
Abstract
TGFβ1 is the most pleiotropic of all known cytokines and thus, to avoid uncontrolled TGFβ-activated processes, its activity is tightly regulated. Studies in fibrosis have led to the discovery that αv integrins are the major regulators of the local activation of latent TGFβ in our tissues. Since all cells can express one or more types of αv integrins, this raises the possibility that, in the complex milieu of a developing cancer, multiple cell types including both cancer cells and stromal cells activate TGFβ. In normal tissues, TGFβ1 is a tumour suppressor through its ability to suppress epithelial cell division, whereas in cancer, in which tumour cells develop genetic escape mechanisms to become resistant to TGFβ growth suppression, TGFβ signalling creates a tumour-permissive environment by activating fibroblast-to-myofibroblast transition, by promoting angiogenesis, by suppressing immune cell populations and by promoting the secretion of both matrix proteins and proteases. In addition, TGFβ drives epithelial-to-mesenchymal transition (EMT) increasing the potential for metastasis. Since αv integrins activate TGFβ, they almost certainly drive TGFβ-dependent cancer progression. In this review, we discuss the data that are helping to develop this hypothesis and describe the evidence that αv integrins regulate the TGFβ promotion of cancer.

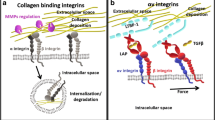
Mechanisms of integrin-mediated transforming growth factor beta (TGFβ) activation and its effect on stromal processes. 1 Matrix-bound latent LAP-TGFβ1 binds αv integrins expressed by epithelial cells or fibroblasts (LAP latency-associated peptide). TGFβ1 becomes exposed. 2 Active TGFβ1 binds the TGFβ receptor in an autocrine or paracrine fashion. 3 TGFβ1 signalling increases integrin expression, LAP-TGFβ1 secretion and trans-differentiation of fibroblasts into contractile cells that secrete collagens and collagen cross-linking proteins. By contracting the matrix, latent TGFβ1 is stretched making the activation of latent TGFβ1 easier and creating a continuous cycle of TGFβ1 signalling. TGFβ1 promotes cancer progression by promoting angiogenesis, immune suppression and epithelial-to-mesenchymal transition (EMT)
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transforming growth factor β1 (TGFβ1) is a pleiotropic cytokine that can function as both tumour suppressor and tumour promoter (for reviews, see Inman 2011; Yang and Moses 2008; Roberts and Wakefield 2003). TGFβ1-dependent tumour suppression is attributable mostly to the direct effects of the inhibition of epithelial cell division by the activation of cyclin-dependent kinase inhibitors (p21, p15 Ink4b). Once tumour cells develop mutations that render them refractile to the growth-suppressive effects of TGFβ1, then the cytokine can become tumour-promoting by acting directly on the tumour cells to drive an invasive programme but also indirectly by promoting a tumour-permissive microenvironment (Inman 2011).
In the last 20 years, it has become clear that the αv family of RGD-binding integrins are major regulators of TGFβ1-dependent processes in both normal and pathological processes (for reviews, see Wipff and Hinz 2008; Margadant and Sonnenberg 2010; Sheppard 2015). Some of these same integrins have also been strongly implicated in the promotion of the growth and spread of many different types of cancer. Therefore, integrins that are able to activate TGFβ1 in vivo are probably also likely to modulate tumour progression indirectly via the local production of active TGFβ1. Strong data also describe the way that TGFβ1 regulates the tumour microenvironment (TME) to regulate cancer progression. In this review, we will combine those data and extrapolate from a combination of in vitro and clinical observations to infer the likely roles of integrin-dependent TGFβ1 regulation of the tumour stroma. First, we will review briefly the integrin-dependent activation of latent-TGFβ1 and those integrins that are candidates for the activation of TGFβ1 in cancer.
TGFβ is deposited in extracellular matrix as an inactive latent complex
The TGFβ family of cytokines and receptors includes activins, bone morphogenetic proteins and the TGFβ cytokines. In this review, we will concentrate on TGFβ1 but direct readers to a recent excellent review of other members of the family (Wakefield and Hill 2013). Three isoforms of TGFβ (TGFβ1, TGFβ2 and TGFβ3) are widely expressed in tissues but have different functions. Knockout of the TGFβ1 gene in mice (tgfb1) results in the infiltration of tissues with mononuclear cells eventually killing the animal within weeks of birth (Shull et al. 1992; Kulkarni et al. 1993), whereas the knockout of TGFβ2 (Sanford et al. 1997) or TGFβ3 (Kaartinen et al. 1995) causes developmental defects. All three isoforms of TGFβ bind to the same cell surface receptor TGFBRII, which exists as a dimer, a constitutively active serine-threonine kinase that, upon binding a ligand, dimerises with and phosphorylates a dimer of TGFBRI (also known as ALK5) forming a quaternary complex (Kang et al. 2009). This complex recruits and activates via phosphorylation the SMAD2 and SMAD3 transcription factors, which now can bind SMAD4 promoting their translocation to the nucleus in which the complex drives the transcription of a wide variety of genes, including the αv integrins (Levy and Hill 2005; Margadant and Sonnenberg 2010). TGFβ also activates multiple SMAD-independent signalling pathways that can promote tumour progression (Inman 2011).
TGFβ1 is a highly pleiotropic cytokine, which is probably the reason that its synthesis and activation is so tightly controlled. As described previously (Annes et al. 2004), the TGFβ1 gene product (TGFB1) is generated as a homo-dimer that is post-translationally cleaved by Furin-like enzymes into a larger pro-peptide, called the latency-associated peptide (LAP), which associates with the smaller TGFβ cytokine, the last-mentioned remaining non-covalently bound; this is referred to as the small latent complex (SLC). Two such processed proteins form a homo-dimer, which become covalently linked to one of four latent TGFβ-binding proteins (LTBP-1,-2,-3 and -4) within the Golgi prior to being secreted as a large latent complex (LLC) of LAP-TGFβ-LTBP. The LLC becomes anchored in the matrix via covalent linkage of LTBPs to fibrillin-1 (for a review, see Robertson et al. 2015). The concentration of TGFβ1 embedded in the extracellular matrix of normal organs has been estimated to be far in excess of that required to promote most TGFβ1-mediated processes (Sheppard 2015). However, while embedded within the LLC, the TGFβ1 remains inert. Thus, TGFβ1-dependent processes are controlled locally in the tissues by the regulation of the activation of latent-TGFβ1, which is, effectively, the exposure or release of the TGFβ1 cytokine to access by TGFβ1 receptors. This is important as uncontrolled TGFβ activity can lead to chronic fibrosis (Margadant and Sonnenberg 2010), which can be a significant risk factor for the development of some cancers (Hubbard et al. 2000; Bataller and Brenner 2005).
Integrin activation of latent TGFβ in vivo
Munger and colleagues reported that the RGD-binding integrin αvβ6 activates latent TGFβ1 by binding to the RGD motif of LAPβ1 and exerting an actin-dependent physical deformation of the LLC, exposing the TGFβ cytokine to TGFβ receptors on adjacent cells (Munger et al. 1999). Of note, αvβ6 also binds TGFβ3 in a similar fashion. The process, which is protease-independent, has been subsequently shown to require fibronectin-anchored LTBP1 (Annes et al. 2004; Dallas et al. 2005). Biologically, the epithelial-specific αvβ6 is required to promote lung fibrosis in response to bleomycin (Munger et al. 1999) and, in separate studies, kidney fibrosis in response to deficiency in the collagen subunit Col4A3 (Hahm et al. 2007) and in both cases, antibodies to αvβ6 suppress fibrosis. Wipff et al. (2007) reported that thrombin-activated contraction by myofibroblasts activates TGFβ1 predominantly via αvβ5 and, less so, via αvβ3 or an unidentified β1 integrin (Wipff et al. 2007). The process of activation is also wholly mechanical, since detergent-treated cells lacking any cell membrane or cytosol can still activate latent TGFβ1 in response to exogenous ATP; again, αvβ5 blockade reduces this TGFβ1 activation by 66 %, twice as much as a blockade of αvβ3 or β1 integrins. Henderson et al. (2013) reported that mice globally deficient for integrin subunit genes, namely itgb3, itgb5 or itgb6 and selectively deficient for itgb8 in haemopoietic cells are not protected from experimental liver fibrosis induced by carbon tetrachloride treatment, whereas mice with myofibroblasts deficient in the αv gene (itgav) are protected (Henderson et al. 2013). Using an αvβ1-specific peptide inhibitor, Henderson and colleagues confirmed that αvβ1 can promote liver fibrosis, possibly identifiying the TGFβ1-activating β1 integrin earlier described by Wipff et al. (2007). These data show that integrin-dependent TGFβ1 activation can vary between tissues and cell types. Henderson and colleagues also concluded that αvβ1 promotes bleomycin-induced lung fibrosis in mice. Since the same group has previously reported αvβ6 as being the driver of bleomycin-induced lung fibrosis (Munger et al. 1999), these results suggest that two or more TGFβ1-activating integrins can operate simultaneously in a single tissue to regulate the stromal response. This has implications for the TGFβ1-dependent modulation of the tumour microenvironment in cancer.
In contrast to other integrins, αvβ8 activates latent-TGFβ1 in a protease-dependent manner by the co-localisation of membrane-bound protease MT1-MMP/MMP14 (Mu et al. 2002). Thus, αvβ8 binds to LAPβ1 or LAPβ3 and brings the latent complexes into proximity of the membrane-bound protease that cleaves the LAP. Integrin αvβ3 might also be capable of using proteases to activate latent TGFβ in vivo, as reports exist that αvβ3 can localise activated matrix metalloproteinase 2 (MMP2) and MMP9 at the cell surface (Brooks et al. 1996; Rolli et al. 2003) and these MMPs have been implicated as activators of TGFβ in vitro (for a discussion, see Jenkins 2008).
