
Abstract
Purpose
Human melanoma is a highly aggressive incurable cancer due to intrinsic cellular resistance to apoptosis, reprogramming, proliferation and survival during tumour progression. Sulforaphane (SFN), an isothiocyanate found in cruciferous vegetables, plays a role in carcinogenesis in many cancer types. However, the cytotoxic molecular mechanisms and gene expression profiles promoted by SFN in human melanoma remain unknown.
Methods
Three different cell lines were used: two human melanoma A375 and 501MEL and human epidermal melanocytes (HEMa). Cell viability and proliferation, cell cycle analysis, cell migration and invasion and protein expression and phosphorylation status of Akt and p53 upon SFN treatment were determined. RNA-seq of A375 was performed at different time points after SFN treatment.
Results
We demonstrated that SFN strongly decreased cell viability and proliferation, induced G2/M cell cycle arrest, promoted apoptosis through the activation of caspases 3, 8, 9 and hampered migration and invasion abilities in the melanoma cell lines. Remarkably, HEMa cells were not affected by SFN treatment. Transcriptomic analysis revealed regulation of genes involved in response to stress, apoptosis/cell death and metabolic processes. SFN upregulated the expression of pro-apoptotic genes, such as p53, BAX, PUMA, FAS and MDM2; promoted cell cycle inhibition and growth arrest by upregulating EGR1, GADD45B, ATF3 and CDKN1A; and simultaneously acted as a potent inhibitor of genotoxicity by launching the stress-inducible protein network (HMOX1, HSPA1A, HSPA6, SOD1).
Conclusion
Overall, the data show that SFN cytotoxicity in melanoma derives from complex and concurrent mechanisms during carcinogenesis, which makes it a promising cancer prevention agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melanoma continues to be the skin disease causing the highest mortality due to its propensity to metastasize. In fact, it is responsible for more than 75% of skin cancer deaths. Epidemiological studies have shown that the incidence of melanoma increases at a faster rate than that of any other cancer worldwide [1, 2]. Unsatisfactory results with single-agent or combination chemotherapy schemes underscore the need for the application of different strategies to cure melanoma. Treatment options have rapidly expanded in the past 5 years with the introduction of targeted therapy (BRAF and MEK inhibitors) and immune checkpoint blockades [3,4,5]. Interestingly, many naturally occurring dietary compounds found in fruits and vegetables consumed daily (e.g., curcumin, indole-3-carbinol, brassinin, sulforaphane (SFN), epigallocatechin-3-gallate, lycopene and quercetin) have been found to inhibit one or more pathways that contribute to malignant transformation and possess cancer-preventive properties against different types of tumours, including melanoma [6, 7]. SFN (R-1-isothiocyanato-4-methylsulfinyl butane), an isothiocyanate found especially in broccoli sprouts, Chinese kale, cabbage and watercress, can prevent or delay preneoplastic lesions as well as act as a therapeutic agent in tumour cell cultures and carcinogen-induced and genetic animal cancer models [8,9,10]. SFN induces phase II carcinogen detoxification enzymes, such as NAD(P)H:quinone oxidoreductase I [11] and heme oxygenase-1 (HMOX-1), via the ARE–NRF2 pathway [12], thereby allowing a different array of electrophilic and oxidative toxicants to be eliminated before they damage critical cellular macromolecules [13, 14]. Studies have shown that SFN induces apoptosis and inhibits the progression and metastasis of many cancers [15,16,17]. Despite a growing number of studies describing the chemopreventive and chemotherapeutic properties of SFN in cancer [18,19,20], little is known about the molecular mechanisms underlying the antitumour effects of SFN in human melanoma cells. Some previous reports have shown that SFN could be effective in treating melanoma, but virtually all promising results have thus far been obtained from animal studies. Specifically, SFN was shown to inhibit the metastasis of B16F-10 mouse melanoma cells both in vivo and in vitro [21]. In a murine model, SFN was shown to induce a significant reduction in the expression of cell proliferation markers (metalloproteinases 2 and 9), leading to an increase in the survival rate of animals bearing metastatic tumours, probably via stimulating a cell-mediated immune response [22, 23]. Moreover, SFN-induced apoptosis in B16F-10 has been shown to be associated with the activation of caspases 3 and 9, BAX and p53 and the downregulation of NF-kB [24].
In this study, we analysed the tumour cytotoxic effects of SFN on human primary melanoma cells (A375), human metastatic melanoma cells (501MEL) and control human epidermal melanocytes (HEMa) using multiple approaches. We also aimed to decipher the A375 cell transcriptome following exposure to SFN using RNA sequencing (RNA-Seq) technology. To the best of our knowledge, the present study is the first to demonstrate the effects of SFN on HEMa cells and on the expression profiles of genes involved in SFN-induced apoptosis, cell cycle arrest, migration and invasion in melanoma.
Materials and methods
Reagents
DL-SFN, dimethyl sulfoxide (DMSO), propidium iodide (PI), Ponceau S and collagenase were purchased from Sigma-Aldrich. Trypan blue, Dulbecco’s modified Eagle medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640, l-glutamine 100×, foetal bovine serum (FBS), penicillin–streptomycin, 0.25% trypsin–EDTA, Medium 254, human melanocyte growth supplement-2 PMA-Free (HMGS-2), RNAse-A, DEPC–water, collagen I bovine, Hank’s balanced salt solution (HBSS), trypsin neutralizer solution and 0.025% trypsin/EDTA solution were obtained from Gibco Life Technologies. Other chemical supplements and compounds were purchased from several companies as follows: CellTiter-Glo luminescent cell viability assay (Promega), turbo DNA free (Ambion, Life Technologies), FITC Annexin V apoptosis detection kit I (BD Pharmingen), TRIzol reagent (Ambion, Life Technologies), methylene blue (Merck), BCA protein assay kit and NP-40 (Pierce, Thermo scientific), ECL Prime Western blotting detection reagents (Amersham), stripping buffer stripAblot and Tween 20 (EuroClone), complete protease inhibitor cocktail (Roche), PageRule prestained protein ladder (Fermentas, part of Thermo Fisher Scientific), superscript III Reverse Transcriptase (Life Technologies) and iQ™ SYBR® green supermix (Bio-Rad).
Cell lines and treatments
A375 is a primary human malignant melanoma cell line that was obtained from American Type Culture Collection (ATCC). The cells were maintained in DMEM medium containing 10% FBS, 1% l-glutamine and 1% penicillin–streptomycin (10,000 units/ml). The 501MEL cell line was obtained from surgically removed tumours from patients with melanoma at the National Cancer Institute in Milan, Italy, and they were maintained in RPMI 1640 medium supplemented with 10% FBS, 1% l-glutamine (200 mM) and 1% penicillin–streptomycin (10,000 units/ml). Human epidermal melanocyte (HEMa) is a cell line isolated from lightly pigmented (LP) adult skin purchased from Invitrogen (Life Technologies). The cells were propagated in Medium 254 supplemented with HMGS-2 and incubated at 37 °C with 5% CO2 until they reached approximately 70% confluence. They were then treated with different concentrations of SFN dissolved in dimethyl sulfoxide (DMSO). The same DMSO concentration used to dilute the SFN was utilized as a negative control (untreated).
