
Abstract
A rare coding variant (rs72824905, p.P522R) conferring protection against Alzheimer’s disease (AD) was identified in the gene encoding the enzyme phospholipase-C-γ2 (PLCG2) that is highly expressed in microglia. To explore the protective nature of this variant, we employed latent process linear mixed models to examine the association of p.P522R with longitudinal cognitive decline in 3595 MCI patients, and in 10,097 individuals from population-based studies. Furthermore, association with CSF levels of pTau181, total tau, and Aβ1-42 was assessed in 1261 MCI patients. We found that MCI patients who carried the p.P522R variant showed a slower rate of cognitive decline compared to non-carriers and that this effect was mediated by lower pTau181 levels in CSF. The effect size of the association of p.P522R with the cognitive decline and pTau181 was similar to that of APOE-ε4, the strongest genetic risk factor for AD. Interestingly, the protective effect of p.P522R was more pronounced in MCI patients with low Aβ1-42 levels suggesting a role of PLCG2 in the response to amyloid pathology. In line with this hypothesis, we observed no protective effect of the PLCG2 variant on the cognitive decline in population-based studies probably due to the lower prevalence of amyloid positivity in these samples compared to MCI patients. Concerning the potential biological underpinnings, we identified a network of co-expressed proteins connecting PLCG2 to APOE and TREM2 using unsupervised co-regulatory network analysis. The network was highly enriched for the complement cascade and genes differentially expressed in disease-associated microglia. Our data show that p.P522R in PLCG2 reduces AD disease progression by mitigating tau pathology in the presence of amyloid pathology and, as a consequence, maintains cognitive function. Targeting the enzyme PLCG2 might provide a new therapeutic approach for treating AD.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is highly heritable and the most common cause of neurodegenerative dementias. The discovery of mutations in amyloid precursor protein (APP), Presenilin1 and Presenilin2 (PSEN1/2) causing rare familial AD laid an important foundation for the ‘amyloid cascade hypothesis’, postulating that the aggregation of amyloid is causative for AD and triggers downstream pathological events such as the formation of tau pathology [38]. The importance of amyloid for the development of the more common, sporadic form of AD has recently been emphasized by two large genome-wide association studies (GWAS) [33, 41]. Besides amyloid, GWAS studies suggest additional causative molecular pathways, including immune system processes [33, 35, 41]. Interestingly, it has been suggested that pathways supported by genetic evidence might provide entry points for drug development with a better chance of clinical success [37, 55]. Unfortunately, for most GWAS-identified loci, the variants responsible for the AD-association are unknown, which hampers their translation into a clear functional consequence.
In 2017, we and others identified a specific rare nonsynonymous coding variant (rs72824905, p.P522R) in the gene encoding the enzyme phospholipase-C-γ2 (PLCG2) that confers protection against the susceptibility to AD [68]. This association has been replicated in multiple independent samples from different populations [10, 13, 78]. PLCG2 is an enzyme mainly expressed in immune cells including microglia which are involved in innate immunity [47, 78]. PLCG2 is related to autoimmune diseases and involved in the activation of platelets in response to amyloid [8, 52, 66]. From a functional perspective, p.P522R induces a slight increase in PLCG2 activity [47]. Thus, therapeutic strategies similarly modulating PLCG2 activity as p.P522R may offer a new treatment option for common sporadic AD forms. To achieve this latter aim, we need first to understand the biological mechanisms linking p.P522R in PLCG2 to the pathophysiological processes of AD. Importantly, van der Lee and colleagues [78] suggested that the p.P522R might also have a protective effect on other neurodegenerative dementias [i.e., Dementia with Lewy Body (DLB) and frontotemporal dementia (FTD)] as well as a positive effect on longevity. However, it is unclear whether these protective effects result from an attenuated protein accumulation process that may be accelerated by microglia activation [4, 5, 28, 70, 72, 79, 85] in carriers of the mutated PLCG2 gene, or whether the response to accumulated protein aggregates is different, which is the suggested modus operandi for other AD-associated genes like TREM2 [34].
To understand the boundary conditions and mechanisms of p.P522R effects, we examined its association with cognitive decline in patients with mild cognitive impairment (MCI) and population-based studies. We also analyzed the effect of p.P522R on cerebrospinal fluid (CSF) biomarkers levels amyloid-beta 1-42 (Aβ1-42), total tau (tTau) protein, and phosphorylated tau (pTau181) protein as well as their interactions in patients with MCI. We also investigated potential biological underpinnings for the effect of PLCG2 within the context of AD pathology. We focused our analysis on the identification of a link between PLCG2 and the known functional relationship between APOE and TREM2. While research identified APOE as a ligand for TREM2 [67, 84] and PLCG2 as an important element of the TREM2 downstream signaling cascade [54], a link between PLCG2 and APOE is currently missing. Consequently, we conducted an unsupervised analysis of trans-co-regulatory network analysis to identify a biological pathway shared between APOE and the TREM2–PLCG2-signaling cascade.
Methods
Study cohorts
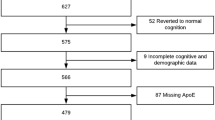
The design, recruitment, and diagnostic procedures are described in detail in the supplementary text 1, online resource. A flowchart of the participant recruitment and their assignment to outcome-specific analyses is presented in Fig. 1. All participants included in the analyses were older than 50 years, since the focus of our study was the effect of p.P522R in late life.

Study design
Longitudinal cognitive decline was analyzed in 3595 MCI patients. These patients were derived from four memory clinic cohorts, namely the Amsterdam dementia cohort (ADC), the German dementia competence network (DCN), the Spanish Fundacio ACE (FACE), and the Alzheimer’s disease neuroimaging initiative (ADNI) cohort that was launched as public–private partnership led by Michael Weiner with the aim to establish measurements of disease progression in MCI and AD (see supplementary text 1, online resource for full description). Also, MCI patients from the prospective, general practitioner-registry-based German study on aging, cognition, and dementia (AgeCoDe) were included. Participants of the AgeCoDe cohort were all non-demented at baseline. Among those, the MCI patients were identified by a standardized diagnostic procedure at each visit and included in this analysis.
10,097 participants from the Three-City Study (3C) and the Longitudinal Aging Study Amsterdam (LASA), as well as all participants from the AgeCoDe study, were included for the analysis of the cognitive decline in population-based samples regardless of MCI or dementia diagnoses.
To analyze the effect of p.P522R on proxy measures of amyloid and tau pathology in the CSF, levels of Aβ1-42, pTau181, and tTau protein were analyzed. CSF data were collected from 1,261 MCI from the DCN, ADC and ANDI cohort as well as from the memory clinic of the University Hospital of Bonn (UHB) and processed at different centers (supplementary text 4, online resource).
Genotyping
In all samples, DNA was extracted using standard procedures. In all cohorts except LASA and ADNI, p.P522R was directly genotyped using the MassARRAY (3C cohort, Agena Bioscience), custom content using the Infinium-global-screening-array-24-v1 (ADC cohort, Illumina), or a TaqMan custom design genotyping assay (other cohorts, Thermo Fisher Scientific), respectively. In LASA, p.P522R genotypes were derived from imputation based on the Infinium-global-screening-array-24-v1(Illumina, imputation quality: R2 = 0.89) or the AXIOM-NL (Affymetrix, imputation quality: R2 = 0.93) array. In ADNI, three different platforms [i.e., Illumina Human610-Quad BeadChip (imputation quality: R2 = 0.87), HumanOmniExpress BeadChip (imputation quality: R2 = 0.83), and Illumina Omni 2.5 M (imputation quality: R2 = 0.93)] were used for deriving p.P522R genotypes from imputed values. In case multiple platforms were used for genotyping the same individual, we retained p.P522R imputations with the highest genotype probability. All procedures are described in detail in supplementary text 2, online resource. For a fraction of the samples included in the analysis, GWAS data were available. In these samples, all individuals of non-European ancestry were discarded and the presence of population stratification was excluded by visually inspecting a plot of the first two principal components (PCs) of the GWAS data (supplementary Fig. 1a–g, online resource). No evidence for population stratification was found during visual inspection. In line with that, adjustment for PCs in those samples with GWAS data did not change the effects observed for p.P522R (supplementary text 3). For this reason, PCs were not used to adjust the analysis maximizing thereby the data set for analysis and thus increasing the statistical power.