The specific importance of RGD-binding integrins as regulators of TGFβ activity in vivo has been confirmed by the creation of mice with LAP but containing the non-integrin binding motif RGE, instead of RGD. These mice have a phenotype that mirrors that of TGFβ1-deficient mice (Yang et al. 2007). However, despite the evidence that all αv integrins appear to activate latent TGFβ, mice deficient in αvβ3 or αvβ5 do not exhibit TGFβ1-deficient phenotypes, whereas mice deficient in αvβ6 exhibit skin and lung inflammation (Munger et al. 1999), the spontaneous development of several types of cancers (Ludlow et al. 2005) and emphysema in older mice (Morris et al. 2003), all phenotypes linked to deficient local TGFβ1 activity. Similarly, whereas the complete ablation of integrin αvβ8 is a lethal phenotype (Zhu et al. 2002), loss in dendritic cells results in chronic inflammation and autoimmunity, almost certainly attributable to the inability of dendritic cells to activate TGFβ1 and thereby regulate the production of TReg cells (Travis et al. 2007). Thus, the apparent ranking in the ability of integrins to activate TGFβ1 in vivo might be linked to their affinity for LAP. The sequence RGDLXXI, as occurs in LAPβ1 and LAPβ3, forms two binding sites for αvβ6: the RGD loop and an adjacent helix that presents a hydrophobic leucine/isoleucine-binding site permitting high affinity binding (DiCara et al. 2007) and probably also explaining the high affinity of αvβ8 for LAPβ1 (Mu et al. 2002). In contrast, no reports are available for a second binding site in LAPβ1 for other αv integrins. However, if the threshold for activating TGFβ1 was lowered, then the weaker binding αv integrins, namely αvβ5, αvβ3 and αvβ1, might play a larger role in local TGFβ1 activation. As discussed below, this is likely to occur.
As mentioned above, researchers at the Hinz laboratory reported that myofibroblasts express several αv integrins that can activate TGFβ1, including αvβ3, αvβ5 and an unidentified β1 integrin. They found that the integrin activation of TGFβ1 does not occur on soft pliable substrates but also established that the stretching of adherent myofibroblasts is sufficient to increase their ability to activate latent TGFβ1 (Wipff et al. 2007). More recently, Klingberg et al. (2014) showed that, when myofibroblasts remodel the extracellular matrix through their contractile activity, they also organise LTBP1 into fibrillar fibronectin deposits, whereas normal fibroblasts are less able to achieve this (Klingberg et al. 2014). Furthermore, myofibroblasts can activate twice as much TGFβ1 on the myofibroblast-organised matrix than the fibroblast-organised matrix, suggesting that placing the matrix under tension primes the latent TGFβ1 for activation. This has been established by using a strain machine that stretches cell-depleted extracellular matrices before cells are added. Myofibroblasts activate TGFβ1 more efficiently on stretched matrices and this correlates with the stretching of the LTBP1-Fn deposits, as observed by fluorescence microscopy. Indeed, a fraction of TGFβ1 is activated independently of integrins if the matrix is sufficiently stretched.
Thus, non-proteolytic TGFβ1 activation by αv integrins is regulated at many levels: (1) sufficient amounts of an appropriate LAP-binding integrin must be present, (2) the integrin must be in the ligand-binding activated state (Wipff et al. 2007), (3) the cytoplasmic tail must be linked directly to the actomyosin cytoskeleton, (4) the extracellular domain is bound to LAPβ1 or LAPβ3 of the LLC, (5) the LLC must be anchored to a strain-resistant matrix in order to permit actin-dependent stretching of the complex to reveal the TGFβ. Finally, if the responding cell is adjacent to these activating events, this maximises the controlled local activation (Munger et al. 1999). Such tight control allows the exquisite regulation of TGFβ1 in our tissues. However, once the process has started, the activation of latent TGFβ1 might be rapidly amplified through integrin-dependent positive feedback loops; this is likely to happen in both fibrosis and cancer. Thus, as described in Fig. 1, the initial activation of TGFβ1 can increase the expression of more TGFβ-activating integrin subunits on both epithelial cells and local fibroblasts through SMAD-dependent signalling (Margadant and Sonnenberg 2010). Moreover, the activated TGFβ1 activates local fibroblasts to become myofibroblasts (discussed below), which develop contractile abilities and contract the extracellular matrix. The contracted matrix stretches the embedded LLC and lowers the threshold for activating more latent TGFβ1, potentially allowing integrins with a lower affinity for LAP (αvβ5, αvβ1, αvβ3) to activate more TGFβ1. The additional TGFβ1 generated further amplifies the process potentially resulting in a cycle of TGFβ1 activation-fibroblast activation-extracellular matrix stiffening-TGFβ1 activation etc. Thus, cancers can progress to a point at which the tumour:stroma interaction develops into an activation loop whereby the upregulated αv integrins, increased matrix stiffness and probably proteases released by both tumour and myofibroblasts result in uncontrolled TGFβ1 activation. This raises the question as to which point(s) within this escalation of TGFβ1 activation therapeutic intervention is still likely to be effective. Logically, the early blockade of a limited number of αv integrins as shown in fibrosis studies (Munger et al. 1999; Hahm et al. 2007) will probably suppress TGFβ1 activation in the tumour microenvironment but, once escalated, TGFβ1 activation might occur via many mechanisms and, thus, multiple interventions will be needed, which will be therapeutically challenging. Notably, whereas the remaining RGD-binding integrins (α5β1, α8β1 and αIIbβ3) have not been reported to activate latent TGFβ1 directly, α5β1 is required for the αvβ6-dependent activation of latent TGFβ1 (Fontana et al. 2005) presumably by binding to the fibronectin trapping the LTBP1.

Cycle of contribution of transforming growth factor β1 (TGFβ1)-activated fibroblasts to extracellular matrix (ECM) composition and stiffness and further TGFβ1 activation (LTBP latent TGFβ-binding proteins, ITGB6 integrin subunit gene 6, LOX lysyl oxidase, POSTN periostin)
TGFβ1-driven changes in the tumour microenvironment can promote cancer progression
Recent reviews have described the role of TGFβ1 in the regulation of the tumour microenvironment (Pickup et al. 2013; Lin and Zhao 2015; Guo et al. 2016). In this review, we will concentrate on the available evidence linking integrin-dependent TGFβ1 activation to many of these microenvironmental changes.
-
1)
Integrin activation of TGFβ drives epithelial-to-mesenchymal transition
The epithelial-to-mesenchymal transition (EMT) is the process by which epithelial cells reduce the expression or function of the proteins that promote cell-cell and cell-basement-membrane adhesion and thus transition towards a more motile and sometimes mesenchymal-like phenotype. This transition is essential during stages of embryogenesis and wound healing but also occurs in pathological processes, possibly for the initiation of metastasis (Mamuya and Duncan 2012). TGFβ1 induces EMT by generating a transcriptional repression of the epithelial gene signatures, including cell-cell adhesion molecule E-cadherin but by elevating mesenchymal genes, such as N-cadherin, αSMA and vimentin in a SMAD-dependent and -independent manner, in part via activation of the transcription factors Snail and Slug (Naber et al. 2013; Medici et al. 2006). Thus, the integrin-dependent activation of TGFβ1 might be expected to promote EMT, as has been reported.
Bates and colleagues (2005) reported that high levels of αvβ6 in colon cancer reduced overall survival from a median of 16.5 months in the αvβ6-low/negative cancers to 5 months in the αvβ6 strongly positive cancers. They also suggested a model of αvβ6-dependent activation of TGFβ1 and subsequent EMT as the potential mechanism for αvβ6-driven colon cancer (Bates et al. 2005).
Workers at the Danen laboratory discovered that the reduction of β1 integrin expression in breast cancer cells suppressed tumour growth but enhanced metastasis to the lungs. In three-dimensional (3D) matrices in vitro, similar treatments with integrin β1 inhibitory antibodies or genetic suppression of β1 expression resulted in a change from cohesive migration to single cell migration; this was dependent upon increased TGFβ1 signalling and was correlated with the loss of E-cadherin. Suppression of TGFβ1 signalling or increasing E-cadherin restored cohesive migration in β1-deficient cells and reduced their metastasis to the lungs (Truong et al. 2014). These observations were in agreement with earlier studies by Giampieri et al. (2009) who reported that activation of TGFβ1 signalling in a tumour cell population promoted a single cell migratory phenotype as opposed to cohesive cell migration in the absence of TGFβ1 signalling (Giampieri et al. 2009). These data link αv integrin activation of latent TGFβ1 directly to the metastatic propensity of cancer (Mamuya and Duncan 2012).
Sometimes, the upregulation of TGFβ1 signalling by integrins is indirect. Mori and colleagues showed that αvβ3 enhanced TGFβ1-induced EMT in MCF10A breast cells by binding directly to fibroblast growth factor 1 (FGF1) and making it available for binding to FGFR1 receptors. This FGF1-αvβ3 engagement and the FGFR1 signalling were necessary for FGF1 to maximise the TGFβ1-dependent EMT (Mori et al. 2015). TGFβ1 also elevates the expression of secreted matricellular glycoprotein fibulin-5, which contains an RGD-motif and is capable of binding α5β1, αvβ3 and αvβ5 (Yanagisawa et al. 2009). Fibulin-5 is expressed during development and is only re-expressed in injured tissues and cells undergoing EMT. Moreover, fibulin-5 stimulates MMP-2 and -9 expression and activity and promotes mammary epithelial cell invasion probably through an enhancement of Twist and reduction in E-cadherin expression. However, the direct link between fibulin-5 and integrins during EMT needs to be further established (Lee et al. 2008).
-
2)
Empirical and inferred data linking integrin activation of TGF to modulation of the tumour stroma
We only recently established the central importance of the many different αv integrins in the activation of latent TGFβ and determined which αv integrins are likely to act when and where (see above). Therefore, relatively few studies have included integrin-blocking antibodies in their in vivo investigations in order to empirically determine which integrin, if any, is responsible for activating TGFβ1 and therefore is responsible for the TGFβ1-dependent effects on tumour cell growth and spread and the tumour microenvironment. In this section, we summarise both empirical data, whereby researchers have directly examined the role of integrin-dependent TGFβ activity in tumour progression and inferred data, whereby we can reasonably assume that integrin-dependent TGFβ1 activation is at play.
Van Aarsen et al. (2008) showed that Detroit-562 oral squamous cell carcinoma (SCC) xenografts developed a strongly positive αvβ6 invasive front and grew into a stroma rich in TGFβ1. Moreover, the xenografts exhibited a reticular αSMA pattern in the stroma at the periphery of the tumour correlating with the expression of αvβ6 and suggesting a relationship between the two proteins (Van Aarsen et al. 2008). A similar accumulation of αSMA-positive myofibroblasts abutting the invasive front of oral cancer, most of which were αvβ6-positive (Van Aarsen et al. 2008; Marsh et al. 2011), had been observed previously (Lewis et al. 2004). Treatment of Detroit-562 xenografts by systemic αvβ6-blocking antibody 6.3G9 or soluble recombinant TGFBRII-Fc protein suppressed tumour growth but did not significantly change the expression of αSMA, CD31-positive blood vessels or collagen deposition detected by Sirius red staining (Van Aarsen et al. 2008). In contrast, Eberlein and colleagues (2013) reported that the treatment of mice bearing Detroit-562 xenografts with 264RAD, a human antibody that inhibits αvβ6, resulted in a dose-dependent reduction in tumour growth that was mirrored by a dose-dependent reduction in fibronectin and SMA, two products of TGFβ-activated fibroblasts, suggesting strongly that αvβ6 was promoting a myofibroblast-rich microenvironment. Why these similar studies resulted in different apparent stromal effects remains unknown. Whether the small amount of αvβ8 blocking activity of the 264RAD antibody had any effect was not explored.