Cell viability and morphology analysis
Cells were seeded at a density of 3 × 105 cells/well onto six-well plates, and after 24 h treated with SFN at final concentrations of 1, 2 and 5 μg/ml. The control cells were treated with the same concentration of DMSO (0.001%). The cell lines were photographed under a phase-contrast microscope at 10× magnification (Nikon Eclipse) to observe morphological changes. For the viability assay, A375 and 501MEL cells were plated at a density of 3 × 105 cells/well and treated with SFN for 24 and 48 h. Dead cells were stained with 0.4% trypan blue dye. In addition, cell viability was evaluated using the Cell Titer-Glo assay. The cells were plated at a density of 5 × 103 cells/well in 100 μL of medium on 96-well white plates (Cellstar, Greiner) and treated with different concentrations of SFN for 24 and 48 h. The cells were subsequently lysed using Cell Titer-Glo luminescent reagent following the manufacturer’s protocol, and the results are expressed as a percentage based on the ratio of the absorbance of treated cells to that of the control cells (100%).
Cell cycle analysis
Cells were plated at a density of 3 × 105 cells/well on six-well plates and treated with SFN for 24 and 48 h. After incubation, the cells were trypsinized, washed with ice-cold phosphate-buffered saline (PBS) and fixed in 70% ethanol overnight. Next, the cells were re-suspended in PBS containing RNAse-A (30 mg/mL) and incubated at 4 °C for 5 min. After FACS buffer (PBS + 2% FBS) and PI (1 mg/mL) were added, the cells were incubated at 4 °C for 30 min and then immediately analysed for DNA content using the FACSCalibur flow cytometer with CellQuest software (Becton–Dickinson). In total, 20,000 events per sample were recorded, and experiments were performed in triplicate. Apoptotic cells with hypodiploid DNA content were measured by quantifying the sub-G1 peak in the cell cycle pattern.
Apoptotic cell death assay
Cells were plated at a density of 3 × 105 cells/well on six-well plates, and 24 h later treated with SFN for 24 and 48 h. Floating cells were collected, combined with the trypsinized adherent cells and stained with Annexin V and PI according to the manufacturer’s recommended protocol. The samples were analysed by FACS within 1 h. For this assay, 20,000 events were counted. The analyses were performed in triplicate.
Western blotting analysis
Cells were washed with PBS and then lysed with lysis buffer [1 M Tris, 2.5 M NaCl, 10% glycerol, 0.5 M glycerophosphate, 1% Tween 20, 0.5% NP-40 and a complete protease inhibitor tablet (Roche)] for 20 min on ice. The cell lysates were separated by 10 or 12% denaturing SDS-PAGE. The proteins were then transferred to nitrocellulose membranes (Whatman), blocked with PBS containing 5% milk and 0.05% Tween and incubated with specific primary antibodies overnight. After being washed with PBS containing 0.05% Tween, the blots were incubated with peroxidase-conjugated secondary antibodies labelled with horseradish peroxidase (HRP) (Sigma, Italy) and developed using ECL according to the manufacturer’s instructions. Rabbit polyclonal anti-GAPDH (ab 9485) and anti-procaspase-9 (ab 32068) were purchased from Abcam (Italy) and diluted 1:500. Rabbit anti-PARP (9542) and anti-p53 (Ser15-9286) were purchased from Cell Signaling Technology (Italy) and diluted 1:500. Anti-cleaved caspase-3 (9661), also purchased from Cell Signaling Technology, was diluted 1:1000. Rabbit anti-Bcl2 (ab 59348) and rabbit polyclonal anti-cyclin B1 (4138), purchased from Santa Cruz Biotechnology (Italy), were diluted 1:500. Rabbit anti-procaspase-8 (ab 49853) was purchased from Sigma and diluted 1:1000.
Scratch wound healing assay
A375 cells were seeded in six-well plates at a concentration of 3 × 105 cells/well in complete medium and incubated for 24 h. After 24 h, the cells were treated with mitomycin (15 µg/ml, Sigma) to prevent cell proliferation for 45 min at 37 °C. Next, the medium was removed, and a vertical scratch was created in the centre of the well using a sterile tip. The cells were subsequently washed with PBS and treated with 2 µg/ml SFN. The groove was monitored and photographed both immediately and 24 h after the scratch was created using a phase-contrast microscope (Evos, Zeiss) with 4× magnification. The migration was evaluated as the residual area of the groove, and three different fields were counted for each condition. The wound area was calculated by tracing a line along the border of the wound using ImageJ software, and the percentage of wound closure was calculated using the following equation: [Wound area (0 h) − Wound area (X h)] × 100/Wound area (0 h) = %Wound closure.
Invasion assay with collagen
The collagen matrix was generated from bovine type I collagen at a final concentration of 1 mg/ml according to the manufacturer’s protocol. Collagen was plated immediately onto 24-well plates and then incubated at 37 °C with 95% humidity for 30 min. After collagen polymerization, cells were seeded at a density of 3 × 104 cells/well and treated with 2 µg/ml SFN. After treatment, viability and invasion were evaluated by analysing and counting the cells in the supernatant, the adherent cells harvested using PBS/EDTA (5 min at 37 °C) from the upper collagen surface and the cells remaining in the collagen matrix after the adherent cells were removed. Collagen was fixed with paraformaldehyde (4%), and migrated cell nuclei were stained with blue methylene (1:10). The samples were analysed using a microscope (Olympus BH-2) to count cell numbers (n = 6 independent fields for each condition), and cell images were captured with an Olympus U-PMTVC camera. The analyses were performed in triplicate for statistical evaluation.
Quantitative real-time PCR (qPCR)
cDNA was synthesized using Superscript III Reverse Transcriptase (Sigma) according to the manufacturer’s instructions. qPCR was performed in 96-well plates using iQ™ SYBR Green Supermix (Bio-Rad) and an I-cycler iQ Real-Time PCR instrument (Bio-Rad). Measurements were performed in triplicate with a variability <0.5 Ct. mRNA expression was analysed by normalizing to that of the housekeeping gene GADPH. Primers were designed with PERL primer software using NCBI EntrezGene reference sequences as templates and synthesized by Sigma. The primers used are listed in Supplementary Table S4.
RNA sequencing (RNA-seq) and data analysis
A375 cells were seeded at a density of 3 × 105 cells/well on six-well plates and treated with SFN for 2, 6, 24 and 48 h. Total RNA was isolated using a standard TRIzol (Life Technologies) extraction protocol. Contaminating DNA was removed by Turbo DNase treatment (Life Technologies), and RNA was purified using the RNeasy Mini Kit (Qiagen). RNA quality was measured by the RNA 6000 Bioanalyzer Nano Kit (Agilent Technologies) according to the manufacturer’s instructions. Only samples with a minimum RIN score of 9.8 were used for further analyses. Double-stranded cDNA libraries were generated from 1 μg of total RNA using a TruSeq RNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions. Libraries were validated using an Agilent High-Sensitivity DNA Kit on an Agilent 2100 Bioanalyzer (Agilent Technologies) and quantified by qPCR using a KAPA Library Quantification Kit (Kapa Biosystems) on a StepOne Real-Time PCR system (Applied Biosystem) at 95 °C (5 min), followed by 35 cycles of 95 °C (30 s) and 60 °C (45 s). After being quantified, the libraries were normalized to a final concentration of 10 nM. The libraries (2 nM) were pooled at final concentrations of 9 and 7 pM and loaded into a HiSeq SR flow cell v3 (Illumina) using a cBot (Illumina) according to the manufacturer’s instructions. The loaded flow cell was sequenced on a HiSeq 1500 platform (Illumina, by PoloGGB) to perform a 100 bp single-read run following the manufacturer’s instructions. The images were processed using the Illumina RTA base calling pipeline, converted in FASTQ using Casava 1.8 software and evaluated for quality using FastQC (v 0.10.1). The reads were trimmed with Trimmomatic (v 0.32) and all reads with >Q20 were selected and aligned with STAR (v 2.3.0) to hg19/GRCh37 in the UCSC Genome Browser. Bam and GTF files were used to create a matrix associating all mapped read numbers to each sample and gene using Htseq. The obtained matrix was processed with the DeSeq R package to select for all differential expressed genes (DEGs) that had a false discovery rate (FDR) <10%. Expression differences were considered significant if their p value was <0.05 and their induction (or repression) ratio was ≥1.5. All graphs were produced with R software (v 3.0.0). Functional clustering was performed with DAVID 6.7 and the Web Gene Set Analysis Toolkit (WebGestalt) for enrichment analysis of the differentially expressed genes. DEGs were screened using enrichment analysis based on the hypergeometric distribution WebGestalt algorithm.
