
Abstract
Anti-cytokine autoantibodies may cause immunodeficiency and have been recently recognized as ‘autoimmune phenocopies of primary immunodeficiencies’ and are found in particular, but not exclusively in adult patients. By blocking the cytokine’s biological function, patients with anti-cytokine autoantibodies may present with a similar clinical phenotype as the related inborn genetic disorders. So far, autoantibodies to interferon (IFN)-γ, GM-CSF, to a group of TH-17 cytokines and to IL-6 have been found to be causative or closely associated with susceptibility to infection. This review compares infectious diseases associated with anti-cytokine autoantibodies with primary immunodeficiencies affecting similar cytokines or related pathways.
Similar content being viewed by others
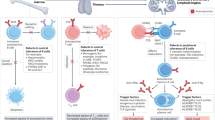


Introduction
Genetically defined, primary immunodeficiencies that impair specific cytokine pathways cause increased susceptibility for selective infectious diseases, and present mostly early in infancy or childhood. Mutations in the cytokine itself, its cognate receptor or downstream signaling molecules may interrupt their biological function, which leads to an impaired immune response. Anti-cytokine autoantibodies were recognized as phenocopies of primary immunodeficiencies (Tangye et al. 2020), and are found in particular, but not exclusively in adult patients. Autoantibodies, produced by auto-reactive B cells, may bind to cytokines. In sufficient concentration, anti-cytokine autoantibodies could block the signaling and neutralize the biological function of target cytokines, by preventing the direct binding to its receptor and (or) depleting the cytokine through forming a cytokine/autoantibodies complex (Piccoli et al. 2015). Autoantibodies against cytokines are, however, not necessarily associated with a respective neutralizing activity (Karner et al. 2016; von Stemann et al. 2017). By blocking the cytokine’s biological function, patients with anti-cytokine autoantibodies may present with a similar clinical phenotype as the related inborn genetic disorders. Although the exact mechanism is largely unknown, the production of autoantibodies may require external exposure to cross-reactive antigens and multiple steps to break tolerance in the adaptive immune response. This may explain why most (but not all) patients with anti-cytokine autoantibodies present later in life. So far, autoantibodies to interferon (IFN)-γ, GM-CSF, to a group of TH-17 cytokines comprising IL-17A, IL-17F, IL-22, IL-23, and to IL-6 have been found to be causative or closely associated with susceptibility to infection. In contrast, high levels of neutralizing autoantibodies may not cause any expected effects in vivo, as, e.g., shown by patients with autoantibodies to type I IFNs (IFNα and IFNω), which do not present with increased susceptibility to viral infections (Weiler et al. 2018). It has been suggested that this may be because of a large number of redundant type I IFN species, resulting in incomplete neutralization of the overall antiviral activity of IFNs by the autoantibodies (Puel et al. 2010).
Anti-interferon-γ autoantibodies as an etiology in mycobacterial infections in adults
Interferon-γ is a key cytokine produced by activated T cells, natural killer cells, and group I innate lymphoid cells. IFN-γ receptors are expressed widely on most cell types, but especially on myeloid cell (such as macrophages and dendritic cells). The identification of IFN-γ receptor deficiencies (IFNGR1 and IFNGR2) in patients with Mendelian susceptibility to mycobacterial disease (MSMD) demonstrated that IFN-γ plays a critical and non-redundant role in controlling mycobacterial infections (Rosain et al. 2019). Isolated and syndromic MSMD comprises now a group of genetic disorders caused by mutations in 16 published genes (see Table 1), all of them being involved in IFN-γ-mediated immunity, including impaired IFN-γ production (IL12B, IL12RB1, IL12RB2, IL23R, IRF8, TYK2, ISG15, IKBKG, RORC) or impaired cellular responses to IFN-γ (IFNGR1, IFNGR2, JAK1, STAT1, IRF8, CYBB, SPPL2A) (Bustamante 2020; Martinez-Barricarte et al. 2018; Rosain et al. 2019). More recently, inborn IFN-γ deficiency has been reported in two patients with mycobacterial infection (Kerner et al. 2020). Patients with complete loss of IFN-γ activity present with early onset, severe, disseminated infections caused by low-virulence mycobacteria, such as bacillus Calmette–Guérin (BCG) vaccines and nontuberculous environmental mycobacteria (NTM). Some patients with MSMD may also develop infections with Mycobacterium tuberculosis, non-typhoidal salmonella (NTS), candidiasis and symptoms of tuberculosis (see Table 1) (Bustamante et al. 2014).
High titers of highly neutralizing anti-IFN-γ autoantibodies (nAIGAs) were initially reported by several groups in sporadic patients or small case series with NTM infections (Doffinger et al. 2004; Hoflich et al. 2004; Kampmann et al. 2005; Patel et al. 2005). In recent years, however, larger cohorts of nAIGA patients were reported from Southeast Asia, with the majority from Thailand, Hong Kong, Taiwan and Japan (Aoki et al. 2018; Browne et al. 2012; Chi et al. 2013, 2016). Only few of the reported cases did not originate from this region (Hanitsch et al. 2015; Kampmann et al. 2005; O'Connell et al. 2014; van de Vosse et al. 2017). Around 500 patients with nAIGAs have been reported up to now in the literature but the exact prevalence rate of nAIGAs-related disease is unknown (Aoki et al. 2018; Barcenas-Morales et al. 2016, 2019; Browne 2014; Browne et al. 2012; Chi et al. 2013, 2016; Chruewkamlow et al. 2016; Doffinger et al. 2004; Hoflich et al. 2004; Jutivorakool et al. 2018; Kampmann et al. 2005; Patel et al. 2005; Wipasa et al. 2018; Wongkulab et al. 2013; Wu et al. 2018).
Similar to patients with MSMD, mycobacterial infections are the main clinical presentations for patients with nAIGAs, and a considerable proportion of these infections (95%) is severe and disseminated (Aoki et al. 2018; Browne et al. 2012; Chi et al. 2016). Both, slowly-growing and rapidly-growing NTMs, are isolated from patients with nAIGAs, but the distribution of species varies widely and depends on the geographical characteristics (Aoki et al. 2018; Browne et al. 2012; Chi et al. 2016; Wongkulab et al. 2013). Every organ system of the body can be infected by NTM species, but lymph nodes, bones/joints, and lungs are the leading organ systems affected (Aoki et al. 2018; Browne et al. 2012; Chi et al. 2016; Wongkulab et al. 2013). Almost all patients are adults in the range from 40 to 70 years with no sex predominance. However, more recently, two young patients presenting at 10 years and 16 years, respectively, have been reported (Liew et al. 2019). In addition to NTM infections, some nAIGAs patients also were developing Tuberculosis, which may precede, be concomitant with, or follow the diagnosis of NTM infections (Browne et al. 2012; Chi et al. 2016; Kampitak et al. 2011). However, the role of nAIGAs in the pathogenesis of tuberculosis remains to be elucidated.