Moore and colleagues reported that αvβ6 co-operated with HER2 to promote HER2-driven invasive breast cancer (Moore et al. 2014). Treatment of mice with 264RAD suppressed the growth of both HER2-over-expressing BT474 and MCF7/HER2-18 tumours, an effect enhanced by combination therapy with the anti-HER2 antibody Trastuzumab. Analysis of tumours harvested mid-therapy showed that, in both tumour types, 264RAD therapy alone was sufficient to reduce total- and phospho-SMAD2 in the tumour and to reduce the number of αSMA-positive and endomucin-positive endothelial cells suggesting a direct link between αvβ6-dependent TGFβ1 signalling, the induction of myofibroblasts and the upregulation of angiogenesis.
Integrin activation of TGFβ1 influences cancer progression not only on the luminal component of breast tissue. Myoepithelial cells are the contractile cells adjacent to luminal breast cells that promote milk secretion from ducts. Allen et al. (2014) identified de novo expression of αvβ6 on breast myoepithelial cells as being a key biomarker associated with an invasive breast phenotype in ductal carcinoma in situ (DCIS; Allen et al. 2014). Moreover, a relationship existed between αvβ6-positive myoepithelial cells in DCIS and the subsequent recurrence of breast cancer. Since only 50 % of women with DCIS develop life-threatening invasive ductal carcinoma but still receive surgery and other therapies, this is an important observation with potential clinical ramifications. To determine the mechanism, Allen and colleagues showed that, when αvβ6 was over-expressed on normal myoepithelial cells, they could activate TGFβ1 to which they responded by upregulating MMP2 and MMP9. Blockade of TGFβ or MMPs inhibited the ability of αvβ6-expressing myoepithelial cells to promote breast cancer invasion. Analysis of DCIS samples confirmed a strong correlation of αvβ6 and MMP9 expression in myoepithelial cells. In further studies, the injection of αvβ6-positive myoepithelial cells together with MDA MB231 breast carcinoma cells resulted in a significantly faster growth of tumours compared with normal myoepithelial cells. Again, the data were consistent with the αvβ6 activation of TGFβ1 driving tumour cell invasion except that, this time, the αvβ6-positive “microenvironmental” myoepithelial cell promoted the invasive phenoptype.
Marsh and colleagues reported that, compared with the more indolent types of basal cell carcinoma (BCC), αvβ6 was selectively upregulated on the morphoeic type of BCC, a tumour that has a distinctly invasive and fibrotic phenotype (Marsh et al. 2008). As BCC is associated with the upregulation of Gli 1 and Gli 2 transcription factors, they were transfected into keratinocytes to generate an in vitro model of BCC. These cells were able to activate latent TGFβ1, which activated fibroblasts to become αSMA-positive myofibroblasts and which, in turn, secreted HGF that acted as a pro-invasive chemoattractant for the BCC cells. The data were consistent with increased αvβ6 expression by morphoeic BCC cells being significantly responsible for the invasive and fibrotic phenotype of this disease. Another skin carcinoma study also linked αvβ6 to the modulation of the TME. Patients lacking collagen 7 fibrils easily develop skin blisters that further develop into chronic fibrotic inflammatory lesions on bony joints and can often develop into invasive and metastatic squamous carcinomas (Fine and Hintner 2009). Martins et al. (2016) developed 3D tumour-mimetic organotypic gels in vitro combining a squamous cell carcinoma (SCC) cell line that had been engineered to lack collagen 7 gene (Col7A1) with fibroblasts in a collagen-based gel. Small-hairpin-RNA-treated SCC cells (shCol7 SCC) admixed with skin fibroblasts invaded significantly more than control cells and upregulated αvβ6 and the associated matrix exhibited a significant increase in fibronectin. The increased invasion, αvβ6 expression and fibronectin were all suppressed by co-incubation with a TGFβ1 receptor inhibitor. When the organotypic gels were implanted under the skin of mice to form tumours, again the loss of collagen 7 resulted in significant upregulation of αvβ6 expression specifically at the invasive front and a significant increase in fibronectin expression in the associated stroma and cancer. Moreover, increased phospho-Smad2/3 signalling was present in the stroma of the Col7-deificient SCC tumours. The authors suggested that Collagen 7 functioned as a TGFβ1 suppressor and, again, the data are consistent with an αvβ6-dependent activation of TGFβ1 activating a fibroblast-to-myofibroblasts transition, which subsequently increases the secretion of fibronectin.
Eberlein et al. (2015) investigated the effects of the co-culture of a panel of non-small cell lung cancer cell lines with fibroblasts and found that those cancer lines that promoted trans-differentiation of fibroblasts to myofibroblasts (αSMA–positive) expressed E-cadherin, EpCAM and αvβ6. Blockade of αvβ6 with 264RAD or of TGFBR1 with a small molecule inhibitor blocked myofibroblast formation. Interestingly, if the inhibitors were added 3 days after co-culture, only the TGFBR1 inhibitor suppressed SMA expression, suggesting that, whereas αvβ6 initiates the process, once started, αvβ6-independent processes maintain TGFβ activation (Eberlein et al. 2015).
Dutta et al. (2015) reported the novel αvβ6 regulation of TGFβ1 signalling in prostate cancer cells. Generating a panel of PC3 prostate cancer cells expressing αvβ6 or αvβ6/β3cyto or αvβ3/β6cyto chimeras, these authors reported that αvβ6 co-precipitated with TGFBRII, a relationship that required the cytoplasmic tail of β6 and that physical association was required for the TGFβ1-dependent upregulation of SMAD3, which in turn upregulated MMP2. This is the first report suggesting that αvβ6 regulates TGFβ1 signalling via physical association with the TGFβ1 receptor complex. The determination of whether this unique observation is seen in other cell models will be of interest.
Thus, numerous examples link the αvβ6-dependent activation of TGFβ1 as a promoter of pre-clinical tumour progression. Therefore, we must consider it likely that, in clinical studies in which poor overall survival has been linked to the over-expression of αvβ6, TGFβ1 activation by αvβ6 is a tumour-driver. Since Bates et al. (2005) reported that high αvβ6 correlates with poor survival in colon cancer, similar observations have been reported for non-small cell lung cancer (Elayadi et al. 2007), cervical cancer (Hazelbag et al. 2007) and breast cancer (Moore et al. 2014). Each of these studies had sufficient clinical samples and follow-up data to establish survival relationships. Other studies with smaller numbers of tissue samples also reported high fractions of carcinomas positive for αvβ6 including ovarian (100 %, Ahmed et al. 2002; 33 %, Van Aarsen et al. 2008), pancreatic (100 %, Sipos et al. 2004), oesophageal (68 %, Van Aarsen et al. 2008) and skin (84 %, Van Aarsen et al. 2008). Eventually, we will establish the role of αvβ6 and, indeed, the role of other αv integrins in activating TGFβ1 in each of these cancers and the subsequent effects on tumour growth and spread.
Notably, if cancer tissues retain the tumour-suppressive responsiveness to TGFβ1, then the blockade of αvβ6 could potentially promote cancer, as was described in a transgenic mouse model of pancreatic cancer (Hezel et al. 2012). In fact, early studies showed that, in normal tissues, αvβ6 was tumour-suppressive through its activation of TGFβ1. Thus, knockout of the β6 gene (itgb6) in mice was associated with a mild inflammatory response in skin and lung (Huang et al. 1996) and with a propensity for the spontaneous formation of certain cancers in up to 25 % of itgb6-null mice (Ludlow et al. 2005), patterns that mimicked a mild form of global TGFβ1 knockout mouse (Shull et al. 1992). These unrelated observations might have indicated that the αvβ6-dependent activation of TGFβ1 in part regulated tumour immune surveillance, as shown subsequently for αvβ8 (Travis et al. 2007). Certainly, a recent study reported that αvβ6 and αvβ8 on keratinocytes regulated the retention time of dendritic cells and memory T cells in the skin and colon by locally activating TGFβ1 (Mohammed et al. 2016).
Therefore, overall, whereas some data are conflicting, strong evidence has been provided that, in some cancer tissues, αvβ6 is associated with tumour promotion, in part via the activation of latent TGFβ1, which subsequently regulates elements in the stroma, including the number of myofibroblasts and blood vessels. In addition, some αv integrins clearly regulate the behaviour of inflammatory and immune cell populations. It is through these three processes, described below, that the αv integrin-dependent activation of TGFβ1 can impact most strongly on the stroma. Of note, the principal reason that most of the available data linking αv integrins to TGFβ-regulation of cancer involve αvβ6 is probably attributable to the absence of widely available reagents that specifically detect and inhibit integrins αvβ8 and αvβ1; this means their distribution and activity in cancer remains unknown. Moreover, whereas αvβ3 has been reported to regulate TGFβ1 signalling negatively in endothelial cells of healing wounds (Reynolds et al. 2005), the widespread expression of αvβ3 and αvβ5 on multiple cell types makes the determination of their specific activities difficult to prove by using global antibody blockade and will require genetic ablation in specific cell types.
-
3)
Myofibroblasts/CAFs promote cancer progression by multiple mechanisms
TGFβ1 is the most powerful activator of the trans-differentiation of fibroblasts into myofibroblasts. Thus, those integrins that activate TGFβ1 can also induce myofibroblast formation and therefore can be considered responsible for myofibroblast-dependent activities that include matrix-remodelling, matrix-stiffening and cancer promotion.
All tissues have resident fibroblasts that maintain the tissue homeostasis of matrix content and whose presence primes tissues to respond to wounding or infection. However, TGFβ1 induces fibroblasts to adopt a contractile wound-repair phenotype and genotype termed a myofibrobast. When this trans-differentiation occurs in the tumour microenvironment, the myofibroblasts are also known as activated fibroblasts, cancer-associated fibroblasts (CAFs) or tumour-associated fibroblasts (TAFs). Not all CAFs are derived from resident cells. Thus, TGFβ1 can induce some non-fibroblast cells, including adipocytes (Jotzu et al. 2010) and circulating bone-marrow-derived suppressor cells (Weber et al. 2015), to trans-differentiate into cancer-associated myofibroblasts.