Constructing the protein interaction network
Using the online database Search Tool for the Retrieval of Interacting Genes (STRING) v. 9.1 ([25]; http://string-db.org), interactions between the DEGs were predicted. The interactions include direct (physical) and indirect (functional) associations derived from four sources: genomic context, high-throughput, co-expression and prior knowledge.
Statistical analysis
All experiments were performed at least three times independently. The data are expressed as the mean ± standard deviation (SD). Data were analysed by the paired two-tailed Student’s t test [p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***)]. The Dunnett’s test was used to assess the significance of the viability assay results. All statistical analyses were performed with Prism 5 Software.
Results
SFN decreased the viability and induced morphological changes in human melanoma cells
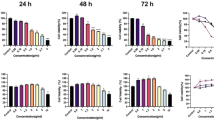
To assess the effects of SFN on cell viability, primary human melanoma A375, metastatic human melanoma 501MEL and HEMa cells were treated with increasing concentrations of SFN (1–5 µg/ml) for 24 and 48 h. SFN treatment significantly reduced the viability of both A375 and 501MEL cells in a dose- and time-dependent manner (Fig. 1a), though A375 cells were more sensitive to SFN treatment than 501MEL cells. In fact, SFN began to significantly reduce the viability of 501MEL cells only at the concentration of 2 µg/ml. After 48 h, 2 µg/ml SFN decreased the viability of the A375 cells by 64% compared to only 46% for the 501MEL cells. As a control, HEMa cells were treated with the same concentrations of SFN, and only the higher dose (5 µg/ml) significantly decreased their viability. These results were also confirmed by cell proliferation analysis using the trypan blue exclusion assay (Supplement Fig. S1a) and phosphorylated AKT (p-AKT) expression analysis (Supplement Fig. S1b). Trypan blue cell counting showed significantly reduced proliferation of both A375 and 501MEL cells at 24 and 48 h post-treatment. Moreover, high levels of p-AKT in both melanoma cell lines were significantly decreased after 24 h of SFN treatment.

SFN inhibits cell viability and induces morphological changes. a HEMa, A375, and 501MEL cells were treated with different doses of SFN for 24 or 48 h. Cell viability was determined by CellTiter-Glo assay. The bars represent the mean values of three experiments plus standard deviation; the significance level compared to the control was specified as *p < 0.05, **p < 0.01 and ***p < 0.001 using one-way ANOVA and the Dunnett’s multiple comparison test. b Representative phase-contrast microscopy (×10) of cell morphology with or without SFN (2 and 5 μg/ml) at 48 h. Scale bar 100 μm
To determine cell death due to reduced viability, cell morphology was observed in the confluent monolayer after treatment with 2 and 5 µg/ml SFN. Specifically, the A375 cells displayed increased size, irregular shape and membrane blebbing after 48 h (Fig. 1b). Morphological alterations were also observed in the 501MEL treated cells, as they retracted into a spherical shape and formed suspended clusters. In contrast to melanoma cells, HEMa cells did not exhibit any significant morphological alterations at the 2 µg/ml dose of SFN, and only high concentrations of SFN induced rounded melanocytes, irregular morphology and membrane blebbing. Because 2 µg/ml SFN had no inhibitory effect on HEMa cells but was extremely effective in both melanoma cells lines, this concentration was used for further analysis.
Sulforaphane induced cell cycle arrest and apoptosis
To further investigate the inhibitory effects of SFN on cell viability, we analysed cell cycle progression and apoptosis by both flow cytometry and Western blot. SFN exposure changed the cell cycle phase distribution in both melanoma cell lines, and, in agreement with the cell viability data, no changes were observed in the HEMa cells (Fig. 2a). A375 and 501MEL cells treated with SFN significantly accumulated in the G2/M phase, as up to 55 and 50% of cells were observed in this phase at 24 h post-treatment, respectively. These numbers shifted down to 40 and 45%, respectively, after 48 h (Fig. 2a). Conversely, the proportion of cells in the G0/G1 phase was markedly decreased in both cell lines, while the percentage of cells in the S phase remained stable.

SFN promotes cell cycle arrest and apoptosis. a Assessment of DNA content in A375, 501MEL and HEMa cells after 24 or 48 h of treatment with 2 μg/ml SFN. DNA content analysis was carried out by flow cytometry using propidium iodide staining. The percentages of A375, 501MEL and HEMa cells in the G0/G1, G2/M and S phases of the cell cycle with or without 2 μg/ml SFN are shown. b Representative plots of the cell cycle analysis histogram by flow cytometry before and after 24 h treatment are reported. The table indicates the percentage of cells with or without 2 µg/ml SFN in the sub-G1 phase. c Annexin V assay of cells at 24 and 48 h after 2 μg/ml SFN exposure. The graphs represent the mean ± SD of apoptotic (early and late) cells at 24 h post-treatment. d Western blot analysis of several proteins involved in the cell cycle and apoptosis in A375 and 501MEL cells after 24 or 48 h exposure to 2 µg/ml SFN. GAPDH was used as loading control. The molecular weight (MW) of the proteins (kDa) is reported. e The representative blots show the protein expression levels of the different proteins analysed and the bar graph represents the results from the photodensitometric analysis of the bands, using GADPH as an internal control. The data are presented as mean values ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 using a paired two-tailed Student’s t test
To evaluate whether SFN induces apoptosis in human melanoma cell lines, we used several experimental approaches. The assessment of cells in the sub-G1 phase using cell cycle analysis showed a time-dependent significant increase in the percentage of cell death after SFN exposure at 48 h in both melanoma cell lines, as up to 29 and 20% of A375 and 501MEL cells were dead, respectively (p = 0.0339 and p = 0.0023, respectively). In contrast, no significant effects were observed in the HEMa cells (Fig. 2b). To validate the pro-apoptotic effects of SFN, both the A375 and the 501MEL cells were analysed by flow cytometry using Annexin V–FITC. The percentage of total (early + late) apoptotic cells increased in a time-dependent manner. At 48 h post-treatment, the number of apoptotic cells reached 38 and 22% in the A375 and 501MEL cells, respectively (Fig. 2c). As previously shown, SFN exhibited a stronger apoptotic effect on the A375 cells than on the 501MEL cells. No effects were observed in the HEMa cells.