CSF collection and biomarker measurement
CSF levels of Aβ1-42, pTau181, and tTau were measured using commercial ELISA immunoassays. Samples were quantified in different laboratories (CSF samples from Erlangen, Bonn, Amsterdam, and the ADNI cohort, see Fig. 1) using different methods described in supplementary text 4.1, online resource. For the present analysis, CSF from different cohorts was harmonized using the method described by Zhou and colleagues [89] (supplementary text 4.2, online resource).
Neuropsychological assessments
In all cohorts, the Mini-Mental State Examination (MMSE) was used as this measure of global cognition is sensitive to change in patients with MCI [59] and available in all cohorts. In the population-based studies, the MMSE might show ceiling effects. Therefore, additional measurements were used for the cognitive function: the 3C cohort also measured episodic memory using the Benton visual retention test, the LASA cohort used the Dutch version of the Adult Verbal Learning Task, and the AgeCoDe cohort used the CERAD word list learning task with delayed recall. Besides, verbal fluency was assessed in the 3C and AgeCoDe study using the Isaac set test and the CERAD animal fluency task, respectively. References and further descriptions for all tests are given in supplementary text 5, online resource.
Statistical analysis
Statistical analyses were conducted in R version 3.4.4 [58] and Mplus version 7.31 [50]. References to all utilized software are provided in supplementary Table 1, online resource. More detailed descriptions of the used methods are provided below and in supplementary text 6, online resource. All p values < 0.05 (two-sided) were considered significant.
Analysis of cognitive decline
The analysis of the effect of p.P522R on longitudinal cognitive decline was performed using latent process linear mixed models [56] (supplementary text 6.1, online resource) as implemented in the R package LCMM [57]. These models jointly estimate a latent process representing the true change of cognition over time and a link function that relates this process to the observed cognitive measurements. This link function takes into account the unequal interval scaling that commonly occurs for cognitive tests and may introduce bias [56] by modeling a monotone but possibly non-linear relationship between the latent process and the outcome. Modeling of the link function also accounted for the skewed distribution of the MMSE indicated ceiling effects in some MCI patients. Moreover, residuals derived from the standard linear mixed model showed deviations from normality which were adjusted by including the appropriate link function. Potential non-linear decline trajectories were assessed using polynomial order of time. The most appropriate link function and polynomial of time were chosen according to the lowest Bayesian information criterion (BIC). Statistical significance of the effect of p.P522R on the decline was assessed using multivariate Wald tests.
For the analysis of MCI patients, median-centered time from MCI diagnosis was used as the time scale to approximate disease progression after cognitive symptom onset. In the AgeCoDe cohort, the baseline for our analysis was defined as the first visit where MCI criteria were met and only subsequent observations were considered as follow-up assessments. In a sensitivity analysis, the follow-up time was restricted up to 6 years to rule out that a possible effect of p.P522R was artificially induced by only a few observations with long follow-up. An integrative data analysis was performed by pooling data from all memory clinic cohorts to maximize the number of p.P522R carriers as recommended for rare predictors [11]. Also, stratified analyses in each cohort were performed. The relationship between p.P522R and APOE-ε4 was investigated by examining the interaction effect on the cognitive decline between the two genotypes. In addition, effect sizes on cognitive change of p.P522R in the absence and presence of APOE-ε4 were compared with the effect of APOE-ε4 alone. Due to potential non-linear decline, effects were calculated at multiple time points on the scale of the latent process, standardized by the predicted variation at the last time point (supplementary text 6.2, online resource).
For the analysis of the effect of p.P522R in three population-based cohorts, median-centered age at assessment was used as the time scale of the linear mixed models representing general age-related cognitive change. Herein, analyses were performed separately for each cohort.
All analyses in all samples were adjusted for age at baseline, as well as gender, education (i.e., dichotomous variable indicating participation in secondary education), and APOE-ε4 status (supplementary text 6.5, online resource). In the pooled analysis of MCI patients from memory clinic cohorts, analyses were additionally adjusted for cohort. Furthermore, center was included as an additional covariate in the 3C study and the genotyping platform was used as a covariate in LASA. In the case of significant associations, analyses were repeated without adjustment to examine the sensitivity of the results to covariate selection. Missing data in the cognitive outcomes were handled using maximum-likelihood estimation [15], so that no participants had to be excluded due to drop-out.
Analysis of CSF biomarkers
For CSF analysis, continuous harmonized CSF biomarker data were log-transformed. Robust regression analysis was used to minimize the influence of outlying observations in the data by applying a down-weighting algorithm [40].
AD biomarkers were analyzed in a joint regression model using data from all CSF samples to maximize the number of available p.P522R carriers. Also, stratified analyses were performed in the samples from the three European memory clinic cohorts (DCN, ADC, and UHB) and the ADNI cohort. All regression analyses were controlled for age, gender, the origin of the CSF samples, and the presence of at least one APOE-ε4 allele (see supplementary text 6.5, online resource). We adjusted p values from tests of multiple outcomes (i.e., three different AD biomarkers) by applying the Bonferroni–Holm correction.
To explore whether p.P522R affects the interplay of AD biomarkers, four AD biomarker categories were constructed as described by Jack and colleagues [29] based on laboratory-specific cut-offs. These categories were derived: AD (Abeta1-42 positive and pTau181 positive, irrespective of tTau), AD pathologic change (Abeta1-42 positive and pTau181 negative, irrespective of tTau), non-AD pathologic change (Abeta1-42 negative and pTau181 positive and/or tTau positive), and no pathologic change (all three biomarkers negative). These categories were used as the outcome in a multinomial regression model with AD as the reference category and Bonferroni–Holm correction for multiple testing (i.e., comparison of four biomarker categories).
In addition, a varying coefficient generalized additive model [82] (supplementary text 6.3, online resource) was used to test whether the effect of p.P522R on continuous pTau181 and tTau levels differs across different Aβ1-42 levels. Thin plate regression splines [81] were used to allow for non-linear, potentially sigmoid relationships between AD biomarkers as proposed by Jack and colleagues [30]. Posterior simulation with simultaneous confidence intervals was used to identify those levels of CSF Aβ1-42 at which p.P522R show its strongest effects.
All analyses in CSF samples were adjusted for age, gender, CSF samples, and APOE-ε4 status (supplementary text 6.5, online resource). In a sensitivity analysis, all analyses were repeated without adjustment for covariates.
Mediation analysis using structural equation models
To evaluate whether or not the associations of p.P522R with CSF biomarkers underlie the association of p.P522R with cognitive decline, a mediation analysis was conducted using structural equation modeling in Mplus version 7.31 [50]. Herein, 1052 MCI patients from the DCN, ADC, and ADNI cohorts were included only if they had CSF AD biomarkers as well as longitudinal MMSE assessments available. To assess mediation, an indirect effect of p.P522R on the cognitive change in the MMSE over 4 years via Aβ1-42 and tTau were estimated, as well as interaction effects between p.P522R, and Aβ1-42 or pTau181. Analyses were repeated using pTau181 instead of tTau in the models. Additional details are provided in supplementary text 6.4, online resource.
Co-regulatory network analysis
The GeneFriends tool was used to generate an unsupervised co-expression gene map based on 4164 Human Microarray data sets containing 26,113 experimental conditions and 19,080 genes. We decided to use the microarrays data available in GeneFriends tool, because the number of experimental conditions available was larger and better annotated compared to the RNA-sequencing data available in the same tool. Further description of the microarray methods is provided elsewhere [77]. Of note, loci around the target genes might co-express due to the existence of common regional co-regulation motifs. Cis-co-regulated genes were deleted from the respective list by removing all transcripts located 200 kb around PLCG2, APOE, and TREM2 loci. Hence, only highly trans-co-regulated and positively co-expressed genes that are collectively upregulated were selected for further analysis (co-expression value > 0.5). Next, the WebGestalt tool [45] was used to identify potential enrichments in identified co-regulated gene lists using overrepresentation enrichment analyses of Gene Ontology non-redundant biological process in humans. Furthermore, genes co-expressed with PLCG2 were tested for enrichment of shared co-expression with APOE and TREM2 using Fisher’s exact test (supplementary text 6.6, online resource). Genes co-expressed in all three candidate genes were selected for additional enrichment analyses using STRING [75] and WebGestalt. Finally, this shared gene set was tested for enrichment of genes differentially expressed in cell-type-specific biomaterials derived from brain, i.e., microglia of AD patients [49] as well as microglia derived from the 5XFAD AD mouse model [39] and the hMAPT-P301S model for tauopathies [19] (supplementary text 6.6, online resource).