Integrins can also potentially regulate TGFβ signalling after TGF activation has occurred. In fibroblasts derived from human oral and dermal tissues, anti-αvβ3 or αvβ5 integrin antibody blockade in the presence of exogenous active TGF-β1 inhibits the expression of α-SMA and collagen gel contraction. Furthermore, in renal fibroblasts, only αvβ5 blockade and not αvβ3 is sufficient to reproduce these same inhibitory effects, suggesting that the differentiation of fibroblasts that are derived from different tissues are not necessarily regulated by the same integrin (Lewis et al. 2004). The mechanisms regarding the reasons that αvβ3 and αvβ5 are required for TGFβ1-induced trans-differentiation to the myofibroblast phenotype have not been established.
Above, we have discussed the way that, by increasing the contraction of the extracellular matrix, myofibroblasts increase the likelihood of activating latent TGFβ1. However, myofibroblasts secrete a huge number of proteins that further enhance cancer progression including extracellular matrix proteins, proteases, growth factors, cytokines and chemokines (for a review, see Miles and Sikes 2014).
Surgeons and pathologists have known for many years that most cancers are stiffer to the touch than the surrounding normal tissues and we now know that the cells responsible for this increased stiffness are the collagen-producing contractile myofibroblasts. Originally, the stiffness was assumed to be an effort on the part of the host to wall off the mutant tissue as a means of protection. Indeed, recent studies of pancreatic cancer suggest that some types of pancreatic fibroblast are tumour-suppressive (Rhim et al. 2014; Ozdemir et al. 2014). However, clearly, tumour cells often secrete latent TGFβ1 and can promote the generation of myofibroblasts and, thus, we can reasonably assume that they derive benefit from activating fibroblasts to myofibroblasts. We have only recently confirmed that a stiff extracellular matrix can signal tumour cells to promote their growth and survival (for a review, see Malik et al. 2015).
Myofibroblasts secrete many different types of collagens (discussed in Miles and Sikes 2014) that they form into fibrils in the matrix. A positive relationship exists between the thickness and orientation of the collagen fibrils and the invasive behaviour of cancers (Provenzano et al. 2006). Myofibroblasts can promote the cross linking of collagen fibres through the secretion of lysyl oxidases (LOX), which can covalently crosslink collagens and elastin. Many studies have reported that levels of LOX correlate with increased invasion, metastasis and poor survival in pancreatic cancer (Miller et al. 2015), astrocytoma (da Silva et al. 2015), breast cancer (Friesenhengst et al. 2014) and liver cancer (Zhu et al. 2015a). Such observations identify LOX and LOX-like enzymes as potential therapeutic targets.
Myofibroblasts also secrete periostin that acts in several ways to promote cancer. First, it can bind to fibronectin through its FAS domain and to bone morphogenetic protein-1 (BMP-1) through its EMI domain. This anchors BMP-1 in the matrix where it can promote the maturation of the pre-Lox propeptide into the mature Lox enzyme, enabling it to generate collagen crosslinks that can contribute to increase matrix stiffening (Maruhashi et al. 2010; Garnero 2012). Periostin is also a ligand for αv integrins. Periostin promotes the proliferation of melanoma cells via binding integrin αvβ3 and αvβ5 and activating the p43/p44 MAPK signalling pathway (Kotobuki et al. 2014). Moreover, the addition of TGFβ-neutralising antibody to cultures of fibroblasts in melanoma-cell-conditioned media prevents the upregulation of periostin (Kotobuki et al. 2014). Periostin also seems to drive oesophageal cancer. Thus, Underwood and colleagues (2015) reported that the number of αSMA-positive myofibroblastic CAFs in oesophageal carcinoma correlated with poor overall survival. Since up to 68 % of oesophageal cancers are reported to be αvβ6-positive (Van Aarsen et al. 2008), one could speculate that αvβ6 integrin induces these αSMA-positive cells via TGFβ1 activation. Moreover, the secretion of periostin by these oesophageal CAFs bind to αvβ3 and αvβ5 on oesophageal cancer cells activating PI3 kinase signalling and promoting oesophageal cancer cell invasion. Genetic ablation of the periostin gene (POSTN) in the CAFs eliminates the pro-invasive effects on the oesophageal cancer cells. Additionally, clinical material has shown an exact correlation between the expression of periostin and αSMA (Underwood et al. 2015). Given its dual role as a component of matrix stiffening and pro-invasive signalling, many recent reports have unsurprisingly linked periostin to cancer promotion and poor survival (Landre et al. 2016; Qin et al. 2016; Sung et al. 2016; Xu et al. 2016; Fukuda et al. 2015) suggesting that it is also a good potential therapeutic target for suppressing cancer development.
TGFβ1-treated fibroblasts can secrete osteopontin (Corallo et al. 2014), an RGD-containing integrin ligand implicated in the promotion of tumour growth, EMT and metastasis (Platzer et al. 2011; Kothari et al. 2016). Thus, increased osteopontin correlates with increased metastasis and often poor survival in multiple types of cancer including laryngeal squamous cell carcinoma (Chen et al. 2015), melanoma (Kiss et al. 2015), nasopharyngeal carcinoma (Hou et al. 2015) and breast cancer (Xu et al. 2015). Lenga et al. (2008) reported that osteopontin was required for TGFβ1 transdifferentiation of cardiac fibroblasts to myofibroblasts, whereas Weber et al. (2015) provided evidence that osteopontin induced bone-marrow-derived mesenchymal suppressor cells to become myofibroblasts.
TGFβ1 induces the transcription of the CCN family of matricellular proteins including CCN1/cysteine-rich protein 61 (CYR61) and CCN2/connective tissue growth factor (CTGF), which affect a variety of cell types, including fibroblasts, through binding to integrins (Leask 2013). CYR61 appears to function as a tumour suppressor or promoter depending on which integrin is engaged.The binding of CYR61 to α6β1 generates senescence-inducing reactive oxygen species in fibroblasts, associated with reduced TGFβ1 expression, or induces p53-dependent apoptosis, therefore protecting against aberrant cell proliferation and fibrosis (Jun and Lau 2010; Lau 2011). In contrast, the binding of CYR61 to αvβ3 or αvβ5 stimulates prostate cancer cell growth and metastasis (Grzeszkiewicz et al. 2001) or fibroblast migration (Sun et al. 2008). In oesophageal cancers, CYR61 mRNA is an independent poor prognostic factor (Huang et al. 2009).
CTGF promotes breast cancer EMT and increases collagen type I fibre deposition (Zhu et al. 2015b). In a separate study, the pro-fibrotic activity of CTGF was observed via a positive feedback loop, binding αvβ3 on fibroblasts and further stimulating αvβ3 upregulation, collagen synthesis and contraction, thereby enhancing the potential for increased TGFβ activation (Hu et al. 2014). In addition, intermediary proteins provide an extra level of regulation of TGFβ1 and integrin autocrine signalling. For example, TGFβ1 induces secretion of protease inhibitor plasminogen activator inhibitor-1 (PAI-1), which in turn can bind to and promote αvβ3 internalisation. Therefore, PAI-1 knockout in mouse embryonic fibroblasts results in reduced αvβ3 endocytosis, elevated transmembrane TGFβ type II receptor expression and enlarged focal adhesion sites, correlating with a three-fold rise in Smad2/3 expression (Pedroja et al. 2009).
Thus, these data and those of others (Beacham and Cukierman 2005; Sherman et al. 2014) suggest that a TGFβ1-activated myofibroblast/CAF-rich tumour stroma is tumour-promoting. However, conflicting data have also been presented. A recent study reported that the global depletion of replicating αSMA-positive myofibroblasts/CAFs in two transgenic mouse models of pancreatic ductal adenocarcinoma (PDAC) enhanced the development of undifferentiated pancreatic cancers. This was associated with a significant reduction in fibrosis, the down-regulation of infiltrating macrophages and the upregulation of immunosuppressive CD4+/FoxP3+ regulatory T cells (TRegs; Ozdemir et al. 2014). In separate studies, Rhim et al. (2014) examined the role of the sonic hedgehog signalling pathway, as sonic hedgehog (Shh) can promote a fibroblast-rich microenvironemnt in cancer. They showed that either genetic ablation of sonic hedgehog protein or pharmacological inhibition of the downstream smoothened protein resulted in the development of more aggressive, highly vascularised, PDAC tumours that had a significant reduction in stromal cell density (Rhim et al. 2014). In contrast, recent work demonstrated that TGFβ1 drove a pro-tumourigenic phenotype in pancreatic cancer (Principe et al. 2016). Using the Elastase-promoter driven KRas G12D mouse, which develops pancreatic neoplasia, Principe et al. (2016) eliminated epithelial TGFβ signalling selectively by over-expression of a dominant negative TGFBR2 (DN TGFBR2) construct or eradicated TGFβ1 signalling globally by generating tgfbr1+/− mice. Mice deficient in TGFβ signalling in their epithelia showed enhanced disease progression, consistent with the loss of TGFβ1-dependent tumour suppression. In contrast, the global knockout mice showed significantly reduced tumour progression, suggesting that TGFβ1 signalling in the stroma normally promoted pancreatic neoplasia. Whereas none of these recent studies included investigations of integrin expression, their contrasting observations concerning the stromal control of pancreatic fibrosis suggest that the genetic background of the transgenic models is a major determining factor for the outcome of such studies and that we still have much to discover.
-
4)
TGFβ1 enhanced angiogenesis
Angiogenesis describes the formation of new blood vessels from pre-existing branches and promotes tumour progression, because blood vessels provide the essential nutrients required by growing tumours and facilitate metastasis (Chung and Ferrara 2011). TGFβ1 can promote angiogenesis (Vinals and Pouyssegur 2001; Li et al. 2001) and thus local activation of TGFβ by αv integrins in cancer is likely to promote the development of blood vessels. However, TGFβ-mediated effects are concentration-dependent, as low concentrations can promote blood vessel formation, whereas higher amounts are anti-angiogenic (Orlova et al. 2011). Moreover, TGFβ-concentration-dependent actions may occur via the stimulation of different TGFβ receptor signalling pathways. Activation of the activin-receptor-like kinase (ALK1) induces endothelial cell proliferation and migration via the phosphorylation of Smad 1/5, whereas the activation of the ALK5/Smad 2 cascade inhibits this effect on endothelial cells (Goumans et al. 2002).
