
Abstract
To investigate the genetic relationships between field strains of iridoviruses gathered from various fish species in Taiwan, viruses that were collected from 2001 to 2009 were analyzed. Open reading frames encoding the viral major capsid protein (MCP) and adenosine triphosphatase (ATPase) were sequenced for phylogenetic analysis. Our results indicated that iridoviruses from Taiwan aquaculture fishes could be classified into two groups: prior to 2005, the viruses were closely related to members of the genus Ranavirus; and after 2005, they were similar to members of the genus Megalocytivirus. Based on the analysis of MCP amino acid sequences, virus isolates were divided into 4 major genotypes that were related to ISKNV, RSIV, FLIV, and GIV, respectively. Pairwise comparisons of MCP genes showed that the ranavirus was an epidemic pathogen for economically important species in the major production regions and cultured marine fish, while the megalocytivirus isolates were sensitive to host range. In addition, the distribution of synonymous and non-synonymous changes in the MCP gene revealed that the iridoviruses were evolving slowly, and most of the variations were synonymous mutations. The Ka/Ks values were lower than one, and hence, the viruses were under negative selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Iridoviruses have been widely reported over the past 20 years in epizootics of finfish and amphibians in many countries. The virus has caused epidemics in Asia through international trade. Piscine iridoviruses that have been reported in Asian countries are classified into three genera: Lymphocystivius, Ranavirus and Megalocytivirus [6, 11, 26, 29, 33]. In Japan, red sea bream iridoviral disease (RSIVD) has been documented in 31 cultured seawater fishes [15]. In China, a molecular epidemiology study indicated that over 50 species of cultured and wild seawater fish were infected by infectious spleen and kidney necrosis virus–like (ISKNV-like) viruses [33]. In Korea, megalocytiviruses were reported in 10 freshwater ornamental fish species and 7 cultured fish species [6, 14].
Several iridovirus infections have been reported in Taiwan. The first one was reported in grouper, with diagnostic evidence provided by electron microscopy observations [1]. According to the histological, genetic, and microscopic characteristics, the epidemic strains were similar to RSIV [3, 32]. In previous studies, the grouper iridovirus TGIV was isolated in Taiwan (TGIV) using the KRE cell line [5], while a GK cell line was suitable for culture of grouper iridovirus (GIV), which was determined by molecular characterization to belong to the genus Ranavirus [19, 30]. Previous studies have shown that Singapore grouper iridovirus (SGIV) was transferred from Taiwan through diseased grouper fry [20].Viruses from cultured seawater fish in Taiwan were recently determined to be more closely related to the RSIV genotype than to the ISKNV genotype [31]. However, these findings were limited to one fish species of grouper and a single-culture environment. The genetic relationships between epidemic strains, geographic distribution, and host susceptibility of seawater and freshwater fish in Taiwan are unclear. In addition, the degree of variation in such iridoviruses is also unclear between different hosts and environments.
The most important gene for the analysis of genetic relationships in members of the family Iridoviridae is the major capsid protein (MCP) gene because this gene product is a structural protein with highly conserved domains [6, 8, 9] that encapsulate the most detailed evolutionary information. In addition, other genes with highly conserved regions, such as those encoding adenosine triphosphatase (ATPase), DNA polymerase, and methyltransferase can provide complementing information [34]. In this study, the genetic diversity of the complete MCP and ATPase genes of iridovirus isolates isolated from seawater and freshwater fishes between 2001 and 2009 in Taiwan was investigated. Furthermore, we evaluate the genetic changes of these isolates and determine the relationships between the epidemic strains. These data could provide important epidemiological information about viral transmission in different fish species and be used to discriminate imported carry-in from endemic circulation.
Materials and methods
Viruses
Twenty-three iridovirus isolates from seawater and freshwater fish from farms distributed throughout all seven Taiwan prefectures (Pingtung, Kaohsiung, Penghu, Tainan, Chiayi, Nantou and Taipei) between 2001 and 2009 were collected. The virus samples were collected from eight species of cultured fish in Taiwan: 11 isolates from giant grouper (Epinephelus lanceolatus), five from orange-spotted grouper (Epinephelus coioides), three from giant seaperch (Lates calcarifer), one from crimson snapper (Lutjanus erythropterus), one from silver sea bream (Rhabdosargus sarba), one from largemouth bass (Micropterus salmoides), one from rock bream (Oplegnathus fasciatus), and one from marble goby (Oxyeleotris marmoratus). Sources of the fish iridoviruses used in this study are listed in Table 1. “GG” refers to fish species of giant grouper; “IV” represents iridovirus, followed by the geographic site of isolation, the isolate number, and the year of isolation.
