basu S, Leonard JC, Desai N et al (2013) Divergence of Erv1-associated mitochondrial import and export pathways in trypanosomes and anaerobic protists. Eukaryot Cell 12:343–355. https://doi.org/10.1128/EC.00304-12
Allen JWA, Ferguson SJ, Ginger ML (2008) Distinctive biochemistry in the trypanosome mitochondrial intermembrane space suggests a model for stepwise evolution of the MIA pathway for import of cysteine-rich proteins. FEBS Lett 582:2817–2825. https://doi.org/10.1016/j.febslet.2008.07.015
Ozer HK, Dlouhy AC, Thornton JD et al (2015) Cytosolic Fe–S cluster protein maturation and iron regulation are independent of the mitochondrial Erv1/Mia40 import system. J Biol Chem 290:27829–27840. https://doi.org/10.1074/jbc.M115.682179
Paddock ML, Wiley SE, Axelrod HL et al (2007) MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci USA 104:14342–14347. https://doi.org/10.1073/pnas.0707189104
Kusminski CM, Holland WL, Sun K et al (2012) MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med 18:1539–1549. https://doi.org/10.1038/nm.2899
Zuris JA, Harir Y, Conlan AR, Shvartsman M, Michaeli D, Tamir S, Paddock ML, Onuchic JN, Mittler R, Cabantchik ZI, Jennings PA, Nechushtai R (2011) Facile transfer of [2Fe–2S] clusters from the diabetes drug target mitoNEET to an apo-acceptorprotein. Proc Natl Acad Sci USA 108(32):13047–13052. https://doi.org/10.1073/pnas.1109986108
Karnkowska A, Vacek V, Zubáčová Z et al (2016) A eukaryote without a mitochondrial organelle. Curr Biol 26:1274–1284. https://doi.org/10.1016/j.cub.2016.03.053
Dupuy J, Volbeda A, Carpentier P et al (2006) Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 14:129–139. https://doi.org/10.1016/j.str.2005.09.009
Maio N, Rouault TA (2016) Mammalian Fe–S proteins: definition of a consensus motif recognized by the co-chaperone HSC20. Metallomics 8(10):1032–1046
Chung J, Anderson SA, Gwynn B et al (2014) Iron regulatory protein-1 protects against mitoferrin-1-deficient porphyria. J Biol Chem 289(11):7835–7843. https://doi.org/10.1074/jbc.M114.547778
Kumánovics A, Chen OS, Li L et al (2008) Identification of FRA1 and FRA2 as genes involved in regulating the yeast iron regulon in response to decreased mitochondrial iron–sulfur cluster synthesis. J Biol Chem 283:10276–10286. https://doi.org/10.1074/jbc.M801160200
Ojeda L, Keller G, Muhlenhoff U et al (2006) Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J Biol Chem 281:17661–17669. https://doi.org/10.1074/jbc.M602165200
Pujol-Carrion N, Belli G, Herrero E et al (2006) Glutaredoxins Grx3 and Grx4 regulate nuclear localisation of Aft1 and the oxidative stress response in Saccharomyces cerevisiae. J Cell Sci 119:4554–4564. https://doi.org/10.1242/jcs.03229
Deponte M (2017) The incomplete glutathione puzzle: just guessing at numbers and figures? Antioxid Redox Signal 27:1130–1161. https://doi.org/10.1089/ars.2017.7123
Kumar C, Igbaria A, D’Autreaux B et al (2011) Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J 30:2044–2056. https://doi.org/10.1038/emboj.2011.105
Rutherford JC, Ojeda L, Balk J et al (2005) Activation of the iron regulon by the yeast Aft1/Aft2 transcription factors depends on mitochondrial but not cytosolic iron–sulfur protein biogenesis. J Biol Chem 280:10135–10140. https://doi.org/10.1074/jbc.M413731200
Sipos K, Lange H, Fekete Z et al (2002) Maturation of cytosolic iron–sulfur proteins requires glutathione. J Biol Chem 277:26944–26949. https://doi.org/10.1074/jbc.M200677200
Rötig A, de Lonlay P, Chretien D et al (1997) Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet 17:215–217. https://doi.org/10.1038/ng1097-215
Filla A, De Michele G, Cavalcanti F et al (1996) The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet 59:554–560
Puccio H, Simon D, Cossée M et al (2001) Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet 27:181–186. https://doi.org/10.1038/84818
Bradley JL, Homayoun S, Hart PE et al (2004) Role of oxidative damage in Friedreich’s ataxia. Neurochem Res 29:561–567
Schulz JB, Dehmer T, Schöls L et al (2000) Oxidative stress in patients with Friedreich ataxia. Neurology 55:1719–1721