We further investigated the mechanisms underlying cell death induced by SFN by assessing proteins that play a crucial role in the cell cycle and apoptosis regulation. Immunoblotting on protein cell extracts revealed that SFN completely inhibited the expression of cyclin B1, a G2/M phase marker, in A375 cells at 48 h post-treatment, which agrees with the accumulation of cells in the G2/M phase. In the 501MEL cells, there was only a partial reduction (Fig. 2d, e). The levels of total p53 and phosphorylated p53 [p-p53 (Ser-15)] were strongly increased in a time-dependent manner in both melanoma cell lines. The levels of Bcl-2, an anti-apoptotic protein and a transcriptional target of p53, were also analysed. In accordance with p53 expression, Bcl-2 expression was decreased in both melanoma cell lines after SFN exposure (Fig. 2d, e). Moreover, to determine whether SFN-induced apoptosis in melanoma cells is caspase-dependent, caspase-8, caspase-9 and caspase-3 expression was assessed. Procaspase-9 was strongly decreased in a time-dependent manner in A375 cells after SFN treatment, suggesting a major role of the intrinsic apoptotic pathway. In contrast, no differences were detected in the 501MEL at any time point. Procaspase-8 was used to assess the extrinsic apoptotic pathway, and its levels were slightly reduced in 501MEL cells at 48 h post-treatment and slightly reduced in A375 cells at both 24 and 48 h post-treatment. Because of caspase-8 and caspase-9 activation, caspase-3 was completely cleaved in A375 cells at 48 h after SFN treatment, while caspase-3 cleavage started later and it was only partially cleaved after 48 h in 501MEL cells (Fig. 2d, e). Because caspase-3 is the most efficient processing enzyme for poly (ADP-ribose) polymerase (PARP), we also analysed its activation. SFN activated PARP cleavage in both melanoma cell lines. PARP was remarkably cleaved at 24 h in A375 cells and persisted until at least 48 h after treatment, while PARP cleavage was only detected in 501MEL cells at 48 h (Fig. 2d, e). These findings highlight the differences in the apoptotic mechanism of SFN between primary and metastatic human melanoma cells.
SFN hampered A375 cell migration and invasion
To evaluate the effects of SFN on melanoma cell migration, we performed a scratch assay. The data demonstrated that SFN treatment strongly reduced wound closure in A375 cells at 24 h. Only 18% of the tumour cells migrated compared to 89% for the untreated cells (p = 0.0022; Fig. 3a). The difference in the migration ability between the treated and untreated cells cannot be ascribed to a difference in their proliferation rates because the cells were treated with mitomycin prior to SFN treatment. To measure the impact of SFN on tumour cell invasion, we counted the number of cells that could penetrate type I collagen matrix. The data showed a significant reduction in the number of tumour cells invading the collagen compared to that of untreated cells (108 vs. 35.5, p = 0.0057) (Fig. 3b). All together, these results clearly suggest that SFN affects the migration and invasion ability of melanoma cells.

SFN reduces cell migration and invasion. a Representative images from the scratch assay of A375 cells captured at 0 and 24 h after treatment with 2 µg/ml SFN by a phase-contrast microscope (10×). The percentage of the closed wound area was calculated by tracing the border around the wound using ImageJ software. Data represent the mean ± SD from four separate experiments. b Representative images of A375 cells treated with 2 µg/ml SFN at 24 h after collagen invasion. Migrated cell nuclei were stained with methylene blue; images were captured by a phase-contrast microscope (4×) and the number of cells that migrated into the collagen was counted. Data represent the mean ± SD of six fields in triplicate. The statistical analysis was performed by paired two-tailed Student’s test (*p < 0.05, **p < 0.01 and ***p < 0.001)
SFN altered the transcriptome profile of melanoma cells
To decipher the antitumour molecular mechanisms driven by SFN, we investigated the whole transcriptome of A375 cells at four different time points (2, 6, 24 and 48 h after SFN treatment) using Illumina RNA-Seq technology. The results from two independent experiments revealed a total of 329 differently expressed genes (DEGs). Among these genes, 219 were affected by SFN treatment (Table 1), while 110 genes were intrinsically modulated during cell growth in culture (without compound treatment) (Supplementary Table S1). Out of the 219 genes, 74 were downregulated (34%) and 145 were upregulated (66%) by SFN at least at one time point (Fig. 4a, b; Table 1). Only two genes were upregulated at 2 h post-treatment, heat shock 70 kDa protein 1A (HSPA1A) and heme oxygenase (decycling) 1 (HMOX1). The number of DEGs modulated by SFN was much higher at 6 and 24 h after exposure compared to that at 48 h post-treatment (Fig. 4a, b; Table 1). Most genes were specifically affected at a unique time point, and only a few genes shared differential expression over several hours post-SFN treatment, specifically between 6 and 24 h (Fig. 4c).

SFN induces changes in gene expression profiles. a Heat map summary reflecting gene expression values of A375 cells treated with 2 µg/ml SFN at different time points (columns). Red indicates high and green indicates low gene expression values. b The histograms represent the number of DEGs at different time points after SFN treatment. c Venn diagram showing the overlap between the differentially expressed genes in the A375 cells treated with 2 µg/ml SFN at different time points
Enrichment cluster analysis of gene functions and biological processes was carried out using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt). Gene ontology analysis based on biological processes, molecular functions and cellular components are presented in Table 2, and the list of genes in each category is presented in Supplement Table S2. Clustering based on the biological process classification revealed that SFN-regulated genes are predominantly involved in response to stress (86 genes, p = 2.43e−12), the apoptotic process/cell death (60 genes, p = 3.44e−11), response to topologically incorrect proteins (16 genes, p = 9.47e−09), response to different stimuli (129 genes, p = 9.47e−08) and positive regulation of metabolic processes (50 genes, p = 0.0001). Molecular function clustering revealed that SFN regulates genes generally involved in protein binding (135 genes, p = 6.97e−08). According to the cellular component classification, SFN induces the expression of genes involved in the intracellular ferritin pathway (2 genes, p = 0.012) and the glutamate–cysteine ligase complex (2 genes, p = 0.012).
Target verification by qPCR
To verify the expression patterns of the DEGs detected by RNA-Seq, 12 genes that are modulated by SFN were selected for quantitative real-time PCR analysis, and GADPH was used as the reference gene. This includes five stress response genes [(HSPA1A, HMOX1, thioredoxin reductase 1 (TXNRD1), glutamate–cysteine ligase, catalytic subunit (GCLC) and modifier subunit (GCLM))], six p53 network genes [early growth response 1 (EGR1), activating transcription factor 3 (ATF3), BCL2-associated X protein (BAX), Fas cell surface death receptor (FAS), growth arrest and DNA damage-inducible beta (GADD45B) and cyclin-dependent kinase inhibitor 1A (CDKN1A))] and integrin beta 4 (ITGB4). The qPCR results showed high concordance with the RNA-Seq data, although the fold change values varied accordingly with the analytical method, suggesting that the RNA-Seq findings are reliable (Fig. 5). However, these data support the observation that SFN regulates the transcription of genes related to the Nrf2-signaling pathway and its involvement in physiological processes in melanoma cells.