Results
The p.P522R variant is associated with slower cognitive decline in MCI patients
Due to the low frequency of the rare variant, it is unlikely to detect statistically significant effects in the individual cohorts. We, therefore, pooled data from all cohorts (Table 1) in a joint analysis in a first step. Here, carriers of the p.P522R variant (n = 61) showed a slower cognitive decline than non-carriers (χ2(2) = 7.83, p = 0.020, Fig. 2a; supplementary Table 2, online resource). Importantly, analyses stratified for cohort demonstrated a highly consistent protective effect of p.P522R in each of the samples (Fig. 2b–e, supplementary Fig. 2, and supplementary Table 3, online resource). This consistency was further confirmed by a non-significant interaction between p.P522R and cohorts concerning the effect on the cognitive decline (χ2(8) = 3.55, p = 0.895). Sensitivity analyses showed no change in the association without adjustment for covariates (χ2(2) = 8.95, p = 0.011) or using a restricted follow-up interval of 6 years (χ2 (2) = 6.58, p = 0.037, supplementary Table 2, online resource).

Effect of p.P522R on the cognitive decline in 3,595 MCI patients. a Predicted trajectories of cognitive decline for p.P522R carrier and non-carrier in the pooled sample of all MCI patients. b Predicted trajectories of cognitive decline for p.P522R carrier and non-carrier in the Fundacio ACE (FACE) cohort. c Predicted trajectories of cognitive decline for p.P522R carrier and non-carrier in the German study on aging, cognition, and dementia (AgeCoDe) cohort. d Predicted trajectories of cognitive decline for p.P522R carrier and non-carrier in the Dementia competence network (DCN) cohort. e Predicted trajectories of cognitive decline for p.P522R carrier and non-carrier in the ADNI cohort. Predicted trajectories for the Amsterdam dementia cohort (ADC) are displayed in supplementary figure 1, online resource due to the unreasonably low number of p.P522R carriers with sufficient follow-up. f Effect of p.P522R and APOE-ε4 on the cognitive change from baseline in the MMSE derived from a linear mixed model with a latent process including an interaction term between APOE-ε4 and p.P522R. Differences on the latent process scale between APOE-ε4 carrier and non-carrier who do not carry the pP522R variant were derived to assess the effect size of APOE-ε4 (red arrow in Fig. 2f and red dots in Fig. 2g, h). The difference between p.P522R carrier and non-carrier in the absence (magenta arrows in Fig. 2f and magenta dots in Fig. 2g) and the presence of APOE-ε4 (green arrows in fig. 2f and green dots in Fig. 2h) were calculated, as well. g The effect size of the association of not carrying any APOE-ε4 allele (red dots) and p.P522R in the presence of APOE-ε4 (green dots) with the cognitive change at different time points. Effect sizes are displayed on the scale of the latent process of the mixed model standardized by the expected variance of the latent process at the last time point considered (see supplementary text 6.2, online resource). h The effect size of the association of not carrying any APOE-ε4 allele (red dots) and p.P522R in the absence of APOE-ε4 (magenta dots) with the cognitive change at different time points. Effect sizes are displayed on the scale of the latent process of the mixed model standardized by the expected variance of the latent process at the last time point considered (see supplementary text 6.2, online resource)
In the same cohorts, the APOE-ε4 allele was, as expected, associated with accelerated cognitive decline (χ2 (2) = 138.33, p = 9.27 × 10–37, supplementary Table 2, online resource), but there was no statistically significant interaction between APOE-ε4 and p.P522R (χ2 (2) = 2.87, p = 0.238, supplementary table 4, online resource). The absence of a significant interaction may arise from limited statistical power due to the small number of carriers for both the p.P522R and the APOE-ε4 alleles (n = 23). However, the protective effect of p.P522R and the detrimental effect of APOE-ε4 on the cognitive change from baseline were similar in magnitude though in opposite directions (Fig. 2f). To directly compare the protective effect of PLCG2 with that of APOE, we expressed the APOE effect on cognitive decline in protective terms, i.e., cognitive changes when APOE-ε4 is absent (Fig. 2g, h). This analysis suggested that the protective effect of p.P522R is as strong as the detrimental APOE-ε4 effect during the first 6 years of follow-up, but this effect decreased at later follow-ups (Fig. 2f, g). This raises the possibility that p.P522R may initially counteract, at least in part, the detrimental effect of the APOE-ε4 allele on cognitive function and could delay deterioration in MCI patients for several years. We also found that the protective effect on cognitive change associated with p.P522R in APOE-ε4 non-carriers was similar to the reduced cognitive change observed in the absence of APOE-ε4 alone, especially at later stages of the cognitive trajectory (Fig. 2h).
Age-related cognitive decline in population-based samples is not associated with p.P522R
Given our observation in MCI and the previously reported effect on longevity [78], we sought to explore whether the effect of p.P522R on cognitive decline can be also extended to the general population (Table 2). As expected from previous case–control studies [68, 78], the frequency of p.P522R in the population-based samples was significantly higher than in the sample of MCI patients from memory clinic cohorts (i.e., FACE, DCN, ADNI, and ADC) who are an at-risk population for dementia (χ2(1) = 10.02, p = 0.002). Importantly, the statistical difference found for this variant between the MCI samples and the population-based studies further support the protective effect of PLCG2. However, p.P522R did not modulate the cognitive decline in any of the neuropsychological tests explored in the population-based samples (supplementary table 5, supplementary Figs. 3–5, online resource).
p.P522R is associated with reduced levels of CSF pTau181 and tTau
To examine the etiology underlying the protective effect of p.P522R, we analyzed AD-related CSF biomarkers in MCI patients (Table 3). Carriers of the p.P522R variant (n = 18) showed a reduction of pTau181 (Est(SE) = − 0.12(0.05), p = 0.015, d = − 0.58) and tTau levels (Est(SE) = − 0.12 (0.05), p = 0.017, d = − 0.57) in the pooled sample of all MCI patients. The association remained statistically significant after Bonferroni–Holm correction for multiple testing (Fig. 3, supplementary table 6, online resource). In contrast, there was no association of p.P522R with Aβ1-42 levels (Est(SE) = − 0.02(0.04), p = 0.686, d = − 0.10). The effect of p.P522R was consistent across MCI patients from both the European MCI cohorts (DCN, ADC, and UHB) and the ADNI cohort (Fig. 3). This was confirmed by non-significant interactions between p.P522R and CSF samples (supplementary table 7, online resource). Sensitivity analyses suggested that unadjusted analysis led to a similar pattern of results (supplementary table 6, online resource).

Associations of p.P522R with CSF biomarkers of Alzheimer’s disease (AD). Black dots and arrows represent Cohen’s d estimates and 95% confidence interval, respectively. For Aβ1-42, negative Cohen’s d estimates indicate more pathology. In the case of pTau181 and tTau, positive Cohen’s d values represent more pathology. N(p.P522R) number of p.P522R carrier, n(wt) number of p.P522R non-carrier, Aβ1-42 amyloid-beta 1-42, pTau181 phosphorylated tau
Again, the effect sizes of p.P522R and APOE-ε4 on pTau181 in the pooled MCI patients were similar, but in opposite directions (p.P522R: d = − 0.58, APOE-ε4: d = 0.56; supplementary table 8, online resource). We did not observe a statistically significant interaction between APOE-ε4 and p.P522R regarding any CSF biomarker (supplementary table 9, online resource). The lack of interaction may, again, follow the same power issue derived from the small-sample size of carriers of both variants (n = 10). Notably, APOE-ε4 showed an effect on all CSF biomarkers, while p.P522R only had an effect on pTau181 and tTau levels (supplementary table 8, online resource).
p.P522R exerts its strongest effect downstream of amyloid pathology
To further explore the role of p.P522R in AD, we examined the interplay of the same CSF biomarkers using the AD biomarker categories proposed by Jack and colleagues [29]. A multinomial regression model using the AD category (Aβ1-42 and pTau181 positive, n = 522) as a reference revealed that p.P522R was associated with the presence of AD pathologic change (Aβ1-42 positive, pTau181 negative, OR(95% CI) 6.28(3.3, 11.9), p = 0.004, n = 279) but not with the presence of non-AD pathologic changes (Aβ1-42 negative, pTau181 and/or tTau positive, OR(95% CI) 0.93(0.39, 2.24), p = 0.938, n = 129) or normal CSF biomarkers (all three biomarkers negative, OR(95% CI) 1.81(0.86, 3.83), p = 0.425, n = 323). Noteworthy, the small number of amyloid-negative individuals might have limited the statistical power to detect an association of p.P522R with the presence of non-AD pathological changes. Unadjusted analyses revealed the same results (supplementary table 10, online resource).