Pandolfo M, Hausmann L (2013) Deferiprone for the treatment of Friedreich’s ataxia. J Neurochem 126:142–146. https://doi.org/10.1111/jnc.12300
Perdomini M, Belbellaa B, Monassier L et al (2014) Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat Med 20:542–547. https://doi.org/10.1038/nm.3510
Drugge U, Holmberg M, Holmgren G et al (1995) Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: a genealogical study. J Med Genet 32:344–347
Holmes-Hampton GP, Crooks DR, Haller RG, Guo S, Freier SM, Monia BP, Rouault TA (2016) Use of antisense oligonucleotides to correct the splicing error in ISCU myopathy patient cell lines. Hum Mol Genet 25(23):5178–5187. https://doi.org/10.1093/hmg/ddw338
Mochel F, Knight MA, Tong W-H et al (2008) Splice mutation in the iron–sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet 82:652–660. https://doi.org/10.1016/j.ajhg.2007.12.012
Saha PP, Kumar SKP, Srivastava S et al (2014) The presence of multiple cellular defects associated with a novel G50E iron–sulfur cluster scaffold protein (ISCU) mutation leads to development of mitochondrial myopathy. J Biol Chem 289:10359–10377. https://doi.org/10.1074/jbc.M113.526665
Kollberg G, Holme E (2009) Antisense oligonucleotide therapeutics for iron–sulphur cluster deficiency myopathy. Neuromuscul Disord 19:833–836. https://doi.org/10.1016/j.nmd.2009.09.011
Spiegel R, Saada A, Halvardson J et al (2014) Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur J Hum Genet 22:902–906. https://doi.org/10.1038/ejhg.2013.269
Farhan SMK, Wang J, Robinson JF et al (2014) Exome sequencing identifies NFS1 deficiency in a novel Fe–S cluster disease, infantile mitochondrial complex II/III deficiency. Mol Genet Genom Med 2:73–80. https://doi.org/10.1002/mgg3.46
Lim SC, Friemel M, Marum JE et al (2013) Mutations in LYRM4, encoding iron–sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet 22:4460–4473. https://doi.org/10.1093/hmg/ddt295
Cavadini P, Biasiotto G, Poli M et al (2007) RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 109:3552–3559. https://doi.org/10.1182/blood-2006-08-041632
Allikmets R, Raskind WH, Hutchinson A et al (1999) Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 8:743–749. https://doi.org/10.1093/hmg/8.5.743
Bekri S, D’Hooghe M, Vermeersch P (1993) X-linked sideroblastic anemia and ataxia. University of Washington, Seattle
Ye H, Jeong SY, Ghosh MC et al (2010) Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest 120:1749–1761. https://doi.org/10.1172/JCI40372
Calvo SE, Tucker EJ, Compton AG et al (2010) High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet 42:851–858. https://doi.org/10.1038/ng.659
Cameron JM, Janer A, Levandovskiy V et al (2011) Mutations in iron–sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet 89:486–495. https://doi.org/10.1016/j.ajhg.2011.08.011
Haack TB, Rolinski B, Haberberger B et al (2013) Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis 36:55–62. https://doi.org/10.1007/s10545-012-9489-7
Seyda A, Newbold RF, Hudson TJ et al (2001) A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14–2p13. Am J Hum Genet 68:386–396. https://doi.org/10.1086/318196
Baker PR, Friederich MW, Swanson MA et al (2014) Variant non-ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137:366–379. https://doi.org/10.1093/brain/awt328
Acknowledgements
This article is based on work from COST Action CA15133 (‘FeSBioNet’) , supported by COST (European Cooperation in Science and Technology). Work in our lab was supported by Start-Up Funds to KT (Scottish Universities Life Sciences Alliance and the University of Glasgow), the Royal Society (Wolfson research merit award Grant Code: WM120111), and the Wellcome Trust (Grant Code: 202308/Z16/Z). MC-R is supported by a PhD studentship from CONACyT (Consejo Nacional de Ciencia y Tecnologia Mexico).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cardenas-Rodriguez, M., Chatzi, A. & Tokatlidis, K. Iron–sulfur clusters: from metals through mitochondria biogenesis to disease. J Biol Inorg Chem 23, 509–520 (2018). https://doi.org/10.1007/s00775-018-1548-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-018-1548-6