Quantitative PCR validation of 12 DEGs in A375 cells treated with SFN. Relative fold expression of candidate genes was normalized against GADPH expression. The data represent the mean ± SD of three independent replicates. The black bars represent untreated cells at time 0, and the grey bars represent the relative fold change after SFN treatment. Statistical analysis was performed by paired two-tailed Student’s test (*p < 0.05, **p < 0.01)
Protein interaction network of differentially expressed proteins (DEPs)
The functional partnerships and interactions that occur between proteins corresponding to the revealed DEGs were predicted by STRING network analysis [25]. The downregulated and upregulated genes were linked into the networks according to their physical and functional associations and their involvement in specific cellular pathways (Fig. 6; Supplement Table S3). Early response to the drug (2 h post-treatment) was clearly associated with overexpression of the HSPA1A and HMOX1 genes. We detected upregulation of the HMOX1 gene across all the time points except the last one (48 h), with the maximum expression being reached at 6 h after exposure. At 6 h after SFN exposure, the number of stimulated genes increased greatly (Fig. 4; Table 1). Out of 114 DEGs, 30 upregulated genes could be organized into a network according to their function: (a) response to stress; (b) protein unfolding response and response to temperature; (c) regulation of cell death. Only five interacting proteins were downregulated, all of which were receptors [coagulation factor II (thrombin) receptor (F2R), lysophosphatidic acid receptor 1 and 3 (LPAR1/3), adenosine A1 receptor (ADORA1) and peroxisome proliferator-activated receptor gamma (PPARG)]. Prolonged SFN treatment (24 h) upregulated the expression of 73 genes and downregulated the expression of 36 genes (Fig. 4; Table 1). The upregulated genes mainly included genes related to apoptosis, including p53-related genes (MDM2, BAX, GADD45A, CDKN1A, ATF3, FAS), and genes involved in the growth arrest and proliferation (EGR1, BTG2). The stress response network was still activated, including the TXNRD1 and SOD1 genes and the histone protein cluster genes HIST1H2BD and HIST1H2BK. The downregulated genes include plasminogen activator tissue (PLAT), ITGB2 and ITGB4 grouped into the network, which could be involved in the regulation of cell migration and tissue remodelling (Fig. 6). Transcription factor-specific protein 1 (SP1) has been shown to be upregulated in several type of cancers; it is associated with poor prognosis and could be implicated in prostate cancer chemoprevention [26]. Our analysis demonstrated that SFN-treated A375 cells showed reduced SP1 expression at 24 h, which orchestrated the coordinated overexpression of 16 other proteins in the network (Fig. 6). The transcriptional co-factor transducin beta-like (TBL1X) 1 has been demonstrated to be involved in cellular proliferation and invasiveness in both human and murine pancreatic ductal adenocarcinoma [27]. Our data also indicated a crucial role for this protein, as it is downregulated at 24 h by SFN and probably controls the expression of ten other genes in the network. At 48 h, SFN upregulated a small number of genes related to the final stage of stress response and p53-related genes that play a role in apoptosis, such as TP53-inducible nuclear protein 1 (TP53INP1), sestrin 2 (SESN2), Fas cell surface death receptor (FAS) and glucose-6-phosphate dehydrogenase (G6PD) (Fig. 6; Table 1).

Protein interaction network analysis using STRING. Representation of a functional protein association network of DEGs from A375 cells treated with 2 µg/ml SFN at 2, 6, 24 and 48 h post-treatment. The coloured nodes represent query proteins and first shell interactions. The line thickness indicates the strength of data support
Discussion
Over the last few years, an increase in research using dietary phytochemicals for targeting cancer has been driven by the partial efficacy of chemotherapy and radiotherapy and the associated side effects. Isothiocyanates and in particular SFN have been investigated in vitro and in vivo studies. SFN has long been known to act as a therapeutic or preventive agent and an inducer of apoptosis in pre-cancerous cells and tumour cells of different origins [18, 28,29,30,31,32,33]. Previous studies have indicated an anticancer activity of SFN in melanoma [24, 34]. SFN has been shown to inhibit the metastasis of murine B16F-10 melanoma cells by inducing apoptosis via caspase activation both in vitro and in vivo. Moreover, partial analysis on the effects of SFN on A375 cell line has been reported indicating a reduction on cell viability, but only at 10 μM onward or after a treatment had been refreshed after 24 h (at 5 μM). No activation of apoptosis mediated by caspase 4, 8, 9 was revealed or other cellular mechanisms such as proliferation, cell cycle progression, invasion and migration analysed. Few reports have shown the pro-apoptotic effects of SFN on human melanoma cells by the p53 and p38 pathways and the anti-proliferative effects associated with loss of the chromatin modifying enzyme EZH2 [35, 36]. Nonetheless, the detailed molecular mechanisms by which SFN affects human melanoma are still unknown. Therefore, we implemented a comprehensive investigation at the cellular and molecular level to evaluate the effects of SFN, for the first time, on both primary and metastatic human melanoma cells and compared with its effects on normal epidermal melanocytes. This study represents an attempt to focus on specific processes and genes involved in response to SFN treatment in melanoma using functional assays and a transcriptomic approach. The results demonstrated that while SFN impairs the viability of both A375 and 501MEL cells in a dose- and time-dependent manner, it does not affect non-cancerous HEMa cells. To identify the main mechanisms responsible for growth inhibition, AKT phosphorylation and apoptosis were detected in all the cell lines tested. In agreement with previous reports [36, 37], our data showed that inhibition of cell viability is correlated with reduced AKT phosphorylation. Many studies have focused on the SFN-driven induction of apoptosis in different cancer cell types [19, 37,38,39]. Consistent with some previous reports [36], we demonstrated that SFN increases the percentage of apoptotic melanoma cells by cleaving PARP, activating p-p53, caspase-3, caspase-8 and caspase-9 and decreasing Bcl2 expression, confirming its pro-apoptotic role. Moreover, our data suggested that metastatic 501MEL cells are more resistant to SFN treatment than primary A375 melanoma cells.
Several studies have demonstrated that SFN causes G1/S and G2/M cell cycle arrest by altering the levels of cyclin A, cyclin B1, cyclin D1 and p21cip1/waf1 in cancer [40, 41]. Indeed, our data indicated that cell cycle arrest occurred in the melanoma cell lines treated with SFN, while no changes were observed in the HEMa cells. In fact, the results showed a strong accumulation of cells in the G2/M phase in a time-dependent manner, followed by a decrease in the number of cells in the G0/G1 phase. G2/M cell cycle arrest is reportedly associated with checkpoint damage, which is regulated by cyclin B1/CDK1 and the cyclin kinase inhibitor p21 in several cell lines [42,43,44]. We demonstrated that SFN induced G2/M arrest via inhibiting cyclin B1, but it did not affect cyclin A (data not shown). SFN strongly reduced the migration and invasion of melanoma A375 cells, which is in agreement with previous studies performed on various cancer cell lines [23, 38, 45,46,47], suggesting the importance of this compound in tumour spreading.
For the first time, we also examined the gene expression profile of A375 cells treated with SFN at different time points (2, 6, 24 and 48 h) using RNA-seq technology. Most of the genes (145) were upregulated, and only 74 genes were downregulated. In the early stages after treatment (from 2 to 6 h), we detected notable coordination of the induction of genes encoding the stress-inducible antioxidant protein Heme oxygenase-1 (HMOX1) and the heat shock-associated genes HSPA1A, HSPA6, HSPA1B, HSPA1L, HSPB8, HSPH1 and HSP90AA1. This was followed by activation of the p53 signalling pathway and p53-dependent apoptotic processes. Heat shock proteins and HMOX1 play an important cytoprotective role during and after exposure to stress, possess powerful anti-apoptotic properties and promote cell survival under various pathological conditions. Thus, the role of these proteins in cancer seems to be ambiguous. Our data showed peak expression of stress-inducible proteins promptly after SFN treatment (from 2 to 6 h), which gradually faded after prolonged SFN treatment (24–48 h) as the apoptotic process began. Consistent with previous reports [48, 49] that associated the Nrf2 pathway with SFN-induced chemoprevention, we found that the Nrf2 target genes HMOX1, TXNRD1, GCLC, GCLM, AKR1B10 and G6PD were upregulated after melanoma treatment.
As expected, a number of cell proliferation gene inhibitors were activated, including GADD45 (encoding for growth arrest) and DNA damage-inducible proteins, which are involved in the G2/M checkpoint and may participate in the regulation of Cdc2 kinase activity and the activation of the p38/Jun N-terminal kinase pathway [43, 50]. Both GADD45A and GADD45B were upregulated in A375 cells treated with SFN for 24 h. Additionally, transcription factor 3 (ATF3), another cell growth repressor and pro-apoptotic gene [51, 52], was significantly upregulated after treatment. In line with the above observation, the anti-apoptotic and pro-survival adenosine receptor ADORA1 [53] was found to be downregulated by SFN. The nuclear transcription factor EGR1 plays a role in signal transduction pathways mediating cellular proliferation and growth arrest and in the control of cell differentiation and death genes through the upregulation of p53 [54, 55] and PTEN [56]. Our data demonstrated that SFN increases the expression of EGR1 in melanoma cells, which is in agreement with recent breast cancer data [57].