We also applied generalized additive models in the MCI data set to model the influence of the non-linear relationship of Aβ1-42 with pTau181 and tTau in the CSF on the effect of p.P522R. We observed that the effect of p.P522R on pTau181 and tTau levels is significantly stronger when CSF levels of Aβ1-42 are low (i.e., more abnormal, pTau181: p = 0.013, tTau: p = 0.020, Fig. 4a, b, supplementary table 11, online resource).

Role of p.P522R in the interrelationship between amyloid pathology, neurodegeneration, and cognitive decline. a Predicted relationship between Aβ1-42 levels in CSF and pTau181 levels in CSF for p.P522R carrier and non-carrier. Red shaded areas indicate significant differences between p.P522R carrier and non-carrier. Significance is based on the estimation of simultaneous confidence intervals that consider the statistical testing at multiple CSF levels. b Predicted relationship between Aβ1-42 levels in CSF total tau levels in CSF for p.P522R carrier and non-carrier. Red shaded areas indicate significant differences between p.P522R carrier and non-carrier. Significance is based on the estimation of simultaneous confidence intervals that consider the statistical testing at multiple CSF levels. c Results from a structural equation model assessing whether the effect of p.P522R on the cognitive change in the normalized MMSE (range 0–100) is mediated Abeta1-42 levels or pTau181 levels in CSF. The model showed a good fit to the data (Model fit indices: RMSEA = 0.017, CFI = 0.996, SRMR = 0.060, see supplementary text 6.4, online resource)
We then sought to conduct a mediation analysis in 1052 MCI patients with both CSF biomarkers and longitudinal follow-up data using structural equation modeling (Fig. 4c, supplementary table 12, online resource). This analysis will allow establishing a link between the findings derived from the cognitive decline in MCI and those obtained from the CSF. This strategy revealed that the effect of p.P522R on the change in cognition from baseline over 4 years was mediated by pTau181 (Est = 1.91, 95% CI 0.12, 4.11) but not by Aβ1-42 (Est = 0.84, 95% CI − 0.54, 2.63, supplementary table 13, online resource). Noteworthy, we found a significant interaction of p.P522R and Aβ1-42 levels on the cognitive decline (χ2(2) = 8.86, p = 0.012), indicating that the protective p.P522R variant is associated with a less pronounced cognitive decline at more abnormal levels of Aβ1-42 (supplementary table 14, supplementary Fig. 6, online resource). This observation is in line with the findings obtained from the generalized additive model. There was no interaction effect between p.P522R and pTau181 levels (χ2(2) = 0.68, p = 0.712) on the cognitive decline. The pattern of results was similar when tTau instead of pTau181was used as a mediator. However, the effect of p.P522R on tTau (Est(SE) = − 0.22(0.12), p = 0.051) and the corresponding mediation effect on the cognitive change over 4 years (Est = 1.94, 95% CI = − 0.13, 4.07) showed only a trend-level association (supplementary tables 12–14, online resource).
Conversely, we observed that the effect of APOE-ε4 on cognition change from baseline over 4 years is mediated by both, Aβ1-42 (Est = − 4.14, 95% CI − 5.67, − 2.57) and pTau181 (Est = − 1.24, 95% CI − 2.15, − 0.56). However, there were no interaction effects between APOE-ε4 and the CSF levels of Aβ1-42 (χ2(2) = 1.29, p = 0.526) and pTau181 (χ2(2) = 2.531, p = 0.282) regarding the cognitive decline.
APOE shares a common co-regulatory network with PLCG2 and TREM2
To explore the biological underpinnings of the interplay of APOE with PLCG2 and the membrane receptor TREM2, we searched for co-regulated genes and pathways. An unsupervised search identified 2748 genes co-expressed with PLCG2 showing enrichment of immune-related pathways (supplementary table 15, online resource). In our analysis, gene-specific networks of APOE and TREM2 were also highly enriched for immunological function (supplementary tables 16–17, online resource). Genes co-expressed with PLCG2 were significantly enriched among those co-expressed with APOE (p = 7.49 × 10–34) or TREM2 (p = 1.37 × 10–33). Furthermore, PLCG2-related genes were disproportionally more likely to be co-expressed with both APOE and TREM2 (p = 3.76 × 10–16). Noteworthy, the shared gene set of 21 loci simultaneously co-regulated with APOE, TREM2, and PLCG2 (Fig. 5) is highly enriched for biological processes related to immune system processes including the complement cascade activation but also tissue remodeling (supplementary table 18, online resource). Furthermore, we also detected a correspondence between the identified network and microglial gene expression in the brain. Herein, we found enrichment for genes differential expressed in microglia from human AD patients (p = 3.57 × 10–12, [49]) as well as in microglia from mouse models of AD (p = 9.47 × 10–8, 5XFAD model [39]) or tauopathies (p = 2.18 × 10–6, hMAPT-P301S model [19], supplementary table 19, online resource).

Co-regulatory network shared between APOE, TREM2, and PLCG2. a Venn diagram showing the number and the overlap of genes highly co-expressed APOE, TREM2, and PLCG2. b Depiction of potential relationships between genes included in the shared co-expression network of APOE, TREM2, and PLCG2. The red circle marks members of the complement cascade. The green circle marks genes involved in tissue remodeling. Green lines between proteins indicate evidence for interaction based on text mining, black lines represent co-expression, blue lines indicate evidence from curated databases, and magenta lines indicate experimentally conformed interactions
Discussion
In this study, we provide the first evidence linking the protective effect of the p.P522R variant in PLCG2 with a slower cognitive decline in patients with MCI and reduced pTau181 and tTau levels. Interestingly, this effect seems to be downstream of Aβ1-42 pathology. Furthermore, we showed that PLCG2 shares a biological co-regulation network with the APOE and TREM2 that is enriched for complement cascade processes and markers of disease-associated microglia. Taken together, our findings strongly support a role of p.P522R in the physiological response to abnormally folded proteins, such as amyloid, and help to characterize the specific function of PLCG2 within the amyloid cascade.
To date, except for CLU and APOE, few of the identified genetic risk variants in case–control GWAS for AD have shown a consistent effect on disease progression in MCI patients [42]. Our study now adds to this list PLCG2 as a strong protector of cognitive function at the MCI stage. This effect is mediated by reduced pTau181 pathology but not by Aβ1-42 pathology. Thus, the p.P522R variant ameliorates cognitive decline by acting downstream of amyloid accumulation, making this accumulation less detrimental. This hypothesis receives additional supports from the observation that p.P522R displays its strongest effect on tau pathology and cognitive decline when amyloid pathology is present. Recent research has further shown that tau pathology depends on Aβ1-42-evoked neuroinflammation [28]. Likewise, the neuronal protection conferred by p.P522R in AD may also operate in neurodegenerative diseases caused by the accumulation of other protein aggregates which trigger downstream damaging effects via neuroinflammation. Supporting this hypothesis, p.P522R in PLCG2 might also have a protective effect on DLB and FTD [78]. For all three diseases (i.e., AD, FTD, and DLB), a genetic overlap has been reported suggesting shared pathogenic pathways which may include pathways related to PLCG2 [16, 24, 36, 64]. Furthermore, patients with DLB frequently show amyloid pathology [53] that contributes to fast disease progression and cognitive impairment [1] suggesting that microglial reaction to amyloid pathology could mediate the protective role of p.P522R on both AD and DLB. In contrast, amyloid pathology is rarely observed in FTD indicating a slightly different mechanism. However, mutations in TREM2 have been consistently reported in FTD patients involving, thus, the TREM2 signal cascade in the pathological events occurring in FTD [20, 61]. Besides, functional studies in genes involved in FTD have shown that several of these genes can modulate microglia function either by increasing production of neurotoxic factors and neuroinflammation, or by altering microglial phagocytosis and related degradation pathways [22]. For example, research has shown that levels of soluble TREM2 (sTREM2) in CSF are increased in familial FTD cases carrying mutations in the progranulin gene (GRN) compared to controls [83]. Likewise, a C9orf72-deficient mouse showed increased expression of Trem2, C1qa, and Tyrobp linking decreased expression of C9orf72 with microglia activity and age-related neuroinflammation [43]. As with GRN and C9orf72, other FTD genes, including TANK-binding kinase 1 (TBK1), Optineurin (OPTN), sequestosome (SQSTM1), and Valosin Containing Protein (VCP), have been linked to neuroinflammation and microglial function, because these genes can regulate one of the crucial regulators of glial activation and neuroinflammation, the nuclear factor-kappa beta (NF-κβ) [22]. Importantly, NF-κβ has been shown to be a downstream effector of PLCG2 activation [65]. It is, therefore, tempting to speculate that downstream neuroinflammatory processes activated by protein aggregation in FTD will lead to activation of a signaling cascade involving PLCG2 wherein the p.P522R-carrying PLCG2 may show its protective effect. In addition, all these findings together may have uncovered a more general effect of PLCG2 in the response to misfolded protein aggregation found in neurodegenerative disorders. At this point, it is important to note that the reported effect of p.P522R on susceptibility to FTD and DLB still requires validation in independent samples [78].