DNA extraction and primer design for PCR
Viral DNA was extracted from pooled organ tissues using a QIAamp DNA Mini Kit (QIAGEN) according to the manufacturer’s instructions. The extracted DNA was eluted with 100 μl of elution buffer. Primers for the genes encoding MCP and ATPase were designed based on the nucleotide sequence of ISKNV (AF371960), RSIV (AY310918), Korean flounder iridovirus (FLIV) (AY633992) and GIV (AY666015). Primers RMCP-F and RMCP-R were derived from the full-length DNA sequences of the MCP gene and were used to amplify approximately 1392 base pairs of the genomes of the isolates belonging to the genus Ranavirus. Primers MMCP-F and MMCP-R were used to amplify the MCP gene of the isolates belonging to the genus Megalocytivirus, and the expected product size was 1362 base pairs. For amplification the ATPase gene, a similar strategy was used for primer design. Both sets of primer sequences were modified slightly, as shown in Table 2, to amplify the expected PCR products.
PCR amplification, TA cloning and sequencing
PCR was carried out in 50-μl reactions containing 1XPCR buffer for Blend Taq (TOYOBO), 0.2 mM dNTPs, 10 pmol of each primer, 1.25 units of Blend Taq DNA polymerase, and 100 ng of extracted genomic DNA. PCR was performed in a PTC-200 DNA thermal cycler (MJ Research Inc., USA) at 95°C for 5 min; 35 cycles of 94°C for 30 sec, 54-58°C for 50 sec, and 72°C for 1 min; and extension at 72°C for 7 min. The PCR products were cloned into the pGEM-T vector (Promega) and sequenced using an ABI PRISM 377 DNA sequencer with the BigDye Terminator Kit (Applied Biosystems, Inc.). Several independent PCR clones (6-8 clones) were analyzed to produce a consensus nucleotide sequence for each virus isolate.
Phylogenetic analysis
Nucleotide and amino acid sequences were analyzed using the BioEdit 7.0.4 software. Multiple sequences were aligned using the ClustalW program. Phylogenetic relationships among species were determined using the neighbor-joining (NJ) method [21], and the reliability of the NJ tree was inferred using the Felsenstein bootstrap method with 1000 replications. This study compared the nucleotide and amino acid sequences of the MCP and ATPase genes with previously reported iridoviruses to determine the relationship between Taiwan iridovirus isolates. Sequences that were retrieved from the GenBank databases are shown in Table 3. Pairwise comparisons of the maximum nucleotide identities for MCP among different hosts using the MEGA 4.0.2 program [27] are shown in Table 4. Ratios of non-synonymous nucleotide substitutions per non-synonymous site (Ka) and synonymous nucleotide substitutions per synonymous site (Ks) were calculated using the MEGA 4.0.2 program. Sequences of the MCP and ATPase genes determined in this study were submitted to GenBank, and the accession numbers are listed in Table 1.
Results
Phylogenetic analysis of MCP
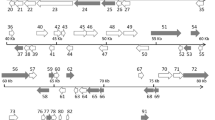
The MCP open reading frame (ORF) of was 1362 bp long and encoded 453 amino acids of the isolates belonging to the genus Megalocytivirus. In isolates belonging to the genus Ranavirus, the ORF was 1392 bp long and coded for a protein of 463 amino acids. In this study, we compared the full-length nucleotide and amino acid sequences of MCP with those previously reported from iridoviruses to determine the relationships among the 23 Taiwanese isolates. Based on the MCP nucleotide phylogenetic tree (Fig. 1), the 23 isolates were divided into 6 groups within the genera Ranavirus and Megalocytivirus. Isolates belonging to the genus Megalocytivirus were divided into 3 major groups: six isolates were closely related to the ISKNV genotype (group I), five isolates clustered with RSIV (group II), and 1 isolate was closely related to TRBIV and FLIV (group III). In the genus Ranavirus, the bootstrap values supported three major groups: three isolates were closely related to GIV (group V), eight isolates were related to SGIV (group VI) and FV3, and TFV was in group IV. In contrast, only five groups were supported in the phylogenetic tree of amino acid sequences (Fig. 2). In the Megalocytivirus cluster, the phylogenetic tree of amino acid sequences is similar to the one based on nucleotide sequences, consisting of three groups; however, all of the isolated strains that were related to ranaviruses from fish were clustered in groupV.