Invasive NTS is another unique clinical picture among patients with nAIGAs. Among MSMD cases, salmonellosis is found in one-third of patients with IL-12/23-related defects (IL-12Rβ1 deficiency and IL-12p40 deficiency) (Bustamante et al. 2014). In the cases with nAIGAs, a history of salmonellosis is found in 29–40% of patients, which is very similar to the proportion of MSMD patients with IL-12/23 defects (Browne et al. 2012; Chi et al. 2016; Wongkulab et al. 2013). However, it should be noted that salmonellosis was found in < 10% of patients with IFN-γR1 and IFN-γR2 deficiency. It is worth to mention that IL-12-related inborn deficiencies show an increased susceptibility to mycobacterial infections; however, the penetrance is not complete. Anti-IL-12 autoantibodies have been reported in a few patients without mycobacterial infection (Sim et al. 2013), which is consistent with the observation of incomplete penetrance for MSMD in IL-12 pathway-related genetic defects (Fieschi and Casanova 2003). Bacteria other than NTS, such as Streptococcus spp., Burkholderia cepacia, B. pseudomallei, Staphylococcus aureus, Legionella pneumophila, and Enterobacteriaceae are occasionally isolated from the nAIGAs patients (Browne et al. 2012; Chi et al. 2016; Tang et al. 2010).
A variety of viral infections have been reported in patients with nAIGAs, but varicella-zoster virus (VZV) is the major causative agent and presents predominantly as shingles (herpes zoster) among this patient population. Most VZV infections are localized, but occasionally disseminated and severe (Browne et al. 2012; Chi et al. 2013, 2016; Jutivorakool et al. 2018). In contrast to patients with nAIGAs, VZV infections are only sporadically reported in patients with IFN-γR1 and IFN-γR2 deficiency. There is no clear explanation for these differences in the clinical presentations between patients with IFN-γR deficiency and patients with nAIGAs, but the age of disease onset might partially explain the divergent phenotypes between the two patient groups. Patients with complete IFN-γR1 and IFN-γR2 deficiency are children and may have no chance to expose to VZV during their lifetime; therefore, these patients are free from herpes zoster. A few cases with nAIGAs have comorbidities, such as diabetes mellitus and autoimmune diseases; nevertheless, the majority cases have no sign of autoimmune disease other than the nAIGAs (Chi et al. 2016; Doffinger et al. 2004; Hung et al. 2018; Jutivorakool et al. 2018).
No familial nAIGAs cases have been reported so far; however, almost all patients with nAIGAs originate from Southeast Asia and Japan, which suggested that genetic factor(s) might be involved. HLA class I and class II molecular typing showed a strong association of nAIGAs and certain HLA class II molecules in Taiwanese patients (DRB1*16:02 and DQB1*05:02) (Chi et al. 2013) and patients from other Southeast Asian countries, including Thailand, the Philippines, Vietnam, and Laos (DRB1*15:02 and DQB1*05:01) (Ku et al. 2016), with the DR/DQ haplotypes being in close vicinity and, therefore, a strong linkage disequilibrium in both cases. DRB1*16:02 and DRB1*15:02 are commonly found in Southeast Asians, Amerindian and Pacific Islanders, but rarely found in Caucasians and Africans (Gonzalez-Galarza et al. 2011; Middleton et al. 2003). The detailed mechanism of these risk alleles in autoantibodies production is still unknown. HLA II class molecules could present the antigens to CD4 T which guide the B-cell activation and maturation. The associations of DRB1*16:02 and DRB1*15:02 are extremely strong. Therefore, these risk alleles might be directly involved in the pathogenic autoantibodies production. However, as only a very small portion of these risk alleles carriers have developed nAIGAs, other factors, including genetic or environmental factors, are likely to be involved. Their identification will be crucial for our understanding of this disease.
Antibodies to GM-CSF are associated not only with pulmonary alveolar proteinosis (PAP) but also with extrapulmonary infections and cerebral Cryptococcosis in the absence of PAP
GM-CSF is a haematopoietic growth factor which in particular promotes the development of macrophages, dendritic cells and neutrophils. In the lung, it is important for differentiation and function of alveolar macrophages. High titer, neutralizing autoantibodies against GM-CSF are the autoimmune correlate of the much rarer primary GM-CSF-receptor deficiency causing PAP by impairing the alveolar macrophage-mediated surfactant lipid and protein metabolism leading to accumulation and respiratory insufficiency. In contrast to anti-IFN-γ autoantibodies, there is no association with specific HLA alleles (Anderson et al. 2019).
Patients with autoimmune PAP may present not only with opportunistic infection, e.g., by intracellular pathogens such as Nocardia and Histoplasma which may be secondary to the underlying lung dysfunction but also with extrapulmonary diseases caused by Cryptococcus, Nocardia and Aspergillus (Punatar et al. 2012; Trapnell et al. 2019). However, it is worth to note that these infections might be secondary to impaired lung function by PAP and/or to therapeutic immuno-suppression.
Recently, anti-GM-CSF autoantibodies have been found in patients with Nocardia infection or cerebral Cryptococcus gattii infection (Kuo et al. 2017; Rosen et al. 2013, 2015). Cryptococcus neoformans is an environmental opportunistic species which causes disease in particular in patients infected by human immunodeficiency virus (HIV). However interestingly, anti-GM-CSF autoantibodies patients mostly suffered from C. gattii, but not from C. neoformans (Kuo et al. 2017; Rosen et al. 2013; Saijo et al. 2014b). Despite carrying neutralizing autoantibodies to GM-CSF, these patients did not manifest PAP at the time of diagnosis, but PAP developed in only few cases at a later time point (Demir et al. 2018; Punatar et al. 2012; Quah et al. 2018; Rosen et al. 2013). It is possible that in those patients, the autoantibodies may have only dampened the function of GM-CSF to some extent, while still allowing sufficient activity of the alveolar macrophages. Piccoli et al. showed that full neutralization needed the synergistic recognition of non-crossreactive epitopes by multiple anti-GM-CSF clones. This could explain the difference between anti-GM-CSF-positive individuals with and without PAP (Piccoli et al. 2015).
Autoantibodies against IL-6 predispose to pyogenic infections
Four patients with autoantibodies against IL-6 who developed severe bacterial infections have been published to date (Bloomfield et al. 2019; Nanki et al. 2013; Puel et al. 2008). Further, patients with autoantibodies against IL-6 and severe bacterial infections have been identified (Doffinger and von Bernuth unpublished data). All patients presented with low C-reactive protein (CrP) despite severe pyogenic infections. In these patients anti-IL-6-auto-antibodies were of high titer and neutralized IL-6 (phosphorylation of STAT3 and / or production of CrP). A similar susceptibility for pyogenic infections has been described in patients with impaired production of IL-6 due to defects in MyD88/IRAK/NEMO/IκBα-dependent signaling or with impaired IL-6-signaling due to defects in the IL-6-receptor/gp130/ZNF341/STAT3-dependent pathway (Beziat et al. 2018, 2020; Courtois et al. 2003; Doffinger et al. 2001; Frey-Jakobs et al. 2018; Minegishi et al. 2007; Nahum et al. 2019; Picard et al. 2003, 2010; Schwerd et al. 2017; Spencer et al. 2019; von Bernuth et al. 2008). The thorough comparison of the infectious phenotype in patients with autoantibodies against IL-6, with impaired IL-6 production or with impaired IL-6 signaling reveals specific overlaps: Patients with autoantibodies against IL-6, with impaired IL-6 signaling and with impaired IL-6 production show increased susceptibility for severe pyogenic infections, in particular but not exclusively by staphylococci and pneumococci (Nanki et al. 2013; Picard et al. 2010; Puel et al. 2008); whereas, increased susceptibility to staphylococcal skin infections seems in particular common in patients with autoantibodies against IL-6 and with selectively impaired IL-6 signaling (Nahum et al. 2019; Puel et al. 2008; Spencer et al. 2019).