Integrins αvβ3 and αvβ5 (Weis and Cheresh 2011) and αvβ8 (Zhu et al. 2002; Arnold et al. 2012) are all reported to regulate angiogenesis but only αvβ8 is described to do so through TGFβ1 activation. The role of αvβ3 and αvβ5 in angiogenesis is under debate. Both integrins are upregulated on endothelial cells of new blood vessels and promote their migration in vitro and their blockade with antibodies have suppressed blood vessel growth in a variety of pre-clinical studies (Gutheil et al. 2000; Friedlander et al. 1995). However, genetic ablation of itgb3, itgb5 or both simultaneously accelerates tumour vasculature growth and development, attributable to the increased expression of vascular endothelial growth factor receptor 2 (VEGFR2) and sensitivity to VEGF-A (discussed by Silva et al. 2008) suggesting that these integrins are not required for blood vessel formation but regulate their rate of growth.
Integrin αvβ8-knock out mice demonstrate severe vascular defects resulting in haemorrhaging, predominantly in the central nervous system, with a large proportion of mice dying shortly after birth and some earlier (Zhu et al. 2002). Knockout of β8 in Muller glia or retinal ganglion cells promotes vascular abnormalities that match retinal TGFβ1-knockout, suggesting αvβ8 is necessary for TGFβ1–dependent retinal maturation (Arnold et al. 2012). Integrin αvβ8 is not expressed on endothelial cells but is expressed on neural peri-vascular astrocytic cells where it binds and activates latent TGFβ1, which promotes increased stability and differentiation of blood vessels (Cambier et al. 2005).
Analysis of αvβ8 function was examined in astrocytoma by using orthotopic transgenic cell lines. Intra-cranial injection of human β8-low astrocytoma cells (U87) resulted in haemorrhagic tumours, whereas β8-positive cells formed microscopic tumours with well-defined vasculature. Orthotopic injection again showed a well-established vasculature in β8 wild-type transformed astrocytes and a disorganised haemorrhagic vasculature in the absence of β8. Moreover, endothelial cells examined from β8−/− mice express approximately two- to three-fold less phospho-Smad 2 and 3 suggesting that less active TGFβ is present in β8-null mice. However, no significant difference was seen in tumour volume in β8−/− and wild-type mice, although the former exhibited reduced survival rates because of haemorrhage (Tchaicha et al. 2010). In separate studies, the over expression of β8 in U87 was shown to result in brain tumours with organised non-haemorrhagic vasculature and this affect required the activation of latent TGFβ1 (Tchaicha et al. 2011).
A key mediator between intracellular TGFβ signalling and transmembrane integrins in endothelial cells is endoglin, a TGFβ superfamily accessory receptor that is expressed in the angiogenic vessels of brain, lung, breast, stomach and colon cancers (Minhajat et al. 2006). In response to murine embryonic endothelial cells binding fibronectin via integrin α5β1, TGF-β1-induced Smad1/5/8 phosphorylation increases in an endoglin-dependent manner. Reciprocally, endoglin is necessary for TGFβ1-induced phosphorylation of β1 integrin threonines 788/789 and FAK, whereas fibronectin/endoglin cross-talk maintains endothelial cell survival (Tian et al. 2012). Perhaps unsurprisingly, endoglin is a prognostic marker in colorectal cancers with correlation to lymph node and liver metastases (Saad et al. 2004).
-
5)
TGFβ regulates inflammatory and immune cells to modulate cancer progression
When integrin-LAP binding is prevented by an RGD to RGE mutation in LAPβ1, mice exhibit a multi-organ inflammatory response that mirrors global TGFβ1-knockout (Yang et al. 2007). Since the effective absence of both αvβ6 and αvβ8 also results in multi-organ inflammation, recapitulating global TGFβ1 deficiency, this suggests that none of the other αv integrins are required to regulate these particular TGFβ1-dependent effects (Aluwihare et al. 2009). However, these studies have only assessed the integrin-dependent activation of latent TGFβ during development and birth but not in adult mice or in pathological situations such as cancer. Thus, the possibility exists that the activation of TGFβ by any αv integrin can influence local inflammatory and immune cells in adult animals. This is important as TGFβ1 promotes immunosuppressive effects on various effector T-cells (for a review, see Travis and Sheppard 2014) and induces tumour-promoting phenotypes in both neutrophils and macrophages.
Travis et al. (2007) reported that the loss of αvβ8 on dendritic cells results in inflammatory bowel disease (Travis et al. 2007). The effect is attributable to an inability of αvβ8-deficient dendritic cells to activate TGFβ locally and thus to promote the differentiation of naïve T-cells into TReg via Foxp3 transcription factor (Chen et al. 2003). TReg cells are a subpopulation of CD4+ CD25+ T cells whose functions encompass the immunosuppression of effector T-cells including the maturation of CD8 cytotoxic T cells (McNally et al. 2011) and maintaining immunological tolerance (discussed in Travis and Sheppard 2014). Moreover, these differentiated cells suppress their production of helper T cell cytokines, instead secreting TGFβ1, further amplifying TGFβ1 signalling and its immunosuppressive effects (Chen et al. 2003). These data correlate with the observation that high levels of TRegs in cancer tissues are associated with poor overall survival from several types of solid cancer (Shang et al. 2015). TReg accumulation correlates with tumour progression in breast, ovarian and pancreatic cancer, although the effect of TRegs on prognosis depends on factors such as tumour location and inflammatory status (Oleinika et al. 2013; Schmidt et al. 2016). Although, to date, no pre-clinical studies have made a detailed examination of immune infiltrates in response to blockade of αv integrins, carcinomas expressing αvβ6, astrocytomas expressing αvβ8 and desmoplastic tumour myofibroblasts expressing αvβ5 and αvβ1, are likely to regulate TReg levels in the TME.
TGFβ1 also promotes the formation of tumour-promoting M2 tumour-associated macrophages and N2 tumour-associated neutrophils (Gong et al. 2012; Fridlender et al. 2009). Macrophages present as having “M1” anti-tumour or “M2” tumour-promoting characteristics. Macrophages co-cultured with ovarian carcinoma cells significantly increase the transcription of TGFβ1, TGFBRI and TGFBRII (Hagemann et al. 2006). TGFβ1 stimulates monocyte recruitment and alters the inflammatory gene expression profile of macrophages by increasing metastasis-associated interleukin-6 (IL-6) and suppressing cytokines such as IL-10 and chemoattractants CCL3 and CCL4 (Krstic and Santibanez 2014). TGFβ1 stimulation of macrophages also promotes angiogenesis under hypoxic conditions by the elevated production of VEGF, MMP-9 and VEGF receptor Flk-1 expression (Jeon et al. 2007). High levels of M2 macrophages correlate with poor survival from cancer including pancreatic and cervical cancer (Petrillo et al. 2015; Hu et al. 2016), gastric cancer spread (Yamaguchi et al. 2015) and relapse after chemotherapy (Hughes et al. 2015). Indeed, pre-clinical studies suggest M2 cells can promote metastasis (Ding et al. 2015; Wu et al. 2015; Yang et al. 2015).
Similarly, tumour-associated neutrophils polarised to the N2 state are also pro-tumourigenic as reviewed recently (Sionov et al. 2015). Thus, depletion of N2 neutrophils within TGFβ1-rich mouse tumours significantly reduces tumour size, supporting the concept that separate populations of anti-tumour and pro-tumour neutrophils are regulated by active TGFβ (Fridlender et al. 2009). Recent studies by Sagiv et al. (2015) suggested neutrophils can be segregated according to their cell density, whereby low-density neutrophils are tumour-promoting and high-density neutrophils are tumour-suppressive. The authors discovered that tumour progression in mice correlated with an increase in the fraction of circulating low-density neutrophils, eventually becoming the dominant fraction; in tumour-free mice, 95 % of circulating neutrophils had the high-density phenotype. Moreover, the authors discovered that TGFβ1 was required and sufficient to promote the transition of the high-density neutrophils into the tumour-supportive low-density neutrophils. Functionally, the high-density neutrophils suppressed tumour growth and were cytotoxic in vitro towards tumour cells. In contrast, the low-density neutrophils were deficient in cytotoxic H2O2, exhibited reduced chemotaxis towards the tumour and were less phagocytic. In addition, low-density neutrophils produced less inflammatory cytokines and were able to suppress the proliferation of CD8+ cytotoxic T-cells and thus effectively promoted tumour growth.
Overall, TGFβ has tumour-promoting and -immunosuppressive effects on several immune cell types in the tumour stroma. However, more studies are needed to determine what proportion of TGFβ activation of immune cells in the tumour stroma is solely integrin-dependent, so that therapeutic targeting of relevant integrins can suppress the pathogenic effects of active TGFβ on tumour-promoting immune components.
Future of drug targeting integrin-dependent activation of TGFβ in cancer
In this review, we discussed the indirect effects on tumour progression exerted by αv integrins because of their ability to activate latent TGFβ1. However, all αv integrins can signal via their cytoplasmic tails and some (e.g., αvβ3 and αvβ6) can generate pro-tumourigenic signals including those for growth, survival and the secretion of proteases, factors that can enhance tumour progression. Thus, the fraction of an integrin’s ability to promote cancer via its cytoplasmic tail versus its ability to activate TGFβ is unclear. Regardless, the direct inhibition of the ligand-binding site of αv integrins is sufficient to block their capacity to activate TGFβ1 and to generate survival signals and has been used in pre-clinical and clinical studies (Eberlein et al. 2013; Marsh et al. 2008).
Of the integrins that can potentially activate TGFβ1, only αvβ3-specific antibodies and peptides have been used in humans as part of anti-cancer therapies (Posey et al. 2001; Hersey et al. 2010; Khasraw et al. 2016) but none has achieved significant clinical success. Whereas some cancers express high levels of αvβ3 (e.g., melanoma and glioblastoma), the discovery that αvβ3 negatively regulates the endothelial cell growth factor receptor VEGFR2 means that blockade of αvβ3 will result in increased angiogenesis and tumour growth (see above). However, Reynolds et al. (2009) reported that sub-therapeutic concentrations of Cilengitide, an αvβ3/αvβ5 peptide inhibitor, enhanced angiogenesis and, as new blood vessels tend to be leakier, Wong et al. (2015) used this to show that pre-dosing with Cilengitide improved drug accessibility to a variety of experimental tumours. Thus, targeting αvβ3 may still be therapeutic but possibly not by inhibiting TGFβ1 activation. Recently, Merck published results of the POSEIDON trial. Their pan-αv blocking humanised antibody Abituzumab was compared with the standard of care (cetuximab, an EGFR inhibitor and irinotecan, a topoisomerase 1 inhibitor) in metastatic colorectal cancer (CRC; Elez et al. 2015). Data from the whole cohort showed that combining Abituzumab with cetuximab and irinotecan provided no significant survival benefit versus the cetuximab and irinotecan combination alone. However, when the expression levels of the different αv inetrgins on the tumour tissue were assessed, the data showed that the risk of death for patients with high αvβ6 (above the median histological score) was reduced by 59 % by the addition of Abituzumab to the treatment regimen. Overall, the data suggest that targeting αvβ6 with inhibitory antibodies could be positively therapeutic for patients whose cancer over-expresses αvβ6. It is worth recalling that, in those studies with large enough patient cohorts, high αvβ6 correlated with poor survival and, thus, the identification of those patients by the simple immunohistochemistry of biopsies might be sufficient to select only the likely responders to αvβ6-blocking therapy.