Besides the effect downstream of Aβ1-42 pathology, p.P522R might also contribute to the initial formation and amplification of pathological protein aggregates itself instead of modulating their downstream effects. Herein, research has shown that microglia contribute to an accelerated formation of amyloid plaques and tau aggregation [4, 5, 70, 72, 85] which is probably dependent on the activation of the inflammasome [28, 73, 79]. The protective effect of p.P522R in PLCG2 could modulate the microglial pathways leading to this accelerated pathology. Our results, however, do not support this hypothesis, because we could not find evidence for an association of p.P522R with Abeta1-42 pathology or pTau181 and tTau in amyloid-negative individuals. Albeit a negative finding, this result should be interpreted with caution, because our AD-focused study design and the limited number of amyloid-negative MCI patients in our sample might have rendered our sample underpowered to detect significant effects of p.P522R on neurodegenerative markers in the absence of amyloidosis. Consequently, additional studies in samples enriched for non-AD dementias are now needed to test our observation.
Interestingly, the stronger effect of p.P552R on tau pathology and cognitive decline in the presence of amyloid pathology offers, in turn, a possible explanation for the lack of association between p.P522R and cognitive decline in population-based studies. The cognitive decline observed in older individuals from the general population is thought to derive from an increased vulnerability of the brain to the initiation of neurodegenerative and non-pathological processes [6, 18, 25, 31]. However, in populations of MCI patients, the prevalence of amyloid and other neuropathologies is increased and exerts a stronger effect on the cognitive decline as compared to cognitively unimpaired participants [26, 32] who form the majority of participants in population-based studies [62]. In fact, MCI cases show a 20–30% higher prevalence of amyloid positivity compared to cognitively normal individuals, independently of age range analyzed [32, 63], as well as a higher prevalence of tau and Lewy body pathology [2]. Our line of arguments receives further support from the observation that the prevalence of amyloid and tau pathology increases with age [7] as well as the probability of being carrier of the p.P522R variant [78]. Thus, it is tempting to speculate that slower cognitive decline in MCI patients and prolonged survival of p.P522R carriers in older ages is due to a modulation of the neuroinflammatory response to progressive pathological protein aggregation, such as amyloid, which leads, in turn, to increased neuronal survival and, finally, improved cognition contributing to longevity [23].
We also examined the potential biological mechanisms underlying the observed effects of p.P522R. Given the compelling evidence supporting the link between TREM2 and APOE at the molecular level, we search for potential shared pathways between TREM2, APOE, and PLCG2. In supporting this hypothesis, the three genes are involved in microglial response within various neurodegenerative conditions. Importantly, APOE and TREM2 might be linked to PLCG2 at the molecular level, as APOE is a ligand of TREM2 [84] and TREM2 is a surface receptor upstream of PLCG2-signaling cascade [54]. Further supporting their functional connection, all three genes modulate similar AD endophenotypes. This includes the effect described here for the APOE-ε4 allele and p.P522R in PLCG2 on the cognitive decline. In addition, in 2019, a rare protective coding variant in APOE, different from the APOE-ε2 allele, was found to mitigate downstream effects of amyloid-beta formation [3], as observed for p.P522R in our study. Furthermore, similar to p.P522R, CSF levels of sTREM2 and the p.R47H variant in TREM2 are both strongly associated with tau pathology [46, 60]. In the case of sTREM2, its CSF levels also show specific alterations in amyloid positive individuals without tau pathology [74]. In line with these molecular and phenotypic links, our unsupervised trans-coregulatory network analysis based on microarray data from multiple tissues and experimental conditions suggested that APOE, PLCG2, and TREM2 share a common, general co-expression network. This network contains the AD hub-gene TYROBP [88] and it is highly enriched for the complement cascade and genes differentially expressed in microglia from AD patients [49] or mouse models of AD (FXFAD model; [39]) or tauopathies (hMAPT -P301S model; [19]). Thus, several biological processes that have been identified as crucial for the pathogenesis of AD but also other neurodegenerative dementias [14, 21] may not only involve APOE and TREM2, as previously described [67] but also PLCG2. Consequently, taking our present and previous research, it is tempting to speculate that this shared mechanism could include the adaption of microglia to a neurodegenerative environment that may contribute to the protective effect across different neurodegenerative dementias, as hypothesized before. Among the pleiotropy of potential underlying pathways, our co-expression network analysis highlights several mechanisms. Those could involve altered downstream TREM2 signaling including the co-expression network members SPP1 and GPNMB [44, 69]. Alternatively, those mechanisms could include the differential regulation of the signaling of CD14 and Toll-like receptors [86, 87] or complement receptors [9, 48]. Importantly, complement cascade activation is observed in several neurodegenerative diseases, including AD, DLB, and FTD [21, 80] and is strongly involved in synapse loss [27] which is strongly related to cognitive impairments [76]. These candidate pathways require further experimental investigation to establish a role in the mediation of the effect of p.P522R on AD, as well as in other neurodegenerative dementias. In fact, several efforts are currently ongoing to gain insight on the molecular mechanisms of PLCG2 variant in the immune pathway connecting it with other AD risk genes.
From a therapeutic point of view, PLCG2-directed therapeutic approaches might, therefore, offer a novel opportunity to complement drugs directed at amyloid clearance or production by modulating the downstream effects of amyloid pathology mediated by microglia [12, 28, 51, 71]. Particular interest, herein, deserves the observation that the protective effect of p.P522R can apparently counteract the deleterious effect of APOE-ε4 on cognitive decline for several years and on tau pathology in MCI. Both effects probably derive from the similar effect sizes of the two genetic variants. Of note, APOE-ε4 showed its strongest effect on amyloid pathology and only a smaller but independent effect on tau pathology, suggesting that APOE-ε4 influences AD pathogenesis more upstream in the amyloid cascade than p.P522R. Thus, therapeutic approaches targeting PLCG2 function might complement those aiming at APOE function. However, our data also suggest that the buffering properties of p.P522R against APOE-ε4-induced deficits seem to decrease as the disease progresses. This reduction in effect may be produced by chronical exposure of microglia to deleterious insult undermining the protective function of microglia including the protective effect given by p.P522R in PLCG2. Understanding the pathways underlying the protective effect of PLCG2 and its buffering properties may help to develop therapeutic targets which can counteract this progressive collapse of the protection delaying or even preventing the beginning of the dementia stage.
Strength and limitations
A considerable strength of our study is the recruitment and analysis of one of the largest samples of MCI patients with longitudinal follow-up and CSF biomarkers. This data set offers the unique opportunity to study the effect of p.P522R at this disease stage and to define the link between its effects on clinical data and the underlying pathophysiological process. Besides, we used an agnostic co-expression network analysis that does not rely on specific cell or tissue expression profile experiments that are usually based in very low numbers and, therefore, prone to measurement error. Importantly, using a general approach relying on a wide range of experiments led us to replicate the previous findings on expression network [10, 68, 88] and also led us to the discovery of a novel links between APOE and TREM2–PLCG2 signaling. Furthermore, the integrative analysis of biomarker, cognitive, and gene-expression data provides the most comprehensive description of the role of PLCG2 in AD pathogenesis to date.