Autoantibodies to TH17 cytokines (IL-17A/F, IL-22, IL-23) are associated with chronic mucocutaneous candidiasis
Autoantibodies to Th17 cytokines can be found in patients suffering from type I Autoimmune polyglandular syndrome (APS1) (Bruserud et al. 2016) and thymoma (Wolff et al. 2014). A common denominator between those conditions may be impaired tolerance induction caused by primary (in APS1) or secondary structural (in thymoma) disruption of thymus function (Barcenas-Morales et al. 2016; Cheng and Anderson 2018). In particular, APS1 patients and to a lesser degree patients with thymoma may present with neutralizing autoantibodies to TH17 cytokines including IL-17A/F, IL-22 and IL-12/IL-23, which are associated with CMC (Kisand et al. 2010; Puel et al. 2010). APS1 is a complex auto-immune syndrome with CMC as its only infectious manifestation (Li et al. 2017, 2018). A subset of patients with thymoma, a thymic epithelial cancer, may as well present with CMC which may be in the context of wider infectious complications (Burbelo et al. 2010; Kisand et al. 2010; Rosenberg et al. 2016). The cytokines IL-17A, IL-17F, and IL-22 are mainly produced by Th17 cells and play an important role in the mucosal defense against Candida (Okada et al. 2016). IL-23 is mainly produced by dendritic cells and macrophages and is required for the development of TH17 cells (Gaffen et al. 2014; Langrish et al. 2004). Primary deficiencies in the IL17 axis including IL-17F, IL-17RA, IL17RF and the intracellular adaptor ACT1 have been found to predispose to CMC (Puel 2020). Patients with impaired IL-12/IL-23 signaling show as well an increased incidence of CMC (Okada et al. 2016). Surprisingly, however, patients with IL-23R deficiency were not reported with CMC (Martinez-Barricarte et al. 2018). Furthermore, CMC is part of the infectious spectrum found in syndromic primary deficiencies of CARD9, STAT3, STAT1, RORT, ZNF341, and JNK1 which all show diminished Th17 immunity (Beziat et al. 2018; Frey-Jakobs et al. 2018; Li et al. 2019; Puel 2020).
Concluding remarks
Up to now autoantibodies to four major groups of cytokines—IFN-γ, GM-CSF, to a group of TH-17 cytokines comprising IL-17A, IL-17F, IL-22, IL-23, and to IL-6—have been found to be causative or closely associated with increased susceptibility to selective infection. The largest body of evidence exists for patients with antibodies against anti-IFN-γ, as more than 500 patients with predominant susceptibility to NTM infections and non-typhoid salmonellosis are described. This clinical phenotype strongly resembles the one of patients with inborn deficiencies of IFN-γ-production or IFN-γ-response. Similarly, multiple patients with autoantibodies against TH-17 cytokines (comprising IL-17A, IL-17F, IL-22, IL-23) whose selective susceptibility to candidiasis strongly resembles inborn deficiencies with impaired production or response to IL-17 were described. Autoantibodies against GM-CSF seem to impair the defense against a rather broad group of pathogens whose common denominator is being controlled by macrophages: Nocardia, Histoplasma, Cryptococcus gattii, Aspergillus and maybe NTM. Autoantibodies to GM-CSF are also the major cause for PAP. Only in this aspect, anti-GM-CSF autoantibodies cause the same disease as inborn defects of the GM-CSF receptor. It is unknown why inborn defects of the GM-CSF receptor do not predispose to infections as do the respective autoantibodies. Autoantibodies against IL-6 have yet been described in only few patients, but seem to predispose to pneumococcal and staphylococcal infections. This selective susceptibility strikingly resembles inborn defects with impaired production of IL-6 or impaired response to IL-6.
References
Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL (1998a) Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280:1432–1435
Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, Wadhwa M, Dockrell H, Salmon M, Fischer A, Durandy A, Casanova JL, Kumararatne DS (1998b) Inherited interleukin 12 deficiency in a child with bacille Calmette–Guerin and Salmonella enteritidis disseminated infection. J Clin Invest 102:2035–2040. https://doi.org/10.1172/JCI4950
Anderson K, Carey B, Martin A, Roark C, Chalk C, Nowell-Bostic M, Freed B, Aubrey M, Trapnell B, Fontenot A (2019) Pulmonary alveolar proteinosis: an autoimmune disease lacking an HLA association. PLoS ONE 14:e0213179. https://doi.org/10.1371/journal.pone.0213179
Aoki A, Sakagami T, Yoshizawa K, Shima K, Toyama M, Tanabe Y, Moro H, Aoki N, Watanabe S, Koya T, Hasegawa T, Morimoto K, Kurashima A, Hoshino Y, Trapnell BC, Kikuchi T (2018) Clinical significance of interferon-gamma neutralizing autoantibodies against disseminated nontuberculous mycobacterial disease. Clin Infect Dis 66:1239–1245. https://doi.org/10.1093/cid/cix996
Applen Clancey S, Ciccone EJ, Coelho MA, Davis J, Ding L, Betancourt R, Glaubiger S, Lee Y, Holland SM, Gilligan P, Sung J, Heitman J (2019) Cryptococcus deuterogattii VGIIa infection associated with travel to the Pacific Northwest outbreak region in an anti-granulocyte-macrophage colony-stimulating factor autoantibody-positive patient in the United States. MBio. https://doi.org/10.1128/mBio.02733-18
Barcenas-Morales G, Jandus P, Doffinger R (2016) Anticytokine autoantibodies in infection and inflammation: an update. Curr Opin Allergy Clin Immunol 16:523–529. https://doi.org/10.1097/ACI.0000000000000316
Barcenas-Morales G, Cortes-Acevedo P, Doffinger R (2019) Anticytokine autoantibodies leading to infection: early recognition, diagnosis and treatment options. Curr Opin Infect Dis 32:330–336. https://doi.org/10.1097/QCO.0000000000000561
Beziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, Pellier I, Zoghi S, Baris S, Keles S, Gray P, Du N, Wang Y, Zerbib Y, Levy R, Leclercq T, About F, Lim AI, Rao G, Payne K, Pelham SJ, Avery DT, Deenick EK, Pillay B, Chou J, Guery R, Belkadi A, Guerin A, Migaud M, Rattina V, Ailal F, Benhsaien I, Bouaziz M, Habib T, Chaussabel D, Marr N, El-Benna J, Grimbacher B, Wargon O, Bustamante J, Boisson B, Muller-Fleckenstein I, Fleckenstein B, Chandesris MO, Titeux M, Fraitag S, Alyanakian MA, Leruez-Ville M, Picard C, Meyts I, Di Santo JP, Hovnanian A, Somer A, Ozen A, Rezaei N, Chatila TA, Abel L, Leonard WJ, Tangye SG, Puel A, Casanova JL (2018) A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. https://doi.org/10.1126/sciimmunol.aat4956
Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, Staal J, Aschenbrenner D, Roels L, Worley L, Claes K, Gartner L, Kohn LA, De Bruyne M, Schmitz-Abe K, Charbonnier LM, Keles S, Nammour J, Vladikine N, Maglorius Renkilaraj MRL, Seeleuthner Y, Migaud M, Rosain J, Jeljeli M, Boisson B, Van Braeckel E, Rosenfeld JA, Dai H, Burrage LC, Murdock DR, Lambrecht BN, Avettand-Fenoel V, Vogel TP, Undiagnosed Diseases N, Esther CR, Haskologlu S, Dogu F, Ciznar P, Boutboul D, Ouachee-Chardin M, Amourette J, Lebras MN, Gauvain C, Tcherakian C, Ikinciogullari A, Beyaert R, Abel L, Milner JD, Grimbacher B, Couderc LJ, Butte MJ, Freeman AF, Catherinot E, Fieschi C, Chatila TA, Tangye SG, Uhlig HH, Haerynck F, Casanova JL, Puel A (2020) Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. https://doi.org/10.1084/jem.20191804
Bloomfield M, Parackova Z, Cabelova T, Pospisilova I, Kabicek P, Houstkova H, Sediva A (2019) Anti-IL6 autoantibodies in an infant with CRP-less septic shock. Front Immunol 10:2629. https://doi.org/10.3389/fimmu.2019.02629
Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL (2012) Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 337:1684–1688. https://doi.org/10.1126/science.1224026
Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, Puel A, Li X, Casanova JL (2013) An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39:676–686. https://doi.org/10.1016/j.immuni.2013.09.002
Browne SK (2014) Anticytokine autoantibody-associated immunodeficiency. Annu Rev Immunol 32:635–657. https://doi.org/10.1146/annurev-immunol-032713-120222
Browne SK, Burbelo PD, Chetchotisakd P, Suputtamongkol Y, Kiertiburanakul S, Shaw PA, Kirk JL, Jutivorakool K, Zaman R, Ding L, Hsu AP, Patel SY, Olivier KN, Lulitanond V, Mootsikapun P, Anunnatsiri S, Angkasekwinai N, Sathapatayavongs B, Hsueh PR, Shieh CC, Brown MR, Thongnoppakhun W, Claypool R, Sampaio EP, Thepthai C, Waywa D, Dacombe C, Reizes Y, Zelazny AM, Saleeb P, Rosen LB, Mo A, Iadarola M, Holland SM (2012) Adult-onset immunodeficiency in Thailand and Taiwan. N Engl J Med 367:725–734. https://doi.org/10.1056/NEJMoa1111160
Bruserud O, Oftedal BE, Wolff AB, Husebye ES (2016) AIRE-mutations and autoimmune disease. Curr Opin Immunol 43:8–15. https://doi.org/10.1016/j.coi.2016.07.003
Burbelo PD, Browne SK, Sampaio EP, Giaccone G, Zaman R, Kristosturyan E, Rajan A, Ding L, Ching KH, Berman A, Oliveira JB, Hsu AP, Klimavicz CM, Iadarola MJ, Holland SM (2010) Anti-cytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood 116:4848–4858. https://doi.org/10.1182/blood-2010-05-286161
Bustamante J (2020) Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. https://doi.org/10.1007/s00439-020-02120-y
Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, Casanova JL (2011) Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 12:213–221. https://doi.org/10.1038/ni.1992
Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL (2014) Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol 26:454–470. https://doi.org/10.1016/j.smim.2014.09.008
Cheng M, Anderson MS (2018) Thymic tolerance as a key brake on autoimmunity. Nat Immunol 19:659–664. https://doi.org/10.1038/s41590-018-0128-9
Chi CY, Chu CC, Liu JP, Lin CH, Ho MW, Lo WJ, Lin PC, Chen HJ, Chou CH, Feng JY, Fung CP, Sher YP, Li CY, Wang JH, Ku CL (2013) Anti-IFN-gamma autoantibodies in adults with disseminated nontuberculous mycobacterial infections are associated with HLA-DRB1*16:02 and HLA-DQB1*05:02 and the reactivation of latent varicella-zoster virus infection. Blood 121:1357–1366. https://doi.org/10.1182/blood-2012-08-452482
Chi CY, Lin CH, Ho MW, Ding JY, Huang WC, Shih HP, Yeh CF, Fung CP, Sun HY, Huang CT, Wu TS, Chang CY, Liu YM, Feng JY, Wu WK, Wang LS, Tsai CH, Ho CM, Lin HS, Chen HJ, Lin PC, Liao WC, Chen WT, Lo CC, Wang SY, Kuo CY, Lee CH, Ku CL (2016) Clinical manifestations, course, and outcome of patients with neutralizing anti-interferon-gamma autoantibodies and disseminated nontuberculous mycobacterial infections. Medicine (Baltimore) 95:e3927. https://doi.org/10.1097/MD.0000000000003927
Chruewkamlow N, Mahasongkram K, Pata S, Chaiwarith R, Salee P, Supparatpinyo K, Kasinrerk W (2016) Immune alterations in patients with anti-interferon-gamma autoantibodies. PLoS ONE 11:e0145983. https://doi.org/10.1371/journal.pone.0145983
Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, Dupuis-Girod S, Bodemer C, Livadiotti S, Novelli F, Rossi P, Fischer A, Israel A, Munnich A, Le Deist F, Casanova JL (2003) A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest 112:1108–1115. https://doi.org/10.1172/JCI18714
Crum-Cianflone NF, Lam PV, Ross-Walker S, Rosen LB, Holland SM (2017) Autoantibodies to granulocyte-macrophage colony-stimulating factor associated with severe and unusual manifestations of Cryptococcus gattii infections. Open Forum Infect Dis 4:211. https://doi.org/10.1093/ofid/ofx211
de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, Kabel PJ, Draaisma JM, van Dissel JT, Kroon FP, Casanova JL, Ottenhoff TH (1998) Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 280:1435–1438. https://doi.org/10.1126/science.280.5368.1435
Demir S, Chebib N, Thivolet-Bejui F, Cottin V (2018) Pulmonary alveolar proteinosis following cryptococcal meningitis: a possible cause? BMJ Case Rep. https://doi.org/10.1136/bcr-2017-222940
Doffinger R, von Bernuth H (unpublished data).