Antibody targeting of integrin αvβ6 in humans is work in progress. Thus, Biogen-Idec have developed the humanised αvβ6-blocking monoclonal antibody STX-100, which is being used in idiopathic fibrosis studies (clinicaltrials.gov# NCT01371305). AstraZeneca-Medimmune have developed the entirely human monoclonal antibody 264RAD, which blocks αvβ6 but also has some blocking activity of αvβ8 (Eberlein et al. 2013). To date, this antibody has not entered clinical trials but speculation regarding its potential effects is of interest, given the results of the POSEIDON study, which has suggested that the blockade of αvβ6 is likely to be therapeutic in colon cancer (Elez et al. 2015). Thus, Eberlein and colleagues (2013) reported the complete suppression of the growth of xenografts of the oral squamous carcinoma cell line Detroit-562 at doses of 5 and 20 mg/kg 264RAD and this correlates with the loss of expression of the target integrin αvβ6 on the residual tumour. Moore and colleagues (2014) also noted that 264RAD-induced breast tumour xenograft suppression also correlates with loss of αvβ6 expression. Since antibody 264RAD operates as a ligand mimetic, we consider it worth noting that, in a recent study, GSK also reported, from in vitro studies, that the exposure of cells to LAP or A20FMDV2, a high-affinity αvβ6-specific 20-amino-acid peptide derived from foot-and-mouth-disease virus, causes a rapid ligand-induced internalisation of αvβ6 but a slower re-expression (Slack et al. 2016). Thus, perhaps ligand-induced transient loss of cell-surface αv integrins may be a goal of future anti-cancer therapeutics, since this might be more efficient as a means of suppressing local TGFβ1 activation than attempting to achieve high enough concentrations of an extracellular inhibitor that can completely suppress the binding of the integrin to LAP.
Antibody blockade of αvβ6 may not be the only therapeutic strategy for regulating TGFβ1 activity. Merck has generated small-molecule inhibitors of αvβ6, αvβ5 and αvβ3 (Goodman et al. 2002) and GSK have recently produced naphtyridine-derivative small molecule antagonists of αvβ6 (Patent WO 2014154725 A1). Whereas neither company has used their compounds clinically, if these small molecules can also operate as ligand-mimetics, they might compare favourably with inhibitory antibodies that are likely to have better pharmacokinetics.
We should also mention the potential added value of the therapeutic targeting of integrins as a means of regulating TGFβ1 activity in cancer and fibrosis as opposed to directly targeting the TGFβ1 signalling pathway. Thus, the blockade of TGFβ receptors with small-molecule kinase inhibitors or of TGFβ itself with inhibitory antibodies would promote a whole body inhibition of TGFβ activity, whereas integrin blockade produces local control of TGFβ activation. Since, as mentioned above, TGFβ is required for normal tissue regulation, integrin targeting would minimise off-target effects.
Concluding remarks
Integrin activation of latent TGFβ1 can be a significant component of the way that tumours develop and respond to their stroma. First, the ability of αvβ6 and αvβ8 integrins to activate TGFβ1 was established as a major platform for the normal homeostatic regulation of TGFβ1 activity. Second, the discovery that both αvβ5 and αvβ1 on fibroblasts also activated latent TGFβ1 in a biomechanical way (like αvβ6) indicated that stromal cells could also contribute to local TGFβ1 activity. Finally, the additional revelation that increasing stromal stiffness reduced the activation threshold for the force required to activate TGFβ1 introduced the likelihood that pathological stromal fibrosis was driven by an amplification of activation of abundant latent TGFβ1 in the matrix generated by multiple αv integrins. However, although multiple mechanisms exist by which TGFβ1 modulates the tumour microenvironment to promote cancer (Fig. 2; Pickup et al. 2013), we still have a relative paucity of empirical in vivo data firmly establishing those TGFβ1-driven stromal responses that are the direct result of the αv integrin activation of TGFβ. We clearly lack certain reagents and model systems that would allow us fully to characterise those αv integrins that activate TGFβ at various times and stages in cancer progression. Thus, we lack certain tissue-specific inducible αvβ integrin knockout mice (including itgb6) that would permit researchers, over the course of the development of a cancer, temporally to eliminate an αvβ integrin and examine its contribution to TGFβ1-driven tumourigenesis. Subsequently, by introducing therapeutics to these same integrins, we could establish when the most effective therapeutic window occurs and also whether cancers reach a point at which the targeting of integrin-dependent TGFβ1 activation is no longer effective, perhaps because so many processes (multiple integrins, tissue stiffness, proteases) are simultaneously able to activate the latent TGFβ. We have mentioned the absence of antibodies that would allow us to determine the expression of αvβ1 and αvβ8 in tissues and, similarly, no αvβ8 blocking antibodies are available for pre-clinical studies. Therefore, as these reagents become available, we may better be able to delineate an appropriate clinical strategy by the study of the temporal increase in the number of diverse mechanisms for latent TGFβ activation that accumulate during cancer development. However, although we may lack some data that will refine our treatments, αv integrins must now be considered as bona fide targets for anti-TGFβ therapy in fibrosis and cancer.

Contribution of integrin-mediated TGFβ1 activation of pro-tumour mechanisms via regulation of cells of the immune system, promotion of epithelial-mesenchymal transition and angiogenesis. Immune system 1) Active TGFβ stimulates naive T-cell-to-Treg differentiation (top). Tregs exhibit αvβ8-mediated TGFβ1 activation, amplifying the differentiation of other naive T-cells. Tregs suppress anti-tumour T-cell responses. Tumour cell integrins mediate TGFβ1 activation (bottom). Active TGFβ1 stimulates macrophage and neutrophil pro-tumour responses. Epithelial-to-mesenchymal transition TGFβ1 stimulation of epithelial cells represses epithelial gene signatures, e.g., cell-cell adhesion molecule E-cadherin (top). This promotes a more mesenchymal invasive cell phenotype (bottom). Angiogenesis TGFβ1 stimulation of tumour cells increases VEGF expression, which binds to its receptor on local endothelial cells. Integrin αvβ3 promotes activation and recycling of VEGFR (left). The αvβ8-mediated activation of TGFβ1 facilitates TGFβ activation of local endothelial cells, which can promote or inhibit angiogenesis via separate TGFβ receptors (right)
References
Ahmed N, Pansino F, Clyde R, Murthi P, Quinn MA, Rice GE, Agrez MV, Mok S, Baker MS (2002) Overexpression of alpha(v)beta6 integrin in serous epithelial ovarian cancer regulates extracellular matrix degradation via the plasminogen activation cascade. Carcinogenesis 23:237–244
Allen MD, Thomas GJ, Clark S, Dawoud MM, Vallath S, Payne SJ, Gomm JJ, Dreger SA, Dickinson S, Edwards DR, Pennington CJ, Sestak I, Cuzick J, Marshall JF, Hart IR, Jones JL (2014) Altered microenvironment promotes progression of preinvasive breast cancer: myoepithelial expression of alphavbeta6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin Cancer Res 20:344–357
Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS (2009) Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 122:227–232
Annes JP, Chen Y, Munger JS, Rifkin DB (2004) Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol 165:723–734
Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, Reichardt LF (2012) Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphaVbeta8-mediated activation of transforming growth factor-beta. J Neurosci 32:1197–1206
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218
Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM (2005) Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest 115:339–347
Beacham DA, Cukierman E (2005) Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol 15:329–341
Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA (1996) Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85:683–693
Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, Einheber S, Boudreau N, Nishimura SL (2005) Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol 166:1883–1894
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198:1875–1886
Chen J, Zhu C, He Z, Geng M, Li G, Tao X, Zhang F (2015) Association of OPN overexpression with tumor stage, differentiation, metastasis and tumor progression in human laryngeal squamous cell carcinoma. Int J Clin Exp Med 8:7116–7124
Chung AS, Ferrara N (2011) Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol 27:563–584
Corallo C, Volpi N, Franci D, Montella A, Biagioli M, Mariotti G, D’Onofrio F, Gonnelli S, Nuti R, Giordano N (2014) Is osteopontin involved in cutaneous fibroblast activation? Its hypothetical role in scleroderma pathogenesis. Int J Immunopathol Pharmacol 27:97–102
da Silva R, Uno M, Marie SK, Oba-Shinjo SM (2015) LOX expression and functional analysis in astrocytomas and impact of IDH1 mutation. PLoS One 10:e0119781
Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM (2005) Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem 280:18871–18880
DiCara D, Rapisarda C, Sutcliffe JL, Violette SM, Weinreb PH, Hart IR, Howard MJ, Marshall JF (2007) Structure-function analysis of Arg-Gly-Asp helix motifs in alpha v beta 6 integrin ligands. J Biol Chem 282:9657–9665
Ding L, Liang G, Yao Z, Zhang J, Liu R, Chen H, Zhou Y, Wu H, Yang B, He Q (2015) Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 6:36441–36455
Dutta A, Li J, Fedele C, Sayeed A, Singh A, Violette SM, Manes TD, Languino LR (2015) Alphavbeta6 integrin is required for TGFbeta1-mediated matrix metalloproteinase2 expression. Biochem J 466:525–536
Eberlein C, Kendrew J, McDaid K, Alfred A, Kang JS, Jacobs VN, Ross SJ, Rooney C, Smith NR, Rinkenberger J, Cao A, Churchman A, Marshall JF, Weir HM, Bedian V, Blakey DC, Foltz IN, Barry ST (2013) A human monoclonal antibody 264RAD targeting alphavbeta6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 32:4406–4416
Eberlein C, Rooney C, Ross SJ, Farren M, Weir HM, Barry ST (2015) E-Cadherin and EpCAM expression by NSCLC tumour cells associate with normal fibroblast activation through a pathway initiated by integrin αvβ6 and maintained through TGFβ signalling.Oncogene 34:704–716
Elayadi AN, Samli KN, Prudkin L, Liu YH, Bian A, Xie XJ, Wistuba II, Roth JA, McGuire MJ, Brown KC (2007) A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res 67:5889–5895