However, our study has also limitations. Our results are based on a limited number of carriers due to the low frequency of the p.P522R variant. Especially when examining the interaction of p.P522R with APOE-ε4 or Aβ1-42, the limited number of carriers might have rendered our study underpowered for quantifying the exact effect size of the interaction. Thus, investigations of independent samples are now necessary to confirm these observations. Nevertheless, the consistency of the effect of p.P522R across cohorts and phenotypes supports the robustness of our findings. Additionally, amyloid and tau burden from cognitively healthy individuals were not available. Since the trajectory of amyloid pathology is expected to reach a plateau in late disease stages [30], the lack of cognitively normal patients with CSF samples might have limited our ability to detect the effects of p.P522R on CSF-Aβ1-42 levels. We were, however, able to detect an effect for the APOE-ε4 allele. Finally, confirmation of findings from the co-regulatory network analysis is still required using microglia expression data.
In conclusion, our data link the protective effect of p.P522R in PLCG2 to lower CSF concentrations of pTau181 and tTau and slower cognitive decline in MCI patients, particularly in amyloid positive individuals and with an effect size similar to that of APOE. We present converging evidence, suggesting that the rare variant p.P522R in PLCG2 might reduce the effect of amyloidosis upon tau pathology and cognitive decline. Therapies targeting the druggable enzyme PLCG2 [17, 68] might, thus, provide a novel therapeutic approach with the potential for disease modification in AD might as well buffer the deleterious effect of APOE-ε4.
References
Abdelnour C, van Steenoven I, Londos E et al (2016) Alzheimer’s disease cerebrospinal fluid biomarkers predict cognitive decline in lewy body dementia. Mov Disord 31(8):1203–1208
Abner EL, Kryscio RJ, Schmitt FA et al (2017) Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 81(4):549–559
Arboleda-Velasquez JF, Lopera F, O’Hare M et al (2019) Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med 25(11):1680–1683
Asai H, Ikezu S, Tsunoda S et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18(11):1584–1593
Bhaskar K, Konerth M, Kokiko-Cochran ON et al (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68(1):19–31
Boyle PA, Wilson RS, Yu L et al (2013) Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 74(3):478–489
Braak H, Thal DR, Ghebremedhin E et al (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70(11):960–969
Chae JJ, Park YH, Park C et al (2015) Brief report: connecting two pathways through Ca2+ signaling: NLRP3 inflammasome activation induced by a hypermorphic PLCG2 mutation. Arthritis Rheumatol 67(2):563–567
Cohen G, Makranz C, Spira M et al (2006) Non-PKC DAG/Phorbol-Ester receptor (s) inhibit complement receptor-3 and nPKC inhibit scavenger receptor-AI/II-mediated myelin phagocytosis but cPKC, PI3k, and PLCγ activate myelin phagocytosis by both. Glia 53(5):538–550
Conway OJ, Carrasquillo MM, Wang X et al (2018) ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener 13(1):53
Curran PJ, Hussong AM (2009) Integrative data analysis: the simultaneous analysis of multiple data sets. Psychol Methods 14(2):81
Dagher NN, Najafi AR, Kayala KMN et al (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflamm 12(1):139
Dalmasso MC, Brusco LI, Olivar N et al (2019) Transethnic meta-analysis of rare coding variants in PLCG2, ABI3, and TREM2 supports their general contribution to Alzheimer’s disease. Transl Psychiatry 9(1):55
Deczkowska A, Keren-Shaul H, Weiner A et al (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173(5):1073–1081
Enders CK (2010) Applied missing data analysis. Guilford Press, New York
Ferrari R, Wang Y, Vandrovcova J et al (2017) Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry 88(2):152–164
Finan C, Gaulton A, Kruger FA et al (2017) The druggable genome and support for target identification and validation in drug development. Sci Transl Med 9(383):eaag1166
Fjell AM, McEvoy L, Holland D et al (2014) What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog Neurobiol 117:20–40
Friedman BA, Srinivasan K, Ayalon G et al (2018) Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 22(3):832–847
Guerreiro RJ, Lohmann E, Brás JM et al (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia–like syndrome without bone involvement. JAMA Neurol 70(1):78–84
Hammond TR, Marsh SE, Stevens B (2019) Immune signaling in neurodegeneration. Immunity 50(4):955–974
Haukedal H, Freude K (2019) Implications of microglia in amyotrophic lateral sclerosis and frontotemporal dementia. J Mol Biol 431(9):1818–1829
Hayat SA, Luben R, Dalzell N et al (2018) Understanding the relationship between cognition and death: a within cohort examination of cognitive measures and mortality. Eur J Epidemiol 33(11):1049–1062
Heckman MG, Kasanuki K, Brennan RR et al (2019) Association of MAPT H1 subhaplotypes with neuropathology of lewy body disease. Mov Disord 34:1325–1332
Herrup K (2010) Reimagining Alzheimer’s disease—an age-based hypothesis. J Neurosci 30(50):16755–16762
Hohman TJ, Tommet D, Marks S et al (2017) Evaluating Alzheimer’s disease biomarkers as mediators of age-related cognitive decline. Neurobiol Aging 58:120–128
Hong S, Beja-Glasser VF, Nfonoyim BM et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352(6286):712–716
Ising C, Venegas C, Zhang S et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575(7784):669–673
Jack R Jr, Clifford, Bennett DA, Blennow K et al (2018) NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14(4):535–562
Jack CR Jr, Clifford R, Knopman DS, Jagust WJ et al (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12(2):207–216
Jagust W (2013) Vulnerable neural systems and the borderland of brain aging and neurodegeneration. Neuron 77(2):219–234
Jansen WJ, Ossenkoppele R, Knol DL et al (2015) Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. Jama 313(19):1924–1938
Jansen IE, Savage JE, Watanabe K et al (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51:404–413
Jay TR, von Saucken VE, Landreth GE (2017) TREM2 in neurodegenerative diseases. Mol Neurodegener 12(1):56
Jones L, Lambert J-C, Wang L-S et al (2015) Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement 11(6):658–671
Jun G, Ibrahim-Verbaas CA, Vronskaya M et al (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21(1):108
Kamb A, Harper S, Stefansson K (2013) Human genetics as a foundation for innovative drug development. Nat Biotechnol 31(11):975
Karran E, Mercken M, de Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10(9):698
Keren-Shaul H, Spinrad A, Weiner A et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169(7):1276–1290
Koller M, Stahel WA (2017) Nonsingular subsampling for regression S estimators with categorical predictors. Comput Stat 32(2):631–646
Kunkle BW, Grenier-Boley B, Sims R et al (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51(3):414–430
Lacour A, Espinosa A, Louwersheimer E et al (2017) Genome-wide significant risk factors for Alzheimer’s disease: role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol Psychiatry 22(1):153
Lall D, Baloh RH (2017) Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest 127(9):3250–3258
Lee CD, Daggett A, Gu X et al (2018) Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97(5):1032–1048
Liao Y, Wang J, Jaehnig EJ et al (2019) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47(W1):W100–W205
Lill CM, Rengmark A, Pihlstrøm L et al (2015) The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement 11(12):1407–1416
Magno L, Lessard CB, Martins M et al (2019) Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther 11(1):16
Makranz C, Cohen G, Baron A et al (2004) Phosphatidylinositol 3-kinase, phosphoinositide-specific phospholipase-Cγ and protein kinase-C signal myelin phagocytosis mediated by complement receptor-3 alone and combined with scavenger receptor-AI/II in macrophages. Neurobiol Dis 15(2):279–286
Mathys H, Davila-Velderrain J, Peng Z et al (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570(7761):332–337
Muthén LK, Muthén BO (1998–2012). Mplus user’s guide, 7th Edn. Muthén & Muthén, Los Angeles, CA
Olmos-Alonso A, Schetters STT, Sri S et al (2016) Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139(3):891–907
Ombrello MJ, Remmers EF, Sun G et al (2012) Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 366(4):330–338
Ossenkoppele R, Jansen WJ, Rabinovici GD et al (2015) Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. Jama 313(19):1939–1950
Peng Q, Malhotra S, Torchia JA et al (2010) TREM2-and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 3(122):ra38–ra38