Doffinger R, Jouanguy E, Dupuis S, Fondaneche MC, Stephan JL, Emile JF, Lamhamedi-Cherradi S, Altare F, Pallier A, Barcenas-Morales G, Meinl E, Krause C, Pestka S, Schreiber RD, Novelli F, Casanova JL (2000) Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette–Guerin and mycobacterium abscessus infection. J Infect Dis 181:379–384. https://doi.org/10.1086/315197
Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL (2001) X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 27:277–285. https://doi.org/10.1038/85837
Doffinger R, Helbert MR, Barcenas-Morales G, Yang K, Dupuis S, Ceron-Gutierrez L, Espitia-Pinzon C, Barnes N, Bothamley G, Casanova JL, Longhurst HJ, Kumararatne DS (2004) Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis 38:e10–e14. https://doi.org/10.1086/380453
Dorman SE, Holland SM (1998) Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest 101:2364–2369. https://doi.org/10.1172/JCI2901
Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, Holland SM, Schreiber RD, Casanova JL (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293:300–303. https://doi.org/10.1126/science.1061154
Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL (2003) Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 33:388–391. https://doi.org/10.1038/ng1097
Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, Curtis J, Gaspar M, Nowak K, Daza-Cajigal V, Kumararatne D, Doffinger R, Thrasher AJ, Nejentsev S (2016) Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun 7:13992. https://doi.org/10.1038/ncomms13992
Fieschi C, Casanova JL (2003) The role of interleukin-12 in human infectious diseases: only a faint signature. Eur J Immunol 33:1461–1464. https://doi.org/10.1002/eji.200324038
Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, Frucht DM, Christel K, von Bernuth H, Jouanguy E, Feinberg J, Durandy A, Senechal B, Chapgier A, Vogt G, de Beaucoudrey L, Fieschi C, Picard C, Garfa M, Chemli J, Bejaoui M, Tsolia MN, Kutukculer N, Plebani A, Notarangelo L, Bodemer C, Geissmann F, Israel A, Veron M, Knackstedt M, Barbouche R, Abel L, Magdorf K, Gendrel D, Agou F, Holland SM, Casanova JL (2006) X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med 203:1745–1759. https://doi.org/10.1084/jem.20060085
Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, Bulashevska A, Proietti M, Frobel P, Noltner C, Yang L, Rojas-Restrepo J, Langer N, Winzer S, Engelhardt KR, Glocker C, Pfeifer D, Klein A, Schaffer AA, Lagovsky I, Lachover-Roth I, Beziat V, Puel A, Casanova JL, Fleckenstein B, Weidinger S, Kilic SS, Garty BZ, Etzioni A, Grimbacher B (2018) ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. https://doi.org/10.1126/sciimmunol.aat4941
Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14:585–600. https://doi.org/10.1038/nri3707
Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR (2011) Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res 39:D913–D919. https://doi.org/10.1093/nar/gkq1128
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, Menon G, Trouillet C, McDonald D, Carey P, Ginhoux F, Alsina L, Zumwalt TJ, Kong XF, Kumararatne D, Butler K, Hubeau M, Feinberg J, Al-Muhsen S, Cant A, Abel L, Chaussabel D, Doffinger R, Talesnik E, Grumach A, Duarte A, Abarca K, Moraes-Vasconcelos D, Burk D, Berghuis A, Geissmann F, Collin M, Casanova JL, Gros P (2011) IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 365:127–138. https://doi.org/10.1056/NEJMoa1100066
Hanitsch LG, Lobel M, Muller-Redetzky H, Schurmann M, Suttorp N, Unterwalder N, Monnich U, Meisel C, Wittke K, Volk HD, Scheibenbogen C, Kolsch U (2015) Late-onset disseminated Mycobacterium avium intracellulare Complex Infection (MAC), cerebral toxoplasmosis and salmonella sepsis in a german caucasian patient with unusual anti-interferon-gamma IgG1 autoantibodies. J Clin Immunol 35:361–365. https://doi.org/10.1007/s10875-015-0161-5
Hoflich C, Sabat R, Rosseau S, Temmesfeld B, Slevogt H, Docke WD, Grutz G, Meisel C, Halle E, Gobel UB, Volk HD, Suttorp N (2004) Naturally occurring anti-IFN-gamma autoantibody and severe infections with Mycobacterium cheloneae and Burkholderia cocovenenans. Blood 103:673–675. https://doi.org/10.1182/blood-2003-04-1065
Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619. https://doi.org/10.1056/NEJMoa073687
Hong GH, Ortega-Villa AM, Hunsberger S, Chetchotisakd P, Anunnatsiri S, Mootsikapun P, Rosen LB, Zerbe CS, Holland SM (2019) Natural history and evolution of anti-interferon-gamma autoantibody-associated immunodeficiency syndrome in Thailand and the US. Clin Infect Dis. https://doi.org/10.1093/cid/ciz786
Hung TC, Chen SC, Liao KS, Cheng SH, Chang SL (2018) Anti-interferon-gamma auto-anti-body and disseminated non-tuberculous mycobacteria infections in thyroid cancer: a case report. QJM 111:647–648. https://doi.org/10.1093/qjmed/hcy094
Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, Levin M, Blanche S, Seboun E, Fischer A, Casanova JL (1996) Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette–Guerin infection. N Engl J Med 335:1956–1961. https://doi.org/10.1056/NEJM199612263352604
Jouanguy E, Lamhamedi-Cherradi S, Altare F, Fondaneche MC, Tuerlinckx D, Blanche S, Emile JF, Gaillard JL, Schreiber R, Levin M, Fischer A, Hivroz C, Casanova JL (1997) Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette–Guerin infection and a sibling with clinical tuberculosis. J Clin Invest 100:2658–2664. https://doi.org/10.1172/JCI119810
Jouanguy E, Lamhamedi-Cherradi S, Lammas D, Dorman SE, Fondaneche MC, Dupuis S, Doffinger R, Altare F, Girdlestone J, Emile JF, Ducoulombier H, Edgar D, Clarke J, Oxelius VA, Brai M, Novelli V, Heyne K, Fischer A, Holland SM, Kumararatne DS, Schreiber RD, Casanova JL (1999) A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet 21:370–378. https://doi.org/10.1038/7701
Jutivorakool K, Sittiwattanawong P, Kantikosum K, Hurst CP, Kumtornrut C, Asawanonda P, Klaewsongkram J, Rerknimitr P (2018) Skin manifestations in patients with adult-onset immunodeficiency due to anti-interferon-gamma autoantibody: a relationship with systemic infections. Acta Derm Venereol 98:742–747. https://doi.org/10.2340/00015555-2959