Finally, we clearly demonstrated that highly expressed p53 is strongly correlated with SFN-induced apoptosis in melanoma. Further, the pro-apoptotic genes BAX, BBC3 [p53 upregulated modulator of apoptosis (PUMA)] and FAS were also found to be upregulated at the transcriptional level. These three genes are important players in the apoptotic process, because they are directly transactivated by p53 [58,59,60,61]. The upregulation of BAX and PUMA after SFN treatment has been previously demonstrated in different tumours [36, 62,63,64]. In addition, our results indicated that SFN enhances FAS/CD95 gene expression at 24 and 48 h, suggesting that the FAS–FASL signalling pathway also contributes to SFN-induced apoptosis in melanoma cells. Similarly, SFN-induced apoptosis by FAS/CD95 has been reported for human leukaemia [65] and human prostate cancer cells [66].
Migration and invasion are crucial events that occur in the metastasis of primary tumours, including melanoma, and understanding all aspects of this process is essential to prevent cancer-related death. Several genes involved in invasion and migration were affected by SFN, including HTRA3, PLAT, INHBA, FST and ITGB4. In, particular, the well-characterized invasion inhibitor [67] serine protease HTRA3 was shown to be upregulated. In contrast, tissue-type plasminogen activator (PLAT) was found to be downregulated at 24 h; this gene encodes a secreted serine protease that converts the pro-enzyme plasminogen to plasmin, which is a fibrinolytic enzyme that plays a role in cell migration and tissue remodelling [68]. The follistatin (FST) gene, related to lymphangiogenesis and cell growth, was downregulated at 24 h. Activin A plays a role in melanoma migration and reduces lymphatic endothelial sprout formation in vitro, but because of its pleiotropic effects on cell mobility, it is not suitable as a pharmacological target [69]. In addition, integrin β4 (ITGB4) downregulation was also observed. ITGB4 promotes invasion and migration in various cancer cells [70]. As a receptor for the laminins, ITGB4 is essential for the organization and maintenance of epithelial structure and plays a pivotal role in the metastasis of various cancer cells [71].
SFN has demonstrated chemopreventive properties with selective cytotoxicity in several type of cancer based on its ability to target multiple mechanisms within the cell to control carcinogenesis. Pro-apoptotic, anti-inflammatory and histone modulation are some of the known and crucial mechanisms by which SFN exerts chemoprevention. In addition to pre-clinical experiments, SFN has demonstrated promising results also in clinical studies in woman with breast ductal carcinoma in situ (DCIS) [72], glioblastoma cells [38] and prostate cancer [73]. Moreover, it was found that people who consumed cruciferous vegetables at least once a week have reduced risk of pharynx, oesophageal, colorectal, breast, oral cavity and kidney cancers [74]. Recently, the effect of SFN on cancer stem cell is also another emerging area of interest that may contribute to its chemopreventive properties [75]. Moreover, combination therapies that associate SFN with other therapeutic agents support a possible treatment modality for prostate and colorectal cancer [76, 77]. SFN combination with agents other than chemotherapeutics has bee report to play an important role also in bladder cancer and bronchial carcinoid cell lines [76, 78]. Furthermore, SFN is highly tolerable and safe in humans with no genotoxic effect [79]. However, more clinical studies are necessary to fully explore the efficacy of SFN as combination therapy, anti-cancer agent or chemopreventive in humans.
In conclusion, for the first time, our data highlight the effects of SFN on melanoma gene expression profiles and elucidates the mechanisms by which it exerts anticancer activity by suppressing various critical hallmarks of cancer, such as cell growth and proliferation, apoptosis, invasion and migration. Our data provides a global view of the potential complex and concurrent mechanisms by which SFN may fight against melanoma and serve as a resource for future investigations. Our results confirm the favourable toxicological profile and high potential for chemotherapeutic activity of SFN. Moreover, our data indicate that SFN is an attractive multipotent antitumour agent for melanoma therapy, and it could open new avenues for the prevention of tumour progression and/or treatment of human malignancies.
References
Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC Jr, Morton DL, Ross MI, Sober AJ, Sondak VK (2009) Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27(36):6199–6206. doi:10.1200/jco.2009.23.4799
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A (2016) Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 66(4):271–289. doi:10.3322/caac.21349
Bhatia P, Friedlander P, Zakaria EA, Kandil E (2015) Impact of BRAF mutation status in the prognosis of cutaneous melanoma: an area of ongoing research. Ann Transl Med 3(2):24. doi:10.3978/j.issn.2305-5839.2014.12.05
Herzberg B, Fisher DE (2016) Metastatic melanoma and immunotherapy. Clin Immunol 172:105–110
Olszanski AJ (2014) Current and future roles of targeted therapy and immunotherapy in advanced melanoma. J Manag Care Spec Pharm 20(4):346–356. doi:10.18553/jmcp.2014.20.4.346
Cheung KL, Kong AN (2010) Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J 12(1):87–97. doi:10.1208/s12248-009-9162-8
Shu L, Cheung KL, Khor TO, Chen C, Kong AN (2010) Phytochemicals: cancer chemoprevention and suppression of tumor onset and metastasis. Cancer Metastasis Rev 29(3):483–502. doi:10.1007/s10555-010-9239-y
Fimognari C, Hrelia P (2007) Sulforaphane as a promising molecule for fighting cancer. Mutat Res 635(2–3):90–104. doi:10.1016/j.mrrev.2006.10.004
Juge N, Mithen RF, Traka M (2007) Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci 64(9):1105–1127. doi:10.1007/s00018-007-6484-5
Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, Nelson JB, Singh SV (2011) Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst 103(7):571–584. doi:10.1093/jnci/djr029
Misiewicz I, Skupinska K, Kowalska E, Lubinski J, Kasprzycka-Guttman T (2004) Sulforaphane-mediated induction of a phase 2 detoxifying enzyme NAD(P)H: quinone reductase and apoptosis in human lymphoblastoid cells. Acta Biochim Pol 51(3):711–721
Prawan A, Kundu JK, Surh YJ (2005) Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal 7(11–12):1688–1703. doi:10.1089/ars.2005.7.1688
Fahey JW, Talalay P (1999) Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol 37(9–10):973–979
Mithen R, Faulkner K, Magrath R, Rose P, Williamson G, Marquez J (2003) Development of isothiocyanate-enriched broccoli, and its enhanced ability to induce phase 2 detoxification enzymes in mammalian cells. Theor Appl Genet 106(4):727–734. doi:10.1007/s00122-002-1123-x