Phylogenetic analysis of iridovirus MCP. The full-length nucleotide sequences of the MCP genes from 23 Taiwan isolates were compared with previously reported sequences. Full virus names and accession numbers of previously published sequences are described in the legends of Tables 1 and 3. Numbers at the tree nodes indicate bootstrap values of 1000 replicates; bootstrap values below 70 are not shown. The scale bar is a genetic distance marker

Phylogenetic analysis of the MCP amino acid sequences. All of the specifications of the tree are similar to those in Fig. 1
Phylogenetic analysis of ATPase
The full-length ATPase gene of the megalocytiviruses was 720 bp long, encoding a protein of 233 amino acids, whereas in the ranaviruses, it was 972 bp long, encoding a protein of 242 amino acids. In this study, we compared nucleotide and amino acid sequences of the ATPase genes with those previously reported from iridoviruses to determine the relationships among the 23 Taiwanese isolates. Based on the ATPase nucleotide phylogenetic tree (Fig. 3), the 23 isolates could be divided into 5 groups in the genera Ranavirus and Megalocytivirus. The bootstrap values supported three major groups belonging to the genus Megalocytivirus. In group I, six isolates were closely related to the ISKNV genotype, and five isolates were related to RSIV (group II). There was one isolate related to TRBIV (group III). In the genus Ranavirus, the bootstrap values supported two major groups: the previously reported iridoviruses that originated from frogs species were localized in group IV, and all of the isolates that were collected from fish species were localized to group V and group VI. Based on the amino acid sequences (Fig. 4), phylogenetic analysis indicated that the 23 isolates could only be divided into 3 groups. All of the isolated strains that were closely related to members of the genera Ranavirus and Megalocytivirus were in group I and III, respectively.

Phylogenetic analysis of nucleotide sequences of the ATPase gene. All specifications of the tree are similar to those in Fig. 1

Phylogenetic analysis of ATPase amino acid sequences. The specifications of the tree are similar to those in Fig. 1
Identity analysis using MCP
In order to clarify the relationships between fish hosts and virus strains, pairwise comparison methods were used to determine the maximum identity of the MCP gene from the 23 isolates. Representative strains of each group (GIV, ISKNV, and RSIV) were included in our analysis. Comparison of GIV-related isolates from five different fish species revealed that they had a high degree of sequence identity to each other (97.9-100%). ISKNV-related isolates from three different fish species were also found to be highly identical to each other, with a high degree of sequence identity (99.5-99.9%). Finally, RSIV-related isolates that were found in three different fish species had a high degree of sequence identity to each other (97.9-99.9%). Furthermore, RBIV/Tp/45/08 showed a high degree of identity to RSIV (93.32%) (Table 4). These results demonstrate that giant seaperch, giant grouper, and orange-spotted grouper are susceptible to ranaviruses and megalocytiviruses. In Taiwan, the ranavirus was responsible for the epidemic affecting mostly marine cultured fish of the Pingtung, Kaohsiung, Penghu, Tainan, and Chiayi regions. Megalocytivirus isolates were found in both seawater and freshwater fish. Additionally, infections with GIV identified in crimson snapper and RSIV in marble goby were reported for the first time. These results indicated that viral transmission is possible in both seawater and freshwater fish species.
Analysis of genetic mutations in MCP
MCP is located on the surface of viral particles and is under selective pressure from host immune responses. To further investigate the extent of sequence variations in the MCP gene, we calculated the non-synonymous and synonymous substitution rates of 23 isolates and compared them to representative strains of GIV, ISKNV, and turbot reddish body iridovirus (TRBIV). The results are summarized in Table 5. Synonymous substitution rates ranged from 0 to 0.11%, whereas the non-synonymous rates were lower than 0.01%. Overall, synonymous substitutions were more frequent than non-synonymous substitutions in all isolates. The Ka/Ks values were lower than 1, indicating that the viruses are under negative selection. Furthermore, the analysis revealed that the nucleotide mutation sites were located in the first and second nucleotides but did not result in amino acid changes.