Kampitak T, Suwanpimolkul G, Browne S, Suankratay C (2011) Anti-interferon-gamma autoantibody and opportunistic infections: case series and review of the literature. Infection 39:65–71. https://doi.org/10.1007/s15010-010-0067-3
Kampmann B, Hemingway C, Stephens A, Davidson R, Goodsall A, Anderson S, Nicol M, Scholvinck E, Relman D, Waddell S, Langford P, Sheehan B, Semple L, Wilkinson KA, Wilkinson RJ, Ress S, Hibberd M, Levin M (2005) Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-gamma. J Clin Invest 115:2480–2488. https://doi.org/10.1172/JCI19316
Karner J, Pihlap M, Ranki A, Krohn K, Trebusak Podkrajsek K, Bratanic N, Battelino T, Willcox N, Peterson P, Kisand K (2016) IL-6-specific autoantibodies among APECED and thymoma patients. Immun Inflamm Dis 4:235–243. https://doi.org/10.1002/iid3.109
Kerner G, Rosain J, Guerin A, AlKhabaz A, Oleaga-Quintas C, Rapaport F, Massaad MJ, Ding JY, Khan T, Al Ali F, Rahman M, Deswarte C, Martinez-Barricarte R, Geha RS, Jeanne-Julien V, Garcia DST, Chi CY, Yang R, Roynard M, Fleckenstein B, Rozenberg F, Boisson-Dupuis S, Ku CL, Seeleuthner Y, Beziat V, Marr N, Abel L, Al-Herz W, Casanova JL, Bustamante J (2020) Inherited human IFNgamma deficiency underlies mycobacterial disease. J Clin Invest. https://doi.org/10.1172/JCI135460
Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A (2010) Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 207:299–308. https://doi.org/10.1084/jem.20091669
Kong XF, Martinez-Barricarte R, Kennedy J, Mele F, Lazarov T, Deenick EK, Ma CS, Breton G, Lucero KB, Langlais D, Bousfiha A, Aytekin C, Markle J, Trouillet C, Jabot-Hanin F, Arlehamn CSL, Rao G, Picard C, Lasseau T, Latorre D, Hambleton S, Deswarte C, Itan Y, Abarca K, Moraes-Vasconcelos D, Ailal F, Ikinciogullari A, Dogu F, Benhsaien I, Sette A, Abel L, Boisson-Dupuis S, Schroder B, Nussenzweig MC, Liu K, Geissmann F, Tangye SG, Gros P, Sallusto F, Bustamante J, Casanova JL (2018) Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol 19:973–985. https://doi.org/10.1038/s41590-018-0178-z
Kreins AY, Ciancanelli MJ, Okada S, Kong XF, Ramirez-Alejo N, Kilic SS, El Baghdadi J, Nonoyama S, Mahdaviani SA, Ailal F, Bousfiha A, Mansouri D, Nievas E, Ma CS, Rao G, Bernasconi A, Sun Kuehn H, Niemela J, Stoddard J, Deveau P, Cobat A, El Azbaoui S, Sabri A, Lim CK, Sundin M, Avery DT, Halwani R, Grant AV, Boisson B, Bogunovic D, Itan Y, Moncada-Velez M, Martinez-Barricarte R, Migaud M, Deswarte C, Alsina L, Kotlarz D, Klein C, Muller-Fleckenstein I, Fleckenstein B, Cormier-Daire V, Rose-John S, Picard C, Hammarstrom L, Puel A, Al-Muhsen S, Abel L, Chaussabel D, Rosenzweig SD, Minegishi Y, Tangye SG, Bustamante J, Casanova JL, Boisson-Dupuis S (2015) Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med 212:1641–1662. https://doi.org/10.1084/jem.20140280
Ku CL, Lin CH, Chang SW, Chu CC, Chan JF, Kong XF, Lee CH, Rosen EA, Ding JY, Lee WI, Bustamante J, Witte T, Shih HP, Kuo CY, Chetchotisakd P, Kiertiburanakul S, Suputtamongkol Y, Yuen KY, Casanova JL, Holland SM, Doffinger R, Browne SK, Chi CY (2016) Anti-IFN-gamma autoantibodies are strongly associated with HLA-DR*15:02/16:02 and HLA-DQ*05:01/05:02 across Southeast Asia. J Allergy Clin Immunol 137(945–8):e8. https://doi.org/10.1016/j.jaci.2015.09.018
Kuo CY, Wang SY, Shih HP, Tu KH, Huang WC, Ding JY, Lin CH, Yeh CF, Ho MW, Chang SC, He CY, Chen HK, Ho CH, Lee CH, Chi CY, Ku CL (2017) Disseminated cryptococcosis due to anti-granulocyte-macrophage colony-stimulating factor autoantibodies in the absence of pulmonary alveolar proteinosis. J Clin Immunol 37:143–152. https://doi.org/10.1007/s10875-016-0364-4
Langrish CL, McKenzie BS, Wilson NJ, de Waal MR, Kastelein RA, Cua DJ (2004) IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 202:96–105. https://doi.org/10.1111/j.0105-2896.2004.00214.x
Li J, Vinh DC, Casanova JL, Puel A (2017) Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr Opin Microbiol 40:46–57. https://doi.org/10.1016/j.mib.2017.10.016
Li J, Casanova JL, Puel A (2018) Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol 11:581–589. https://doi.org/10.1038/mi.2017.97
Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, Bougarn S, Cypowyj S, Grodecka L, Levy R, Beziat V, Shang L, Payne K, Avery DT, Migaud M, Boucherit S, Boughorbel S, Guennoun A, Chrabieh M, Rapaport F, Bigio B, Itan Y, Boisson B, Cormier-Daire V, Syx D, Malfait F, Zoppi N, Abel L, Freiberger T, Dietz HC, Marr N, Tangye SG, Colombi M, Casanova JL, Puel A (2019) Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol. https://doi.org/10.1126/sciimmunol.aax7965
Liew WK, Thoon KC, Chong CY, Tan NWH, Cheng DT, Chan BSW, Ng MSY, Das L, Arkachaisri T, Huang CH, Kuan JL, Chai LYA, Koh MJA (2019) Juvenile-onset immunodeficiency secondary to anti-interferon-gamma autoantibodies. J Clin Immunol 39:512–518. https://doi.org/10.1007/s10875-019-00652-1
Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova JL, Puel A (2015) Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med 212:619–631. https://doi.org/10.1084/jem.20141065
Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208:1635–1648. https://doi.org/10.1084/jem.20110958jem.20110958
Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F, Latorre D, Mahdaviani SA, Aytekin C, Mansouri D, Bryant VL, Jabot-Hanin F, Deswarte C, Nieto-Patlan A, Surace L, Kerner G, Itan Y, Jovic S, Avery DT, Wong N, Rao G, Patin E, Okada S, Bigio B, Boisson B, Rapaport F, Seeleuthner Y, Schmidt M, Ikinciogullari A, Dogu F, Tanir G, Tabarsi P, Bloursaz MR, Joseph JK, Heer A, Kong XF, Migaud M, Lazarov T, Geissmann F, Fleckenstein B, Arlehamn CL, Sette A, Puel A, Emile JF, van de Vosse E, Quintana-Murci L, Di Santo JP, Abel L, Boisson-Dupuis S, Bustamante J, Tangye SG, Sallusto F, Casanova JL (2018) Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. https://doi.org/10.1126/sciimmunol.aau6759
Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, Moore JP, Tavana G, Lewis LR, Zhu Y, Muzny DM, Gibbs RA, Huston DP (2008) Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med 205:2711–2716. https://doi.org/10.1084/jem.20080759
Middleton AM, Keig P, Wilson R (2003) In vitro models of infection I–human respiratory tissue organ culture. Methods Mol Med 71:277–295. https://doi.org/10.1385/1-59259-321-6:277