Bao Y, Wang W, Zhou Z, Sun C (2014) Benefits and risks of the hormetic effects of dietary isothiocyanates on cancer prevention. PLoS ONE 9(12):e114764. doi:10.1371/journal.pone.0114764
Feitelson MA, Arzumanyan A, Kulathinal RJ, Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML, Nawroth R, Sanchez-Garcia I, Sharma D, Saxena NK, Singh N, Vlachostergios PJ, Guo S, Honoki K, Fujii H, Georgakilas AG, Bilsland A, Amedei A, Niccolai E, Amin A, Ashraf SS, Boosani CS, Guha G, Ciriolo MR, Aquilano K, Chen S, Mohammed SI, Azmi AS, Bhakta D, Halicka D, Keith WN, Nowsheen S (2015) Sustained proliferation in cancer: mechanisms and novel therapeutic targets. Semin Cancer Biol 35(Suppl):S25–S54. doi:10.1016/j.semcancer.2015.02.006
Sestili P, Fimognari C (2015) Cytotoxic and antitumor activity of sulforaphane: the role of reactive oxygen species. Biomed Res Int 2015:402386. doi:10.1155/2015/402386
Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, Gasc N, Tulliez J, Terce F (2000) Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res 60(5):1426–1433
Mondal A, Biswas R, Rhee YH, Kim J, Ahn JC (2016) Sulforaphene promotes Bax/Bcl2, MAPK-dependent human gastric cancer AGS cells apoptosis and inhibits migration via EGFR, p-ERK1/2 down-regulation. Gen Physiol Biophys 35(1):25–34. doi:10.4149/gpb_2015033
Wang L, Tian Z, Yang Q, Li H, Guan H, Shi B, Hou P, Ji M (2015) Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget 6(28):25917–25931. doi:10.18632/oncotarget.4542
Kim SJ, Kim BS, Kyung TW, Lee SC, Rho CW, Choi KR, Hwang HJ, Choi HS (2006) Suppressive effects of young radish cultivated with sulfur on growth and metastasis of B16-F10 melanoma cells. Arch Pharm Res 29(3):235–240
Thejass P, Kuttan G (2006) Antimetastatic activity of sulforaphane. Life Sci 78(26):3043–3050. doi:10.1016/j.lfs.2005.12.038
Thejass P, Kuttan G (2007) Modulation of cell-mediated immune response in B16F-10 melanoma-induced metastatic tumor-bearing C57BL/6 mice by sulforaphane. Immunopharmacol Immunotoxicol 29(2):173–186. doi:10.1080/08923970701511728
Hamsa TP, Thejass P, Kuttan G (2011) Induction of apoptosis by sulforaphane in highly metastatic B16F-10 melanoma cells. Drug Chem Toxicol 34(3):332–340. doi:10.3109/01480545.2010.538694
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447–D452. doi:10.1093/nar/gku1003
Sankpal UT, Goodison S, Abdelrahim M, Basha R (2011) Targeting Sp1 transcription factors in prostate cancer therapy. Med Chem 7(5):518–525
Stoy C, Sundaram A, Rios Garcia M, Wang X, Seibert O, Zota A, Wendler S, Mannle D, Hinz U, Sticht C, Muciek M, Gretz N, Rose AJ, Greiner V, Hofmann TG, Bauer A, Hoheisel J, Berriel Diaz M, Gaida MM, Werner J, Schafmeier T, Strobel O, Herzig S (2015) Transcriptional co-factor Transducin beta-like (TBL) 1 acts as a checkpoint in pancreatic cancer malignancy. EMBO Mol Med 7(8):1048–1062. doi:10.15252/emmm.201404837
Clarke JD, Dashwood RH, Ho E (2008) Multi-targeted prevention of cancer by sulforaphane. Cancer Lett 269(2):291–304. doi:10.1016/j.canlet.2008.04.018
Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E, Argani P, Davidson NE, Talalay P, Kensler TW, Visvanathan K (2007) Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 28(7):1485–1490. doi:10.1093/carcin/bgm049
Moon D-O, Kim M-O, Kang S-H, Choi YH, Kim G-Y (2009) Sulforaphane suppresses TNF-α-mediated activation of NF-κB and induces apoptosis through activation of reactive oxygen species-dependent caspase-3. Cancer Lett 274(1):132–142. doi:10.1016/j.canlet.2008.09.013
Park SY, Kim GY, Bae SJ, Yoo YH, Choi YH (2007) Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol Rep 18(1):181–187
Suppipat K, Park CS, Shen Y, Zhu X, Lacorazza HD (2012) Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PLoS ONE 7(12):e51251. doi:10.1371/journal.pone.0051251
Tseng E, Scott-Ramsay EA, Morris ME (2004) Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and mammary epithelial MCF-12A cell lines. Exp Biol Med (Maywood) 229(8):835–842
Mantso T, Sfakianos AP, Atkinson A, Anestopoulos I, Mitsiogianni M, Botaitis S, Perente S, Simopoulos C, Vasileiadis S, Franco R, Pappa A, Panayiotidis MI (2016) Development of a novel experimental in vitro model of isothiocyanate-induced apoptosis in human malignant melanoma cells. Anticancer Res 36(12):6303–6309
Fisher ML, Adhikary G, Grun D, Kaetzel DM, Eckert RL (2015) The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. Mol Carcinog. doi:10.1002/mc.22448
Rudolf K, Cervinka M, Rudolf E (2014) Sulforaphane-induced apoptosis involves p53 and p38 in melanoma cells. Apoptosis 19(4):734–747. doi:10.1007/s10495-013-0959-7
Yao A, Shen Y, Wang A, Chen S, Zhang H, Chen F, Chen Z, Wei H, Zou Z, Shan Y, Zhang X (2015) Sulforaphane induces apoptosis in adipocytes via Akt/p70s6k1/Bad inhibition and ERK activation. Biochem Biophys Res Commun 465(4):696–701. doi:10.1016/j.bbrc.2015.08.049
Zhang Z, Li C, Shang L, Zhang Y, Zou R, Zhan Y, Bi B (2016) Sulforaphane induces apoptosis and inhibits invasion in U251MG glioblastoma cells. Springerplus 5:235. doi:10.1186/s40064-016-1910-5
Zuryn A, Litwiniec A, Safiejko-Mroczka B, Klimaszewska-Wisniewska A, Gagat M, Krajewski A, Gackowska L, Grzanka D (2016) The effect of sulforaphane on the cell cycle, apoptosis and expression of cyclin D1 and p21 in the A549 non-small cell lung cancer cell line. Int J Oncol 48(6):2521–2533. doi:10.3892/ijo.2016.3444
Jackson SJ, Singletary KW (2004) Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J Nutr 134(9):2229–2236
Liu KC, Shih TY, Kuo CL, Ma YS, Yang JL, Wu PP, Huang YP, Lai KC, Chung JG (2016) Sulforaphane induces cell death through G2/M phase arrest and triggers apoptosis in HCT 116 human colon cancer cells. Am J Chin Med 44(6):1289–1310. doi:10.1142/s0192415x16500725
Porter LA, Singh G, Lee JM (2000) Abundance of cyclin B1 regulates gamma-radiation-induced apoptosis. Blood 95(8):2645–2650
Shih RS, Wong SH, Schoene NW, Zhang JJ, Lei KY (2010) Enhanced Gadd45 expression and delayed G2/M progression are p53-dependent in zinc-supplemented human bronchial epithelial cells. Exp Biol Med (Maywood) 235(8):932–940. doi:10.1258/ebm.2010.010076
Toyoshima FMT, Wada A, Fukuda M, Nishida E (1998) Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J 17(10):2728–2735. doi:10.1093/emboj/17.10.2728 (T1998)
Lee YR, Noh EM, Han JH, Kim JM, Hwang BM, Kim BS, Lee SH, Jung SH, Youn HJ, Chung EY, Kim JS (2013) Sulforaphane controls TPA-induced MMP-9 expression through the NF-kappaB signaling pathway, but not AP-1, in MCF-7 breast cancer cells. BMB Rep 46(4):201–206
Lenzi M, Fimognari C, Hrelia P (2014) Sulforaphane as a promising molecule for fighting cancer. Cancer Treat Res 159:207–223. doi:10.1007/978-3-642-38007-5_12
Shan Y, Zhang L, Bao Y, Li B, He C, Gao M, Feng X, Xu W, Zhang X, Wang S (2013) Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J Nutr Biochem 24(6):1062–1069. doi:10.1016/j.jnutbio.2012.08.004