Plenge RM, Scolnick EM, Altshuler D (2013) Validating therapeutic targets through human genetics. Nat Rev Drug Discov 12(8):581
Proust-Lima C, Dartigues J-F, Jacqmin-Gadda H (2011) Misuse of the linear mixed model when evaluating risk factors of cognitive decline. Am J Epidemiol 174(9):1077–1088
Proust-Lima C, Philipps V, Liquet B (2017) Estimation of extended mixed models using latent classes and latent processes: The R package lcmm. J Stat Softw 78(2):56. https://doi.org/10.18637/jss.v078.i02
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org
Raghavan N, Samtani MN, Farnum M et al (2013) The ADAS-Cog revisited: novel composite scales based on ADAS-Cog to improve efficiency in MCI and early AD trials. Alzheimers Dement 9(1):S21–S31
Rauchmann B-S, Schneider-Axmann T, Alexopoulos P et al (2019) CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol Aging 74:182–190
Rayaprolu S, Mullen B, Baker M et al (2013) TREM2 in neurodegeneration: evidence for association of the p R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener 8(1):19
Roberts R, Knopman DS (2013) Classification and epidemiology of MCI. Clin Geriatr Med 29(4):753–772
Roberts RO, Aakre JA, Kremers WK et al (2018) Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population-based setting. JAMA Neurol 75(8):970–979
Rongve A, Witoelar A, Ruiz A et al (2019) GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci Rep 9(1):7013
Schulze-Luehrmann J, Ghosh S (2006) Antigen-receptor signaling to nuclear factor κB. Immunity 25(5):701–715
Shen M-Y, Hsiao G, Fong T-H et al (2008) Expression of amyloid beta peptide in human platelets: pivotal role of the phospholipase Cγ2-protein kinase C pathway in platelet activation. Pharmacol Res 57(2):151–158
Shi Y, Holtzman DM (2018) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18(12):759–772
Sims R, van der Lee SJ, Naj AC et al (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49(9):1373
Song WM, Joshita S, Zhou Y et al (2018) Humanized TREM2 mice reveal microglia-intrinsic and-extrinsic effects of R47H polymorphism. J Exp Med 215(3):745–760
Sosna J, Philipp S, Albay R et al (2018) Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener 13(1):11
Spangenberg EE, Lee RJ, Najafi AR et al (2016) Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139(4):1265–1281
Spangenberg E, Severson PL, Hohsfield LA et al (2019) Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10(1):1–21
Stancu I-C, Cremers N, Vanrusselt H et al (2019) Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137(4):599–617
Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G et al (2019) Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener 14(1):1
Szklarczyk D, Gable AL, Lyon D et al (2018) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613
Terry RD, Masliah E, Salmon DP et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580
van Dam S, Cordeiro R, Craig T et al (2012) GeneFriends: an online co-expression analysis tool to identify novel gene targets for aging and complex diseases. BMC Genom 13(1):535
van der Lee SJ, Conway OJ, Jansen I et al (2019) A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta neuropathol 138:1–14
Venegas C, Kumar S, Franklin BS et al (2017) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552(7685):355–361
Waegaert R, Dirrig-Grosch S, Parisot F et al (2020) Longitudinal transcriptomic analysis of altered pathways in a CHMP2Bintron5-based model of ALS-FTD. Neurobiol Dis 136:104710
Wood SN (2003) Thin plate regression splines. J R Stat Soc Series B Stat Methodol 65(1):95–114
Wood SN (2011) Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J R Stat Soc Series B Stat Methodol 73(1):3–36
Woollacott IOC, Nicholas JM, Heslegrave A et al (2018) Cerebrospinal fluid soluble TREM2 levels in frontotemporal dementia differ by genetic and pathological subgroup. Alzheimers Res Ther 10(1):79
Yeh FL, Wang Y, Tom I et al (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91(2):328–340
Yoshiyama Y, Higuchi M, Zhang B et al (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53(3):337–351
Zanoni I, Ostuni R, Marek LR et al (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147(4):868–880
Zanoni I, Tan Y, Di Gioia M et al (2017) By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity 47(4):697–709
Zhang B, Gaiteri C, Bodea L-G et al (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153(3):707–720
Zhou HH, Singh V, Johnson SC et al (2018) Statistical tests and identifiability conditions for pooling and analyzing multisite datasets. Proc Natl Acad Sci 115(7):1481–1486
Acknowledgements
Open Access funding provided by Projekt DEAL. This publication was funded in part by the German Federal Ministry of Education and Research (BMBF) (grants KND: 01GI0102, 01GI0420, 01GI0422, 01GI0423, 01GI0429, 01GI0431, 01GI0433, 01GI0434; grants KNDD: 01GI0710, 01GI0711, 01GI0712, 01GI0713, 01GI0714, 01GI0715, 01GI0716, 01ET1006B). Analyses were also funded by the German Federal Ministry of Education and Research (BMBF 01EA1410A) within the project “Diet-Body-Brain: from epidemiology to evidence-based communication”. Part of the work was funded by the JPND EADB grant (German Federal Ministry of Education and Research (BMBF) grant: 01ED1619A). Part of the analysis was funded by the German Research Foundation (DFG) grant: RA 1971/6-1 to Alfredo Ramirez. Research of the Alzheimer Center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. The clinical database structure was developed with funding from Stichting Dioraphte. Genotyping of the Dutch case–control samples was performed in the context of EADB (European Alzheimer DNA biobank) funded by the JPco-fuND FP-829-029 (ZonMW project number 733051061). Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; ElanPharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research and Development, LLC.; Johnson and Johnson Pharmaceutical Research and Development LLC.; Lumosity; Lundbeck; Merck and Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California. The Three-City Study genotyping and analysis was funded by the GENMED Labex, the Joint Programming Initiative on Neurodegenerative Diseases Research (JPND; PERADES project), the Institute Pasteur de Lille, the University of Lille, and the Nord-Pas de Calais Regional Council. This work also benefited from the Lille Métropole Communauté Urbaine Council, the French government’s LABEX DISTALZ program (development of innovative strategies for a transdisciplinary approach to Alzheimer’s disease). In addition, the Three-City Study was performed as part of a collaboration between the Institut National de la Santé et de la Recherche Médicale (Inserm), the Victor Segalen Bordeaux II University and Sanofi-Synthélabo. The Fondation pour la Recherche Médicale funded the preparation and initiation of the study. Additional funding for the 3C Study was also obtained from the Caisse Nationale Maladie des Travailleurs Salariés, Direction Générale de la Santé, MGEN, Institut de la Longévité, Agence Française de Sécurité Sanitaire des Produits de Santé, the Aquitaine and Bourgogne Regional Councils, Fondation de France and the joint French Ministry of Research/INSERM “Cohortes et collections de données biologiques” programme. Lille Génopôle received an unconditional grant from Eisai. Fundacio ACE cohort receives support from the Innovative Medicines Initiative 2 Joint Undertaking which receives support from the European Union’s Horizon 2020 research and innovation programme (ADAPTED Grant No. 115975). A. Ruiz’s research is also supported by Instituto de Salud Carlos III (ISCIII) grants PI13/02434, PI16/01861, and PI19/01301. Acción Estratégica en Salud, integrated in the Spanish National R + D + I Plan and financed by ISCIII-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER-“Una manera de Hacer Europa”), by Fundación bancaria “La Caixa” and Grifols SA (GR@ACE project). For the Longitudinal Aging Study Amsterdam (LASA) supports was largely obtained from a grant from the Netherlands Ministry of Health, Welfare and Sports, Directorate of Long-Term Care. The data collection in 2012–2013 was financially supported by the Netherlands Organization for Scientific Research (NWO) in the framework of the project “New Cohorts of young old in the twenty-first century” (File Number 480-10-014). Genotyping using Axiom-NL array was financially supported by EMGO+ Research Institute. PL was supported by the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement no 115372, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies in kind contribution. JW is supported by an Ilídio Pinho professorship and iBiMED (UID/BIM/04501/2013), at the University of Aveiro, Portugal. JP was supported by a grant from the Swiss National Science Foundation (320030L_141179).