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448:1058–1062. https://doi.org/10.1038/nature06096
Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N (1997) Positional cloning of the APECED gene. Nat Genet 17:393–398. https://doi.org/10.1038/ng1297-393
Nahum A, Sharfe N, Broides A, Dadi H, Naghdi Z, Mandola AB, Vong L, Arbiv A, Dalal I, Brami I, Wormser O, Levy J, Roifman CM (2019) Defining the biological responses of IL-6 by the study of a novel IL-6 receptor chain immunodeficiency. J Allergy Clin Immunol. https://doi.org/10.1016/j.jaci.2019.11.015
Nanki T, Onoue I, Nagasaka K, Takayasu A, Ebisawa M, Hosoya T, Shirai T, Sugihara T, Hirata S, Kubota T, Harigai M, Miyasaka N (2013) Suppression of elevations in serum C reactive protein levels by anti-IL-6 autoantibodies in two patients with severe bacterial infections. Ann Rheum Dis 72:1100–1102. https://doi.org/10.1136/annrheumdis-2012-202768
Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M (1996) A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 335:1941–1949. https://doi.org/10.1056/NEJM199612263352602
O'Connell E, Rosen LB, LaRue RW, Fabre V, Melia MT, Auwaerter PG, Holland SM, Browne SK (2014) The first US domestic report of disseminated Mycobacterium avium complex and anti-interferon-gamma autoantibodies. J Clin Immunol 34:928–932. https://doi.org/10.1007/s10875-014-0073-9
Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CS, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay JP, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, Casanova JL (2015) Immunodeficiencies. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349:606–613. https://doi.org/10.1126/science.aaa4282
Okada S, Puel A, Casanova JL, Kobayashi M (2016) Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology 5:e114. https://doi.org/10.1038/cti.2016.71
Patel SY, Ding L, Brown MR, Lantz L, Gay T, Cohen S, Martyak LA, Kubak B, Holland SM (2005) Anti-IFN-gamma autoantibodies in disseminated nontuberculous mycobacterial infections. J Immunol 175:4769–477610.4049/jimmunol.175.7.4769
Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL (2003) Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299:2076–2079. https://doi.org/10.1126/science.1081902
Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, Creech CB, Ku CL, Ehl S, Marodi L, Al-Muhsen S, Al-Hajjar S, Al-Ghonaium A, Day-Good NK, Holland SM, Gallin JI, Chapel H, Speert DP, Rodriguez-Gallego C, Colino E, Garty BZ, Roifman C, Hara T, Yoshikawa H, Nonoyama S, Domachowske J, Issekutz AC, Tang M, Smart J, Zitnik SE, Hoarau C, Kumararatne DS, Thrasher AJ, Davies EG, Bethune C, Sirvent N, de Ricaud D, Camcioglu Y, Vasconcelos J, Guedes M, Vitor AB, Rodrigo C, Almazan F, Mendez M, Arostegui JI, Alsina L, Fortuny C, Reichenbach J, Verbsky JW, Bossuyt X, Doffinger R, Abel L, Puel A, Casanova JL (2010) Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89:403–425. https://doi.org/10.1097/MD.0b013e3181fd8ec3
Piccoli L, Campo I, Fregni CS, Rodriguez BM, Minola A, Sallusto F, Luisetti M, Corti D, Lanzavecchia A (2015) Neutralization and clearance of GM-CSF by autoantibodies in pulmonary alveolar proteinosis. Nat Commun 6:7375. https://doi.org/10.1038/ncomms8375
Puel A (2020) Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum Genet. https://doi.org/10.1007/s00439-020-02141-7
Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, Mamani-Matsuda M, Jouanguy E, Gendrel D, Casanova JL (2008) Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J Immunol 180:647–654
Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL (2010) Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207:291–297. https://doi.org/10.1084/jem.20091983
Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68. https://doi.org/10.1126/science.1200439
Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR (2012) Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect 65:173–179. https://doi.org/10.1016/j.jinf.2012.03.020
Quah J, Low TB, Fong R (2018) Disseminated Cryptococcus gattii infection preceding onset of pulmonary alveolar proteinosis. Respirol Case Rep 6:e00357. https://doi.org/10.1002/rcr2.357
Rosain J, Kong XF, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, Markle J, Okada S, Boisson-Dupuis S, Casanova JL, Bustamante J (2019) Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol Cell Biol 97:360–367. https://doi.org/10.1111/imcb.12210
Rosen LB, Freeman AF, Yang LM, Jutivorakool K, Olivier KN, Angkasekwinai N, Suputtamongkol Y, Bennett JE, Pyrgos V, Williamson PR, Ding L, Holland SM, Browne SK (2013) Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J Immunol 190:3959–3966. https://doi.org/10.4049/jimmunol.1202526
Rosen LB, Rocha Pereira N, Figueiredo C, Fiske LC, Ressner RA, Hong JC, Gregg KS, Henry TL, Pak KJ, Baumgarten KL, Seoane L, Garcia-Diaz J, Olivier KN, Zelazny AM, Holland SM, Browne SK (2015) Nocardia-induced granulocyte macrophage colony-stimulating factor is neutralized by autoantibodies in disseminated/extrapulmonary nocardiosis. Clin Infect Dis 60:1017–1025. https://doi.org/10.1093/cid/ciu968
Rosenberg JM, Price JV, Barcenas-Morales G, Ceron-Gutierrez L, Davies S, Kumararatne DS, Doffinger R, Utz PJ (2016) Protein microarrays identify disease-specific anti-cytokine autoantibody profiles in the landscape of immunodeficiency. J Allergy Clin Immunol 137(204–13):e3. https://doi.org/10.1016/j.jaci.2015.07.032
Rosenzweig SD, Dorman SE, Uzel G, Shaw S, Scurlock A, Brown MR, Buckley RH, Holland SM (2004) A novel mutation in IFN-gamma receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol 173:4000–4008. https://doi.org/10.4049/jimmunol.173.6.4000
Saijo T, Chen J, Chen SC, Rosen LB, Yi J, Sorrell TC et al (2014a) Anti-granulocyte-macrophage colony-stimulating factor auto-antibodies bare a risk factor for central nervous system infection by Cryptococcus gatii in otherwise immunocompotent patients. mBio 5:e00912–e914
Saijo T, Chen J, Chen SC, Rosen LB, Yi J, Sorrell TC, Bennett JE, Holland SM, Browne SK, Kwon-Chung KJ (2014b) Anti-granulocyte-macrophage colony-stimulating factor autoantibodies are a risk factor for central nervous system infection by Cryptococcus gattii in otherwise immunocompetent patients. mBio 5:e00912–e914. https://doi.org/10.1128/mBio.00912-14
Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, Capitani M, McGowan SJ, Sweeney E, Weber A, Chen L, Bowness P, Riordan A, Cant A, Freeman AF, Milner JD, Holland SM, Frede N, Muller M, Schmidt-Arras D, Grimbacher B, Wall SA, Jones EY, Wilkie AOM, Uhlig HH (2017) A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med 214:2547–2562. https://doi.org/10.1084/jem.20161810
Sim BT, Browne SK, Vigliani M, Zachary D, Rosen L, Holland SM, Opal SM (2013) Recurrent Burkholderia gladioli suppurative lymphadenitis associated with neutralizing anti-IL-12p70 autoantibodies. J Clin Immunol 33:1057–1061. https://doi.org/10.1007/s10875-013-9908-z
Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, Gurel M, Zhang Y, Sun G, Sabroe RA, Greene D, Rae W, Shahin T, Kania K, Ardy RC, Thian M, Staples E, Pecchia-Bekkum A, Worrall WPM, Stephens J, Brown M, Tuna S, York M, Shackley F, Kerrin D, Sargur R, Condliffe A, Tipu HN, Kuehn HS, Rosenzweig SD, Turro E, Tavare S, Thrasher AJ, Jodrell DI, Smith KGC, Boztug K, Milner JD, Thaventhiran JED (2019) Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med 216:1986–1998. https://doi.org/10.1084/jem.20190344
Tanaka T, Motoi N, Tsuchihashi Y, Tazawa R, Kaneko C, Nei T, Yamamoto T, Hayashi T, Tagawa T, Nagayasu T, Kuribayashi F, Ariyoshi K, Nakata K, Morimoto K (2011) Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genet 48:205–209. https://doi.org/10.1136/jmg.2010.082586
Tang BS, Chan JF, Chen M, Tsang OT, Mok MY, Lai RW, Lee R, Que TL, Tse H, Li IW, To KK, Cheng VC, Chan EY, Zheng B, Yuen KY (2010) Disseminated penicilliosis, recurrent bacteremic nontyphoidal salmonellosis, and burkholderiosis associated with acquired immunodeficiency due to autoantibody against gamma interferon. Clin Vaccine Immunol 17:1132–1138. https://doi.org/10.1128/CVI.00053-10
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova JL, Sullivan KE (2020) Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol 40:24–64. https://doi.org/10.1007/s10875-019-00737-x
Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, Wang T, Morgan C, Cottin V, McCarthy C (2019) Pulmonary alveolar proteinosis. Nat Rev Dis Primers 5:16. https://doi.org/10.1038/s41572-019-0066-3
van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 365:54–61. https://doi.org/10.1056/NEJMoa1100102
van de Vosse E, van Wengen A, van der Meide WF, Visser LG, van Dissel JT (2017) A 38-year-old woman with necrotising cervical lymphadenitis due to Histoplasma capsulatum. Infection 45:917–920. https://doi.org/10.1007/s15010-017-1060-x
Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, Mahlaoui N, Gineau L, Alcais A, Lamaze C, Puck JM, de Saint BG, Khayat CD, Mikhael R, Casanova JL (2008) Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med 205:1729–1737. https://doi.org/10.1084/jem.20071987
von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Arostegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yague J, Anton J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Marodi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321:691–696. https://doi.org/10.1126/science.1158298
von Stemann JH, Rigas AS, Thorner LW, Rasmussen DGK, Pedersen OB, Rostgaard K, Erikstrup C, Ullum H, Hansen MB (2017) Prevalence and correlation of cytokine-specific autoantibodies with epidemiological factors and C-reactive protein in 8,972 healthy individuals: results from the Danish Blood Donor Study. PLoS ONE 12:e0179981. https://doi.org/10.1371/journal.pone.0179981
Weiler FG, Peterson P, Costa-Carvalho BT, de Barros DM, Correia-Deur JE, Sader SL, Espindola-Antunes D, Guerra-Junior G, Dias-da-Silva MR, Lazaretti-Castro M (2018) The heterogeneity of autoimmune polyendocrine syndrome type 1: clinical features, new mutations and cytokine autoantibodies in a Brazilian cohort from tertiary care centers. Clin Immunol 197:231–238. https://doi.org/10.1016/j.clim.2018.09.012
Wipasa J, Chaiwarith R, Chawansuntati K, Praparattanapan J, Rattanathammethee K, Supparatpinyo K (2018) Characterization of anti-interferon-gamma antibodies in HIV-negative immunodeficient patients infected with unusual intracellular microorganisms. Exp Biol Med (Maywood) 243:621–626. https://doi.org/10.1177/1535370218764086
Wolff AS, Karner J, Owe JF, Oftedal BE, Gilhus NE, Erichsen MM, Kampe O, Meager A, Peterson P, Kisand K, Willcox N, Husebye ES (2014) Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J Immunol 193:3880–3890. https://doi.org/10.4049/jimmunol.1401068
Wongkulab P, Wipasa J, Chaiwarith R, Supparatpinyo K (2013) Autoantibody to interferon-gamma associated with adult-onset immunodeficiency in non-HIV individuals in Northern Thailand. PLoS ONE 8:e76371. https://doi.org/10.1371/journal.pone.0076371
Wu UI, Chuang YC, Sheng WH, Sun HY, Jhong YT, Wang JY, Chang SC, Wang JT, Chen YC (2018) Use of QuantiFERON-TB Gold In-tube assay in screening for neutralizing anti-interferon-gamma autoantibodies in patients with disseminated nontuberculous mycobacterial infection. Clin Microbiol Infect 24:159–165. https://doi.org/10.1016/j.cmi.2017.06.029
Wu UI, Wang JT, Sheng WH, Sun HY, Cheng A, Hsu LY, Chang SC, Chen YC (2020) Incorrect diagnoses in patients with neutralizing anti-interferon-gamma-autoantibodies. Clin Microbiol Infect. https://doi.org/10.1016/j.cmi.2020.02.030
Xie YL, Rosen LB, Sereti I, Barber DL, Chen RY, Hsu DC, Qasba SS, Zerbe CS, Holland SM, Browne SK (2016) Severe paradoxical reaction during treatment of disseminated tuberculosis in a patient with neutralizing anti-IFNgamma autoantibodies. Clin Infect Dis 62:770–773. https://doi.org/10.1093/cid/civ995
Acknowledgements
C.-L. K. was supported by grants from the Chang Gung Memorial Hospital (CMRPD1A0681-3, CORPD1F0041-3, CMRPD1F0171-3 and BMRP) and Taiwan Ministry of Science and Technology grant 105-2628-B-182-002-MY3. H.v.B. was supported by the German federal Ministry of Education and Research (PID NET-TPA5; 01GM1111D and 01GM1517E).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ku, CL., Chi, CY., von Bernuth, H. et al. Autoantibodies against cytokines: phenocopies of primary immunodeficiencies?. Hum Genet 139, 783–794 (2020). https://doi.org/10.1007/s00439-020-02180-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02180-0