Elez E, Kocakova I, Hohler T, Martens UM, Bokemeyer C, Van Cutsem E, Melichar B, Smakal M, Csoszi T, Topuzov E, Orlova R, Tjulandin S, Rivera F, Straub J, Bruns R, Quaratino S, Tabernero J (2015) Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial. Ann Oncol 26:132–40
Fine JD, Hintner H (2009) Life with epidermolysis bullosa (EB): etiology, diagnosis, multidisciplinary care and therapy. Springer, Vienna
Fontana L, Chen Y, Prijatelj P, Sakai T, Fassler R, Sakai LY, Rifkin DB (2005) Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J 19:1798–1808
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16:183–194
Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA (1995) Definition of two angiogenic pathways by distinct alpha v integrins. Science 270:1500–1502
Friesenhengst A, Pribitzer-Winner T, Schreiber M (2014) Association of the G473A polymorphism and expression of lysyl oxidase with breast cancer risk and survival in European women: a hospital-based case–control study. PLoS One 9:e105579
Fukuda K, Sugihara E, Ohta S, Izuhara K, Funakoshi T, Amagai M, Saya H (2015) Periostin is a key niche component for wound metastasis of melanoma. PLoS One 10:e0129704
Garnero P (2012) The contribution of collagen crosslinks to bone strength. Bonekey Rep 1:182
Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E (2009) Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11:1287–1296
Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N (2012) TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol 13:31
Goodman SL, Holzemann G, Sulyok GA, Kessler H (2002) Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins. J Med Chem 45:1045–1051
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21:1743–1753
Grzeszkiewicz TM, Kirschling DJ, Chen N, Lau LF (2001) CYR61 stimulates human skin fibroblast migration through integrin alpha vbeta 5 and enhances mitogenesis through integrin alpha vbeta 3, independent of its carboxyl-terminal domain. J Biol Chem 276:21943–21950
Guo L, Zhang Y, Zhang L, Huang F, Li J, Wang S (2016) MicroRNAs, TGF-beta signaling, and the inflammatory microenvironment in cancer. Tumour Biol 37:115–125
Gutheil JC, Campbell TN, Pierce PR, Watkins JD, Huse WD, Bodkin DJ, Cheresh DA (2000) Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res 6:3056–3061
Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S, Balkwill FR (2006) Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol 176:5023–5032
Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF 3rd, Sheppard D, Gardner HA, Violette SM (2007) Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol 170:110–125
Hazelbag S, Kenter GG, Gorter A, Dreef EJ, Koopman LA, Violette SM, Weinreb PH, Fleuren GJ (2007) Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J Pathol 212:316–324
Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D (2013) Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19:1617–1624
Hersey P, Sosman J, O’Day S, Richards J, Bedikian A, Gonzalez R, Sharfman W, Weber R, Logan T, Buzoianu M, Hammershaimb L, Kirkwood JM (2010) A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage IV metastatic melanoma. Cancer 116:1526–1534
Hezel AF, Deshpande V, Zimmerman SM, Contino G, Alagesan B, O’Dell MR, Rivera LB, Harper J, Lonning S, Brekken RA, Bardeesy N (2012) TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res 72:4840–4845
Hou X, Wu X, Huang P, Zhan J, Zhou T, Ma Y, Qin T, Luo R, Feng Y, Xu Y, Chen L, Zhang L (2015) Osteopontin is a useful predictor of bone metastasis and survival in patients with locally advanced nasopharyngeal carcinoma. Int J Cancer 137:1672–1678
Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW (2016) The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. doi:10.1007/s13277-015-4741-z
Hu X, Li N, Tao K, Fang X, Liu J, Wang Y, Wang H, Shi J, Wang Y, Ji P, Cai W, Bai X, Zhu X, Han J, Hu D (2014) Effects of integrin ανβ3 on differentiation and collagen synthesis induced by connective tissue growth factor in human hypertrophic scar fibroblasts. Int J Mol Med 34:1323–1334
Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV Jr, Sheppard D (1996) Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 133:921–928
Huang X, Xiao DW, Xu LY, Zhong HJ, Liao LD, Xie ZF, Li EM (2009) Prognostic significance of altered expression of SDC2 and CYR61 in esophageal squamous cell carcinoma. Oncol Rep 21:1123–1129
Hubbard R, Venn A, Lewis S, Britton J (2000) Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med 161:5–8
Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, Tazzyman S, Danson S, Addison C, Clemons M, Gonzalez-Angulo AM, Joyce JA, De Palma M, Pollard JW, Lewis CE (2015) Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res 75:3479–3491
Inman GJ (2011) Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev 21:93–99
Jenkins G (2008) The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol 40:1068–1078
Jeon SH, Chae BC, Kim HA, Seo GY, Seo DW, Chun GT, Kim NS, Yie SW, Byeon WH, Eom SH, Ha KS, Kim YM, Kim PH (2007) Mechanisms underlying TGF-beta1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol 81:557–566
Jotzu C, Alt E, Welte G, Li J, Hennessy BT, Devarajan E, Krishnappa S, Pinilla S, Droll L, Song YH (2010) Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst) 33:61-79
Jun JI, Lau LF (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12:676–685
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J (1995) Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 11:415–421
Kang JS, Liu C, Derynck R (2009) New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol 19:385–394
Khasraw M, Lee A, McCowatt S, Kerestes Z, Buyse ME, Back M, Kichenadasse G, Ackland S, Wheeler H (2016) Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J Neuro-Oncol 128:163–171
Kiss T, Ecsedi S, Vizkeleti L, Koroknai V, Emri G, Kovacs N, Adany R, Balazs M (2015) The role of osteopontin expression in melanoma progression. Tumour Biol 36:7841–7847
Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B (2014) Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J Cell Biol 207:283–297
Kothari AN, Arffa ML, Chang V, Blackwell RH, Syn WK, Zhang J, Mi Z, Kuo PC (2016) Osteopontin-A master regulator of epithelial-mesenchymal transition. J Clin Med 5:39
Kotobuki Y, Yang L, Serada S, Tanemura A, Yang F, Nomura S, Kudo A, Izuhara K, Murota H, Fujimoto M, Katayama I, Naka T (2014) Periostin accelerates human malignant melanoma progression by modifying the melanoma microenvironment. Pigment Cell Melanoma Res 27:630–639
Krstic J, Santibanez JF (2014) Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal 2014:521754
Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S (1993) Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A 90:770–774
Landre V, Antonov A, Knight R, Melino G (2016) p73 promotes glioblastoma cell invasion by directly activating POSTN (periostin) expression. Oncotarget 7:11785–11802
Lau LF (2011) CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci 68:3149–3163
Leask A (2013) Integrin 1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts. Adv Wound Care 2:160–166
Lee YH, Albig AR, Regner M, Schiemann BJ, Schiemann WP (2008) Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 29:2243–2251
Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R (2008)Osteopontin expression is required for myofibroblast differentiation.Circ Res 102:319–327
Levy L, Hill CS (2005) Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol 25:8108–8125
Lewis MP, Lygoe KA, Nystrom ML, Anderson WP, Speight PM, Marshall JF, Thomas GJ (2004) Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer 90:822–832
Li C, Guo B, Bernabeu C, Kumar S (2001) Angiogenesis in breast cancer: the role of transforming growth factor beta and CD105. Microsc Res Tech 52:437–449
Lin RL, Zhao LJ (2015) Mechanistic basis and clinical relevance of the role of transforming growth factor-beta in cancer. Cancer Biol Med 12:385–393
Ludlow A, Yee KO, Lipman R, Bronson R, Weinreb P, Huang X, Sheppard D, Lawler J (2005) Characterization of integrin beta6 and thrombospondin-1 double-null mice. J Cell Mol Med 9:421–437
Malik R, Lelkes PI, Cukierman E (2015) Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol 33:230–236
Mamuya FA, Duncan MK (2012) aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med 16:445–455
Margadant C, Sonnenberg A (2010) Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11:97–105
Marsh D, Dickinson S, Neill GW, Marshall JF, Hart IR, Thomas GJ (2008) Alpha vbeta 6 integrin promotes the invasion of morphoeic basal cell carcinoma through stromal modulation. Cancer Res 68:3295–3303
Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, Upile T, Kalavrezos N, Violette SM, Weinreb PH, Chester KA, Chana JS, Marshall JF, Hart IR, Hackshaw AK, Piper K, Thomas GJ (2011) Stromal features are predictive of disease mortality in oral cancer patients. J Pathol 223:470–481
Martins VL, Caley MP, Moore K, Szentpetery Z, Marsh ST, Murrell DF, Kim MH, Avari M, McGrath JA, Cerio R, Kivisaari A, Kahari VM, Hodivala-Dilke K, Brennan CH, Chen M, Marshall JF, O’Toole EA (2016) Suppression of TGFbeta and angiogenesis by type VII collagen in cutaneous SCC. J Natl Cancer Inst 108:djv293
Maruhashi T, Kii I, Saito M, Kudo A (2010) Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 285:13294–13303
McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ (2011) CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci U S A 108:7529–7534
Medici D, Hay ED, Goodenough DA (2006) Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol Biol Cell 17:1871–1879
Miles FL, Sikes RA (2014) Insidious changes in stromal matrix fuel cancer progression. Mol Cancer Res 12:297–312
Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, Bailey P, Chang D, Oien K, Karim S, Au A, Steele C, Carter CR, McKay C, Anderson K, Evans TR, Marais R, Springer C, Biankin A, Erler JT, Sansom OJ (2015) Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med 7:1063–1076
Minhajat R, Mori D, Yamasaki F, Sugita Y, Satoh T, Tokunaga O (2006) Organ-specific endoglin (CD105) expression in the angiogenesis of human cancers. Pathol Int 56:717–723
Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, Welty NE, Igyarto BZ, Wijeyesinghe S, Thompson EA, Matte C, Bartholin L, Kaplan A, Sheppard D, Bridges AG, Shlomchik WD, Masopust D, Kaplan DH (2016) Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol 17:414–421
Moore KM, Thomas GJ, Duffy SW, Warwick J, Gabe R, Chou P, Ellis IO, Green AR, Haider S, Brouilette K, Saha A, Vallath S, Bowen R, Chelala C, Eccles D, Tapper WJ, Thompson AM, Quinlan P, Jordan L, Gillett C, Brentnall A, Violette S, Weinreb PH, Kendrew J, Barry ST, Hart IR, Jones JL, Marshall JF (2014) Therapeutic targeting of integrin alphavbeta6 in breast cancer. J Natl Cancer Inst 106:dju169