Discussion
In phylogenetic analysis, iridoviruses from Taiwanese fish clustered into two different genera. Prior to 2005, the epidemic strains were genetically similar to members of the genus Ranavirus; however, after 2005, they were closely related to members of the genus Megalocytivirus (Fig. 1). In previous studies, classification of the ranaviruses was based on host species and geographical range, as they are mostly detected in distinctive hosts and geographical regions [4, 12, 13, 18, 35]. Our results support this, since 11 strains of viruses in our study, collected from different fish species in different years, belonged to the same group and were similar to ranaviruses (Figs. 1, 2). According to the phylogenetic tree profiles of viral genomic DNA in the MCP genes, the genotypes of these isolates are closely related to SGIV and GIV. It appears that the viruses gradually adapted and evolved into the current local strains. Based on the phylogenetic tree profiles of amino acid sequences and nucleotide sequences of MCP, Taiwanese isolates belong to three genotypes in the genus Megalocytivirus, which include ISKNV, RSIV, and FLIV. An emerging strain (RBIV/Tp/45/08) invaded Taiwanese fish farms, and its genotype was found to be closely related to that of FILV and TRBIV (Figs. 1, 2). This strain originated from diseased rock bream that was imported as larvae from Korea, causing an epidemic point source in the farm. The cumulative mortality rate was 100% at 7 days after infection. Fortunately, there were no other farms near the outbreak.
Many studies have evaluated the genetic relationships between the epidemic strains, geographic distribution, and host susceptibility of members of the genus Megalocytivirus [23, 24]. In this study, we found six viruses of the genotype I group that were isolated from three different host fish species in both freshwater and marine environments between 2005 and 2008. The genotype II group consisted of five viruses isolated from three different host fish species isolated between 2006 and 2009. Interestingly, a new strain of MGIV/Nt/546/09 was detected from the marble goby in 2009, and this virus was genetically distinct from other viruses belonging to genotype II. Our findings confirm previous results [7, 33] that suggested freshwater and marine fish species are susceptible to ISKNV. The results also demonstrate that giant seaperch, giant grouper, and orange-spotted grouper are more sensitive to members of the genera Ranavirus and Megalocytivirus. In Taiwan, ranaviruses are epidemic in Pingtung, Kaohsiung, Penghu, Tainan, and Chiayi, mostly in marine fish. In this study, the megalocytivirus isolates from both seawater and freshwater fish species were mostly collected from Pingtung County. When comparing affected locations identified in previous studies in Taiwan [2, 33], the endemic areas are the same for both the megalocytiviruses and the ranaviruses. We have demonstrated that the main production areas are centralized in Taiwan. Moreover, the production chain is divided into several segments in the larvae and juvenile stages, resulting in the fry being constantly subjected to stress due to movement of the fish in commercial transactions, forcing the fry to adapt to different rearing environments in a short period of time. Consequently, multiple strains of GIV, RSIV, and ISKNV exited simultaneously from the same geographic locations, and the strain variations of these isolates may not be related to geography in Taiwan.
Comparison of nucleotide and amino acid sequences of iridovirus strains from 23 isolates shows high sequence identity in GIV, RSIV, ISKNV, and FLIV. Most of the variations observed were synonymous mutations, and the nucleotide substitutions in these isolates occurred at the first and second nucleotide positions and did not cause amino acid changes. Mutations in some isolates caused amino acid changes, particularly in the first-found strains in this study. Moreover, genomic variations occurred more often in freshwater fish species like giant seaperch, largemouth bass, and marble goby. We propose that the more frequent occurrence of non-synonymous mutations in freshwater fish species may be related to the fact that the references strains are most often detected in the seawater fish species or that these strains are involved in cross-species infections. However, this phenomenon requires more research. The Ka/Ks ratio of MCP segments from iridoviruses were lower than one, indicating that the viruses are under negative selection. These results suggest that the viruses are stable in the environment and could infect in both seawater and freshwater fish hosts.