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data, but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: https://adni.loni.usc.edu/wp-2content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Addresses: Luca Kleineidam, Ilker Karaca, Michael T. Heneka, Wolfgang Maier, Anja Schneider, Michael Wagner—Universitätklinikum Bonn, Klinik für Neurodegenerative Erkankungen und Gerontopsychiatrie, Sigmund-Freud-Straße 25, 53127 Bonn, Germany; Vincent Chouraki, Phillipe Amouyel, Jean-Charles Lambert—Univ. Lille, Inserm, CHU Lille, Institut Pasteur de Lille, U1167-RID-AGE-Facteurs de risque et déterminants moléculaires des maladies liées au vieillissement, F-59000 Lille 59019 France; Tomasz Próchnicki, Eicke Latz—Institute of Innate Immunity, Universitätklinikum Bonn, Sigmund-Freud-Straße 25, 53127 Bonn, Germany; Sven J. van der Lee, Iris E. Jansen, Marc Hulsman, Philip Scheltens, Wiesje M van der Flier, Henne Holstege—Alzheimer Center Amsterdam, Department of Neurology, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam UMC, De Boelelaan 1117, 1081 HV, Amsterdam, The Netherlands; Laura Madrid-Márquez, Antonio González-Pérez, Mª Eugenia Sáez—Andalusian Bioinformatics Research Centre (CAEBi), Río de la Plata 2, Bajo 4, 41013 Seville, Spain; Holger Wagner-Thelen, Pamela V. Martino Adami, Frank Jessen, Alfredo Ramirez—Universitätklinik Köln, Klinik für Psychiatrie und Psychtherapie, Kerpener Strasse 62, 50937 Köln, Germany; Leonie Weinhold, Matthias Schmid—Institute of Medical Biometry, Informatics and Epidemiology (IMBIE), Universitätklinikum Bonn, Sigmund-Freud-Str. 25, 53127, Bonn, Germany; Steffen Wolfsgruber, Frederic Brosseron—German Center for Neurodegenerative Diseases (DZNE), Sigmund-Freud-Straße 27, 53127 Bonn, Germany; Anne Boland, Jean-Francois Deleuze—Centre National de Recherche en Génomique Humaine, CP 5721, 2 rue Gaston Crémieux, 91057 EVRY Cedex; Piotr Lewczuk, Johannes Kornhuber—Universitätsklinikum Erlangen, Psychiatrische und Psychotherapeutische Klinik, Schwabachanlage 6, 91054 Erlangen, Germany; Julius Popp—Department of Psychiatry, Lausanne University Hospital, Route du Mont, 1008, Prilly, Switzerland; Oliver Peters—Charité-Universitätsmedizin Berlin, Klinik für Psychiatrie und Psychtherapie, Hindenburgdamm 30, 12203 Berlin, Germany; Claudine Berr—Inserm U1061, Hôpital La Colombière, 39 av. Charles Flahault, B.P. 34493, 34093 Montpellier Cedex 5, France; Reinhard Heun—Universtitätklinikum Bonn, Klinik für Psychiatrie und Psychotherapie, Sigmund-Freud-Straße 25, 53127 Bonn, Germany; Lutz Frölich—Zentralinstitut für Seelische Gesundheit (ZI), Quadrat J5, 68159 Mannheim, Germany; Christophe Tzourio, Jean-François Dartigues—Bordeaux Population Health (BPH Center), INSERM U1219, Université de Bordeaux, 146 rue Léo Saignat, 33076 Bordeaux cedex; Michael Hüll—Zentrum für Psychiatrie Emmendingen, Klinik für Alterspsychiatrie- und psychotherapie, Neubronnstr. 25, 79312 Emmendingen, Germany; Ana Espinosa, Isabel Hernández, Itziar de Rojas, Adelina Orellana, Sergi Valero, Agustin Ruiz, Lluis Tarraga, Merce Boada—Research Center and Memory Clinic. Fundació ACE. Institut Català de Neurociències Aplicades-Universitat Internacional de Catalunya – Barcelona, Gran Via de Carles IIII. 85bis. 08028, Barcelona, Spain; Najada Stringa, Natasja M. van Schoor, Martijn Huisman—Amsterdam Public Health Research Institute, Department of Epidemiology and Biostatistics, Amsterdam UMC – Vrije Universiteit Amsterdam, De Boelelaan 1089A, 1081 HV, Amsterdam, the Netherlands; Eckart Rüther, Jens Wiltfang—Universitätmedizin Göttingen, Klinik für Psychiatrie und Psychotherapie, Von-Siebold-Straße 5, 37075 Göttingen, Germany; Martin Scherer—Institut für Allgemeinmedizin, Universitätsklinikum Hamburg-Eppendorf, Martinistraße 52, 20246 Hamburg; Steffi Riedel-Heller—Institut für Sozialmedizin, Arbeitsmedizin und Public Health (ISAP), Medizinische Fakultät, Universität Leipzig, Ph.-Rosenthal-Str. 55, 04103 Leipzig, Germany.
Author information
Authors and Affiliations
Consortia
Contributions
LK designed and conceptualized the study, analyzed and interpreted the data, and drafted the manuscript. VC contributed to the design of the study, analyzed the longitudinal data from the 3C study, and interpreted the data. SvdL contributed to the design of the study, analyzed the longitudinal data from the LASA cohort, and interpreted the data. LM-M contributed to the design, acquisition, and analysis of gene co-expression data, and interpreted the data. HWT contributed to the analysis of CSF biomarkers, data acquisition, and interpretation. TP, IK, LW, SW, MS, and AG-P contributed to the design of the study and interpreted the data. AB, PVMA, PL, JP, FB, IJ, MH, AE, IH, IR, AO, SV, NS, NMS, PA, J-FDe, and AS contributed to the data acquisition. JK, CB, LF, OP, CT, J-FDa, MHül, MHui, PS, ER, JW, RH, LT, MS, SR-H, MTH, MB, WM, and WMF contributed to the design of the study and data acquisition. MW, FJ, J-CL, HH, MES, and EL contributed the design, acquisition, and interpretation of the data. AR designed and conceptualized the study, contributed to the acquisition of data, interpreted the data, conducted the gene co-expression analyses, and contributed to the writing of the manuscript. AR supervised the study, wrote the manuscript, designed and conceptualized the study, and acquired and interpreted the data. All authors critically revised the manuscript for important intellectual content and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors report no conflict of interest. Piotr Lewczuk received a consultation and/or lecture honoraria from IBL International, Fujirebio Europe, AJ Roboscreen, and Roche. Jens Wiltfang received honoraria for consulting activities, lectures or advisory board participation from Pfizer, Eli Lilly, Hoffmann-La-Roche, MSD Sharp + Dome, Janssen-Cilag GmbH, Immungenetics AG, Boehringer Ingelheim. Lutz Frölich received honoraria for consulting activities, lectures, or advisory board participation from Allergan, Eli Lilly, Avraham Pharmaceuticals, Axon Neuroscience, Axovant, Biogen, Boehringer Ingelheim, Eisai, Functional Neuromodulation, Lundbeck, MerckSharpe and Dohme, Novartis, Pfizer, Pharnext, Roche, Schwabe Pharma; he served on Data and Safety Monitoring boards or endpoint committees with Avraham Pharmaceuticals, Axon Neuroscience, Forschungszentrum Jülich, Novartis, Pharmatropix. Julius Popp received honoraria for consulting activities, lectures or advisory board participation from Fujirebio Europe, Ono Pharma, Eli Lilly, and Nestlé Institute of Health Sciences. Agustin Ruiz reports consulting Fees: Landsteiner Genmed, Grifols, Araclon biotech. And Lecture Fees: Araclon Biotech.PA reports personal fees from Servier, Total, Genoscreen, Takeda, and Foundation Alzheimer. Oliver Peters received research funding, consultancy fees, or speech honoraria from Axovant, Biogen, Genentech, Lilly, Lundbeck, Novartis, Pharmatrophix, Piramal, Probiodrug, Roche, Takeda, and TauRx Pharmaceuticals. Merce Boada receives fees or has received for consulting from Lab. Servier, Roche, Lilly, Avid, Bayer, Elan, Janssen, Neuroptix, and Sanofi. She receives or has received fees for lectures from Lilly, Nutricia, Roche, Schwabe, Araclon, Esteve, Grifols, Janssen, Novartis, Piramal, Pfizer-Wyett, and Servier. She receives fees for being part of the Advisory Board of Lilly and Schwabe. She reports grants/research funding from Abbvie, Araclon, Biogen Research Limited, Bioiberica, Grifols, Lilly, S.A, Merck Sharp and Dohme, Kyowa Hakko Kirin, Laboratorios Servier, Nutricia SRL, Oryzon Genomics, Piramal Imaging Limited, Roche Pharma SA, and Schwabe Farma iberica SLU, all outside the submitted work. She has not received personal compensations from these organizations.
Ethical approval
All procedures performed in studies involving human participants were following the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. For this retrospective study, all patients’ clinical, genetic, and biomarker data were already available at the time point of this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The members of the Alzheimer’s Disease Neuroimaging Initiative (ADNI) group are listed in Acknowledgements section. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kleineidam, L., Chouraki, V., Próchnicki, T. et al. PLCG2 protective variant p.P522R modulates tau pathology and disease progression in patients with mild cognitive impairment. Acta Neuropathol 139, 1025–1044 (2020). https://doi.org/10.1007/s00401-020-02138-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-020-02138-6