Mori S, Kodaira M, Ito A, Okazaki M, Kawaguchi N, Hamada Y, Takada Y, Matsuura N (2015) Enhanced expression of integrin alphavbeta3 induced by TGF-beta is required for the enhancing effect of fibroblast growth factor 1 (FGF1) in TGF-beta-induced epithelial-mesenchymal transition (EMT) in mammary epithelial cells. PLoS One 10:e0137486
Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D (2003) Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422:169–173
Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL (2002) The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157:493–507
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D (1999) The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319–328
Naber HP, Drabsch Y, Snaar-Jagalska BE, ten Dijke P, van Laar T (2013) Snail and Slug, key regulators of TGF-beta-induced EMT, are sufficient for the induction of single-cell invasion. Biochem Biophys Res Commun 435:58–63
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR (2013) Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 171:36–45
Orlova VV, Liu Z, Goumans MJ, ten Dijke P (2011) Controlling angiogenesis by two unique TGF-beta type I receptor signaling pathways. Histol Histopathol 26:1219–1230
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R (2014) Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25:719–734
Pedroja BS, Kang LE, Imas AO, Carmeliet P, Bernstein AM (2009) Plasminogen activator inhibitor-1 regulates integrin alphavbeta3 expression and autocrine transforming growth factor beta signaling. J Biol Chem 284:20708–20717
Petrillo M, Zannoni GF, Martinelli E, Pedone Anchora L, Ferrandina G, Tropeano G, Fagotti A, Scambia G (2015) Polarisation of tumor-associated macrophages toward M2 phenotype correlates with poor response to chemoradiation and reduced survival in patients with locally advanced cervical cancer. PLoS One 10:e0136654
Pickup M, Novitskiy S, Moses HL (2013) The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer 13:788–799
Platzer G, Schedlbauer A, Chemelli A, Ozdowy P, Coudevylle N, Auer R, Kontaxis G, Hartl M, Miles AJ, Wallace BA, Glatter O, Bister K, Konrat R (2011) The metastasis-associated extracellular matrix protein osteopontin forms transient structure in ligand interaction sites. Biochemistry 50:6113–6124
Posey JA, Khazaeli MB, DelGrosso A, Saleh MN, Lin CY, Huse W, LoBuglio AF (2001) A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother Radiopharm 16:125–132
Principe DR, DeCant B, Mascarinas E, Wayne E, Diaz A, Akagi N, Hwang RF, Pasche B, Dawson DW, Fang D, Bentrem DJ, Munshi HG, Jung B, Grippo PJ (2016) TGFbeta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res (in press)
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ (2006) Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 4:38
Qin X, Yan M, Zhang J, Wang X, Shen Z, Lv Z, Li Z, Wei W, Chen W (2016) TGFbeta3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci Rep 6:20587
Reynolds AR, Hart IR, Watson AR, Welti JC, Silva RG, Robinson SD, Da Violante G, Gourlaouen M, Salih M, Jones MC, Jones DT, Saunders G, Kostourou V, Perron-Sierra F, Norman JC, Tucker GC, Hodivala-Dilke KM (2009) Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med 15:392–400
Reynolds LE, Conti FJ, Lucas M, Grose R, Robinson S, Stone M, Saunders G, Dickson C, Hynes RO, Lacy-Hulbert A, Hodivala-Dilke K (2005) Accelerated re-epithelialization in beta3-integrin-deficient-mice is associated with enhanced TGF-beta1 signaling. Nat Med 11:167–174
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ (2014) Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25:735–747
Roberts AB, Wakefield LM (2003) The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A 100:8621–8623
Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB (2015) Latent TGF-beta-binding proteins. Matrix Biol 47:44–53
Rolli M, Fransvea E, Pilch J, Saven A, Felding-Habermann B (2003) Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc Natl Acad Sci U S A 100:9482–9487
Saad RS, Liu YL, Nathan G, Celebrezze J, Medich D, Silverman JF (2004) Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in colorectal cancer. Modern Pathol 17:197–203
Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV, Ariel A, Hovav AH, Henke E, Fridlender ZG, Granot Z (2015) Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 10:562–573
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124:2659–2670
Schmidt A, Zhang XM, Joshi RN, Iqbal S, Wahlund C, Gabrielsson S, Harris RA, Tegner J (2016) Human macrophages induce CD4Foxp3 regulatory T cells via binding and re-release of TGF-beta. Immunol Cell Biol (in press)
Shang B, Liu Y, Jiang SJ (2015) Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 5:15179
Sheppard D (2015) Epithelial-mesenchymal interactions in fibrosis and repair. Transforming growth factor-beta activation by epithelial cells and fibroblasts. Ann Am Thorac Soc 12 (Suppl 1):S21–S23
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM (2014) Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159:80–93
Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359:693–699
Silva R, D’Amico G, Hodivala-Dilke KM, Reynolds LE (2008) Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol 28:1703–1713
Sionov RV, Fridlender ZG, Granot Z (2015) The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron 8:125–158
Sipos B, Hahn D, Carceller A, Piulats J, Hedderich J, Kalthoff H, Goodman SL, Kosmahl M, Kloppel G (2004) Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 45:226–236
Slack RJ, Hafeji M, Rogers R, Ludbrook SB, Marshall JF, Flint DJ, Pyne S, Denyer JC (2016) Pharmacological characterization of the alphavbeta6 integrin binding and internalization kinetics of the foot-and-mouth disease virus derived peptide A20FMDV2. Pharmacology 97:114–125
Sun ZJ, Wang Y, Cai Z, Chen PP, Tong XJ, Xie D (2008) Involvement of Cyr61 in growth, migration, and metastasis of prostate cancer cells. Br J Cancer 99:1656–1667
Sung PL, Jan YH, Lin SC, Huang CC, Lin H, Wen KC, Chao KC, Lai CR, Wang PH, Chuang CM, Wu HH, Twu NF, Yen MS, Hsiao M, Huang CY (2016) Periostin in tumor microenvironment is associated with poor prognosis and platinum resistance in epithelial ovarian carcinoma. Oncotarget 7:4036–4047
Tchaicha JH, Mobley AK, Hossain MG, Aldape KD, McCarty JH (2010) A mosaic mouse model of astrocytoma identifies alphavbeta8 integrin as a negative regulator of tumor angiogenesis. Oncogene 29:4460–4472
Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH (2011) Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res 71:6371–6381
Tian H, Mythreye K, Golzio C, Katsanis N, Blobe GC (2012) Endoglin mediates fibronectin/alpha5beta1 integrin and TGF-beta pathway crosstalk in endothelial cells. EMBO J 31:3885–3900
Travis MA, Sheppard D (2014) TGF-beta activation and function in immunity. Annu Rev Immunol 32:51–82
Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, Bluestone JA, Sheppard D (2007) Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449:361–365
Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE, Balcioglu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, Meerman JH, van de Water B, Danen EH (2014) Beta1 integrin inhibition elicits a prometastatic switch through the TGFbeta-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal 7:ra15
Underwood TJ, Hayden AL, Derouet M, Garcia E, Noble F, White MJ, Thirdborough S, Mead A, Clemons N, Mellone M, Uzoho C, Primrose JN, Blaydes JP, Thomas GJ (2015) Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J Pathol 235:466–477
Van Aarsen LA, Leone DR, Ho S, Dolinski BM, McCoon PE, LePage DJ, Kelly R, Heaney G, Rayhorn P, Reid C, Simon KJ, Horan GS, Tao N, Gardner HA, Skelly MM, Gown AM, Thomas GJ, Weinreb PH, Fawell SE, Violette SM (2008) Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res 68:561–570
Vinals F, Pouyssegur J (2001) Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol Cell Biol 21:7218–7230
Wakefield LM, Hill CS (2013) Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 13:328–341
Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MA, Franzen CA, Gupta GN, Osipo C, Zlobin A, Syn WK, Zhang J, Kuo PC, Mi Z (2015) Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 34:4821–4833
Weis SM, Cheresh DA (2011) AlphaV integrins in angiogenesis and cancer. Cold Spring Harbor Perspect Med 1:a006478
Wipff PJ, Hinz B (2008) Integrins and the activation of latent transforming growth factor beta1—an intimate relationship. Eur J Cell Biol 87:601–615
Wipff PJ, Rifkin DB, Meister JJ, Hinz B (2007) Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 179:1311–1323
Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, Cereser B, Crnogorac-Jurcevic T, McDonald S, Elia G, Hagemann T, Kocher HM, Hodivala-Dilke KM (2015) Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 27:123–137
Wu Q, Chen GL, Li YJ, Chen Y, Lin FZ (2015) Paeoniflorin inhibits macrophage-mediated lung cancer metastasis. Chin J Nat Med 13:925–932
Xu X, Chang W, Yuan J, Han X, Tan X, Ding Y, Luo Y, Cai H, Liu Y, Gao X, Liu Q, Yu Y, Du Y, Wang H, Ma L, Wang J, Chen K, Fu C, Cao G (2016) Periostin expression in intra-tumoral stromal cells is prognostic and predictive for colorectal carcinoma via creating a cancer-supportive niche. Oncotarget 7:798–813
Xu YY, Zhang YY, Lu WF, Mi YJ, Chen YQ (2015) Prognostic value of osteopontin expression in breast cancer: a meta-analysis. Mol Clin Oncol 3:357–362
Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, Miyashita T, Tajima H, Ninomiya I, Munesue S, Harashima A, Harada S, Yamamoto H, Ohta T (2015) Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer (in press)
Yanagisawa H, Schluterman MK, Brekken RA (2009) Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal 3:337–347
Yang L, Moses HL (2008) Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res 68:9107–9111
Yang M, Liu J, Piao C, Shao J, Du J (2015) ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis 6:e1780
Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS (2007) Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol 176:787–793
Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF (2002) Beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 129:2891–2903
Zhu J, Huang S, Wu G, Huang C, Li X, Chen Z, Zhao L, Zhao Y (2015a) Lysyl oxidase is predictive of unfavorable outcomes and essential for regulation of vascular endothelial growth factor in hepatocellular carcinoma. Dig Dis Sci 60:3019–3031
Zhu X, Zhong J, Zhao Z, Sheng J, Wang J, Liu J, Cui K, Chang J, Zhao H, Wong S (2015b) Epithelial derived CTGF promotes breast tumor progression via inducing EMT and collagen I fibers deposition. Oncotarget 6:25320–25338
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Khan, Z., Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res 365, 657–673 (2016). https://doi.org/10.1007/s00441-016-2474-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-016-2474-y