The sequence identities between Taiwanese isolates in the MCP and ATPase genes demonstrate that they belong to different species in the genera Ranavirus and Megalocytivirus, according to the phylogenetic tree profiles of viral amino acids in the MCP proteins, which clustered into distinct genotypes (I, II, III, andV). The MCP is located on the viral surface and is exposed to environmental pressures, and it therefore can be used as an indicator of relationships within the family Iridoviridae. Our results show that the amino acid sequence of the ATPase is more conserved than that of the MCP. Presently, the function of the ATPase is poorly understood. Therefore, the high variability of the MCP gene may be a good tool to study virus strain evolution, classification, and taxonomic differentiation. Continuous monitoring of the iridovirus genotypes with a worldwide database may improve the understanding of local viral genotype shifts and their relationship to worldwide epidemiology. This information will be useful for disease diagnosis and control, including vaccine development and future cures.
References
Chao CB, Pang VF (1997) An outbreak of an iridovirus-like infection in cultured grouper (Epinephelus spp.) in Taiwan. J Chin Soc Vet Sci 23:411–422
Chao CB, Chen CY, Lai YY, Lin CS, Huang HT (2004) Histological, ultrastructural, and in situ hybridization study on enlarged cells in grouper Epinephelus hybrids infected by grouper iridovirus in Taiwan (TGIV). Dis Aquat Organ 58:127–142
Chao CB, Yang SC, Tsai HY, Chen CY, Lin CS, Huang HT (2002) A nested PCR for the detection of grouper iridovirus in Taiwan (TGIV) in cultured hybrid grouper, giant seaperch and largemouth bass. J Aquat Anim Health 14:104–113
Chinchar VG (2002) Ranaviruses (family Iridoviridae): emerging cold-blooded killers. Arch Virol 147:447–470
Chou HY, Hsu CC, Peng TY (1998) Isolation and characterization of a pathogenic iridovirus from cultured grouper (Epinephelus sp.) in Taiwan. Fish Pathol 33:201–206
Do JW, Cha SJ, Kim JS, An EJ, Lee NS, Choi HJ, Lee CH, Park MS, Kim JW, Kim YC, Park JW (2005) Phylogenetic analysis of the major capsid protein gene of iridovirus isolates from cultured flounders Paralichthys olivaceus in Korea. Dis Aquat Organ 64:193–200
Dong C, Weng S, Luo Y, Huang M, Ai H, Yin Z, He J (2010) A new marine megalocytivirus from spotted knifejaw, Oplegnathus punctatus, and its pathogenicity to freshwater mandarin fish, Siniperca chuatsi. Virus Res 147:98–106
Go J, Lancaster M, Deece K, Dhungyel O, Whittington R (2006) The molecular epidemiology of iridovirus in Murray cod (Maccullochella peelii) and dwarf gourami (Colisa lalia) from distant biogeographical regions suggests a link between trade in ornamental fish and emerging iridoviral diseases. Mol Cell Probes 20:212–222
He JG, Deng M, Weng SP, Li Z, Zhou SY, Long QX, Wang XZ, Chan SM (2001) Complete genome analysis of the mandarin fish infectious spleen and kidney necrosis iridovirus. Virology 291:126–139
He JG, Lu L, Deng M, He HH, Weng SP, Wang XH, Zhou SY, Long QX, Wang XZ, Chan SM (2002) Sequence analysis of the complete genome of an iridovirus isolated from the tiger frog. Virology 292:185–197
Hossain M, Song JY, Kitamura SI, Jung SJ, Oh MJ (2008) Phylogenetic analysis of lymphocystis disease virus from tropical ornamental fish species based on a major capsid protein gene. J Fish Dis 31:473–479
Hyatt AD, Gould AR, Zupanovic Z, Cunningham AA, Hengstberger S, Whittington RJ, Kattenbelt J, Coupar BE (2000) Comparative studies of piscine and amphibian iridoviruses. Arch Virol 145:301–331
Jancovich JK, Mao J, Wyatt C, Chinchar VG, Case ST, Kumar S, Valente G, Subramanian S, Davidson EW, Collins JP, Jacobs BL (2003) Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mortalities in North America. Virology 316:90–103
Jeong JB, Cho HJ, Jun LJ, Hong SH, Chung JK, Jeong HD (2008) Transmission of iridovirus from freshwater ornamental fish (pearl gourami) to marine fish (rock bream). Dis Aquat Organ 82:27–36