Gerhauser C, You M, Liu J, Moriarty RM, Hawthorne M, Mehta RG, Moon RC, Pezzuto JM (1997) Cancer chemopreventive potential of sulforamate, a novel analogue of sulforaphane that induces phase 2 drug-metabolizing enzymes. Cancer Res 57(2):272–278
Greaney AJ, Maier NK, Leppla SH, Moayeri M (2016) Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J Leukoc Biol 99(1):189–199. doi:10.1189/jlb.3A0415-155RR
Salvador JM, Brown-Clay JD, Fornace AJ Jr (2013) Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol 793:1–19. doi:10.1007/978-1-4614-8289-5_1
Fan F, Jin S, Amundson SA, Tong T, Fan W, Zhao H, Zhu X, Mazzacurati L, Li X, Petrik KL, Fornace AJ Jr, Rajasekaran B, Zhan Q (2002) ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 21(49):7488–7496. doi:10.1038/sj.onc.1205896
Huang X, Li X, Guo B (2008) KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J Biol Chem 283(44):29795–29801. doi:10.1074/jbc.M802515200
Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, Borea PA (2009) Adenosine receptors and cancer. Handb Exp Pharmacol 193:399–441. doi:10.1007/978-3-540-89615-9_14
Liu J, Grogan L, Nau MM, Allegra CJ, Chu E, Wright JJ (2001) Physical interaction between p53 and primary response gene Egr-1. Int J Oncol 18(4):863–870
Nair P, Muthukkumar S, Sells SF, Han SS, Sukhatme VP, Rangnekar VM (1997) Early growth response-1-dependent apoptosis is mediated by p53. J Biol Chem 272(32):20131–20138
Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3(12):1124–1128. doi:10.1038/ncb1201-1124
Yang M, Teng W, Qu Y, Wang H, Yuan Q (2016) Sulforaphene inhibits triple negative breast cancer through activating tumor suppressor Egr1. Breast Cancer Res Treat 158(2):277–286. doi:10.1007/s10549-016-3888-7
Guo B, Cao S, Toth K, Azrak RG, Rustum YM (2000) Overexpression of Bax enhances antitumor activity of chemotherapeutic agents in human head and neck squamous cell carcinoma. Clin Cancer Res 6(2):718–724
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80(2):293–299
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188(11):2033–2045
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7(3):683–694
Choi S, Singh SV (2005) Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent. Cancer Res 65(5):2035–2043. doi:10.1158/0008-5472.can-04-3616
Chu WF, Wu DM, Liu W, Wu LJ, Li DZ, Xu DY, Wang XF (2009) Sulforaphane induces G2-M arrest and apoptosis in high metastasis cell line of salivary gland adenoid cystic carcinoma. Oral Oncol 45(11):998–1004. doi:10.1016/j.oraloncology.2009.05.641
Fimognari C, Nusse M, Cesari R, Iori R, Cantelli-Forti G, Hrelia P (2002) Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 23(4):581–586
Shang HS, Shih YL, Lee CH, Hsueh SC, Liu JY, Liao NC, Chen YL, Huang YP, Lu HF, Chung JG (2016) Sulforaphane-induced apoptosis in human leukemia HL-60 cells through extrinsic and intrinsic signal pathways and altering associated genes expression assayed by cDNA microarray. Environ Toxicol. doi:10.1002/tox.22237
Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, Lee YJ, Xiao H, Herman-Antosiewicz A (2005) Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem 280(20):19911–19924. doi:10.1074/jbc.M412443200
Chien J, Campioni M, Shridhar V, Baldi A (2009) HtrA serine proteases as potential therapeutic targets in cancer. Curr Cancer Drug Targets 9(4):451–468
Lu P, Takai K, Weaver VM, Werb Z (2011) Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a005058
Heinz M, Niederleithner HL, Puujalka E, Soler-Cardona A, Grusch M, Pehamberger H, Loewe R, Petzelbauer P (2015) Activin A is anti-lymphangiogenic in a melanoma mouse model. J Invest Dermatol 135(1):212–221. doi:10.1038/jid.2014.328
Wan X, Kim SY, Guenther LM, Mendoza A, Briggs J, Yeung C, Currier D, Zhang H, Mackall C, Li WJ, Tuan RS, Deyrup AT, Khanna C, Helman L (2009) Beta4 integrin promotes osteosarcoma metastasis and interacts with ezrin. Oncogene 28(38):3401–3411. doi:10.1038/onc.2009.206
Zhang X, Rozengurt E, Reed EF (2010) HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Sci Signal 3(149):ra85. doi:10.1126/scisignal.2001158
Zhang Z, Atwell LL, Farris PE, Ho E, Shannon J (2016) Associations between cruciferous vegetable intake and selected biomarkers among women scheduled for breast biopsies. Public Health Nutr 19(7):1288–1295. doi:10.1017/S136898001500244X
Alumkal JJ, Slottke R, Schwartzman J, Cherala G, Munar M, Graff JN, Beer TM, Ryan CW, Koop DR, Gibbs A, Gao L, Flamiatos JF, Tucker E, Kleinschmidt R, Mori M (2015) A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs 33(2):480–489. doi:10.1007/s10637-014-0189-z
Wu QJ, Yang Y, Vogtmann E, Wang J, Han LH, Li HL, Xiang YB (2013) Cruciferous vegetables intake and the risk of colorectal cancer: a meta-analysis of observational studies. Ann Oncol 24(4):1079–1087. doi:10.1093/annonc/mds601
Vyas AR, Moura MB, Hahm ER, Singh KB, Singh SV (2016) Sulforaphane inhibits c-Myc-mediated prostate cancer stem-like traits. J Cell Biochem 117(11):2482–2495. doi:10.1002/jcb.25541
Islam SS, Mokhtari RB, Akbari P, Hatina J, Yeger H, Farhat WA (2016) Simultaneous targeting of bladder tumor growth, survival, and epithelial-to-mesenchymal transition with a novel therapeutic combination of acetazolamide (AZ) and sulforaphane (SFN). Target Oncol 11(2):209–227. doi:10.1007/s11523-015-0386-5
Kallifatidis G, Labsch S, Rausch V, Mattern J, Gladkich J, Moldenhauer G, Buchler MW, Salnikov AV, Herr I (2011) Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol Ther 19(1):188–195. doi:10.1038/mt.2010.216
Mokhtari RB, Kumar S, Islam SS, Yazdanpanah M, Adeli K, Cutz E, Yeger H (2013) Combination of carbonic anhydrase inhibitor, acetazolamide, and sulforaphane, reduces the viability and growth of bronchial carcinoid cell lines. BMC Cancer 13:378. doi:10.1186/1471-2407-13-378
Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, Ye L, Talalay P (2006) Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr Cancer 55(1):53–62. doi:10.1207/s15327914nc5501_7
Acknowledgements
This work was supported by the PRIN project “SKINFLAM” (GA number 2010C2LKKJ_001). FR was supported by an individual fellowship granted by the Umbria Region with funding from the European Social Fund. EK’s fellowship was supported by the European Community in the framework of the Seventh Framework Programme under the Marie Curie Actions COFUND project “I-Move” 8267232.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict interest.
Financial disclosure
I certify that no party having a direct interest in the results of the research supporting this article has or will confer a benefit on me or on any organization with which I am associated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Arcidiacono, P., Ragonese, F., Stabile, A. et al. Antitumor activity and expression profiles of genes induced by sulforaphane in human melanoma cells. Eur J Nutr 57, 2547–2569 (2018). https://doi.org/10.1007/s00394-017-1527-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-017-1527-7