Kawakami H, Nakajima K (2002) Cultured fish species affected by red sea bream iridoviral disease from 1996 to 2000. Fish Pathol 37:45–47
Kurita J, Nakajima K, Hirono I, Aoki T (1998) Polymerase chain reaction (PCR) amplification of DNA of red sea bream iridovirus (RSIV). Fish Pathol 33:17–23
Lu L, Zhou SY, Chen C, Weng SP, Chan SM, He JG (2005) Complete genome sequence analysis of an iridovirus isolated from the orange-spotted grouper, Epinephelus coioides. Virology 339:81–100
Marsh IB, Whittington RJ, O’Rourke B, Hyatt AD, Chisholm O (2002) Rapid differentiation of Australian, European and American ranaviruses based on variation in major capsid protein gene sequence. Mol Cell Probes 16:137–151
Murali S, Wu MF, Guo IC, Chen SC, Yang HW, Chang CY (2002) Molecular characterization and pathogenicity of a grouper iridovirus (GIV) from yellow grouper, Epinephelus awoara (Temminck & Schlegel). J Fish Dis 25:91–100
Qin QW, Shi C, Gin KY, Lam TJ (2002) Antigenic characterization of a marine fish iridovirus from grouper, Epinephelus spp. J Virol Methods 106:89–96
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Shi CY, Wang YG, Yang SL, Huang J, Wang QY (2004) The first report of an iridovirus-like agent infection in farmedturbot, Scophthalmus maximus, in China. Aquaculture 236:11–25
Shinmoto H, Taniguchi K, Ikawa T, Kawai K, Oshima S (2009) Phenotypic diversity of infectious red sea bream iridovirus isolates from cultured fish in Japan. Appl Environ Microbiol 75:3535–3541
Song JY, Kitamura S, Jung SJ, Miyadai T, Tanaka S, Fukuda Y, Kim SR, Oh MJ (2008) Genetic variation and geographic distribution of megalocytiviruses. J Microbiol 46:29–33
Song WJ, Qin QW, Qiu J, Huang CH, Wang F, Hew CL (2004) Functional genomics analysis of Singapore grouper iridovirus: complete sequence determination and proteomic analysis. J Virol 78:12576–12590
Sudthongkong C, Miyata M, Miyazaki T (2002) Viral DNA sequences of genes encoding the ATPase and the major capsid protein of tropical iridovirus isolates which are pathogenic to fishes in Japan, South China Sea and Southeast Asian countries. Arch Virol 147:2089–2109
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tan WG, Barkman TJ, Gregory CV, Essani K (2004) Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 323:70–84
Tidona CA, Darai G (1997) The complete DNA sequence of lymphocystis disease virus. Virology 230:207–216
Tsai CT, Ting JW, Wu MH, Wu MF, Guo IC, Chang CY (2005) Complete genome sequence of the grouper iridovirus and comparison of genomic organization with those of other iridoviruses. J Virol 79:2010–2023
Wang CS, Chao SY, Ku CC, Wen CM, Shih HH (2009) PCR amplification and sequence analysis of the major capsid protein gene of megalocytiviruses isolated in Taiwan. J Fish Dis 32:543–550
Wang CS, Shih HH, Ku CC, Chen SN (2003) Studies on epizootic iridovirus infection among red sea bream, Pagrus major (Temminck & Schlegel), cultured in Taiwan. J Fish Dis 26:127–133
Wang YQ, Lu L, Weng SP, Huang JN, Chan SM, He JG (2007) Molecular epidemiology and phylogenetic analysis of a marine fish infectious spleen and kidney necrosis virus-like (ISKNV-like) virus. Arch Virol 152:763–773
Whittington RJ, Becker JA, Dennis MM (2010) Iridovirus infections in finfish—critical review with emphasis on ranaviruses. J Fish Dis 33:95–122
Williams T, Barbosa-Solomieu V, Chinchar VG (2005) A decade of advances in iridovirus research. Adv Virus Res 65:173–248
Acknowledgments
This study was supported by grants from the Council of Agriculture, Taiwan, Republic of China, numbers 94AS-13.2.5-HI-H4, 95AS-13.2.4-HI-H8, 96AS-14.2.4-HI-H6, 97AS-14.2.4-HI-H4, and 98AS-9.2.4-HI-H1.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Huang, SM., Tu, C., Tseng, CH. et al. Genetic analysis of fish iridoviruses isolated in Taiwan during 2001–2009. Arch Virol 156, 1505–1515 (2011). https://doi.org/10.1007/s00705-011-1017-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-011-1017-9