
Abstract
The first clinical trials of the safety and efficacy of interferon-alpha2 (IFN-alpha2) were performed about 30 years ago. Since then, several single-arm studies have convincingly demonstrated that IFN-alpha2 is a highly potent anti-cancer agent in several cancer types but unfortunately not being explored sufficiently due to a high toxicity profile when using non-pegylated IFN-alpha2 or high dosages or due to competitive drugs, that for clinicians at first glance might look more attractive. Within the hematological malignancies, IFN-alpha2 has only recently been revived in patients with the Philadelphia-negative myeloproliferative neoplasms—essential thrombocytosis, polycythemia vera, and myelofibrosis (MPNs)—and in patients with chronic myelogenous leukemia (CML) in combination with tyrosine kinase inhibitors. In this review, we tell the IFN story in MPNs from the very beginning in the 1980s up to 2018 and describe the perspectives for IFN-alpha2 treatment of MPNs in the future. The mechanisms of actions are discussed and the impact of chronic inflammation as the driving force for clonal expansion and disease progression in MPNs is discussed in the context of combination therapies with potent anti-inflammatory agents, such as the JAK1–2 inhibitors (licensed only ruxolitinib) and statins as well. Interferon-alpha2 being the cornerstone treatment in MPNs and having the potential of inducing minimal residual disease (MRD) with normalization of the bone marrow and low-JAK2V617F allele burden, we believe that combination therapy with ruxolitinib may be even more efficacious and hopefully revert disease progression in many more patients to enter the path towards MRD. In patients with advanced and transforming disease towards leukemic transformation or having transformed to acute myeloid leukemia, “triple therapy” is proposed as a novel treatment modality to be tested in clinical trials combining IFN-alpha2, DNA-hypomethylator, and ruxolitinib. The rationale for this “triple therapy” is given, including the fact that even in AML, IFN-alpha2 as monotherapy may revert disease progression. We envisage a new and bright future with many more patients with MPNs obtaining MRD on the above therapies. From this stage—and even before—vaccination strategies may open a new horizon with cure being the goal for some patients.
Similar content being viewed by others

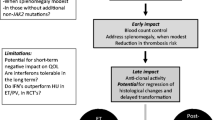

Introduction
About 60 years ago, interferon (IFN) was discovered by Isaacs and Lindenmann [1] who described this cytokine to be able to interfere with virus replication. Later, the IFN receptor was identified and shortly after the JAK/STAT-signal transduction pathway as described in several recent reviews [2,3,4,5,6]. It early became apparent that one of the mechanisms of action of IFN-alpha2 involved stimulation of immune cells [7, 8]. Due to all the other properties of IFN, including its antiproliferative, immunomodulatory, and antiangiogenic effects, great interest in the potential use of IFN in the treatment of several malignancies was soon raised. The production and purification of human leukocyte IFNs [9] were followed by the first clinical study in the late 1970s on the efficacy of IFN-alpha2 in multiple myeloma (MM) [10]. Soon after, IFN-alpha2 was cloned, allowing large amounts of IFNs to be produced for experimental research and clinical trials, opening an exciting era of several years, in which the safety and efficacy of IFN was tested in a variety of hematological malignancies. Among these are multiple myeloma, hairy-cell leukemia (HCL), chronic myelogenous leukemia (CML), the classical Philadelphia-negative chronic myeloproliferative neoplasms and essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF) (MPNs), the hypereosinophilic syndromes, and systemic mastocytosis (SM). Outstanding breakthroughs in the treatment of HCL and CML with IFN-alpha2 were confirmed in several large clinical trials. Thus, a large proportion of patients with HCL achieved long-lasting complete remissions with normalization of peripheral blood values and the bone marrow in concert with a marked improvement in their immune defense towards infections. Likewise, IFN-alpha2 proved to be the first agent with the potential of inducing complete and sustained cytogenetic remissions with disappearance of Philadelphia chromosome in CML and—in addition—in some patients even the induction of major molecular remissions with a significant and sustained reduction of the BCR-ABL transcript in a subset of patients. These results were historical IFN milestones in the treatment of hematological malignancies (HCL and CML), which otherwise had a dismal prognosis with severe and often lethal atypical infections (HCL) or increasing genomic instability with terminal fatal leukemic transformation within a few years from the time of diagnosis in the large majority unless a bone marrow transplantation was an option (CML). Accordingly, IFN-alpha2 remained the best medical treatment of CML during the next decades until the targeted treatment with the tyrosine kinase inhibitor (TKI) imatinib mesylate substituted IFN-alpha2 about 20 years ago and later other TKIs (e.g., dasatinib and nilotonib) have entered as second-generation TKIs. As in CML, the mechanisms of action of IFN in patients with the Philadelphia-negative MPNs are likely multifactorial. In CML, IFN-alpha2 has been shown to restore the adhesion of CML primitive progenitor cells to marrow stroma, downregulate the expression of the BCR-ABL1, and activate several transcriptional factors that regulate cell proliferation, maturation, and apoptosis. In addition, very early in the IFN-era in CML, immune studies unraveled IFN-alpha to have very potent immune enhancing capacity, inducing recognition and elimination of CML cells by the immune system [11, 12]. Importantly, in 2009, a novel mechanism on hematopoietic stem cells (HSC) was described by Essers et al., implying induction of cell cycling in quiescent HSC and early progenitors by IFN-alpha2 [13]. One year later, they also showed that chronic administration of IFN-alpha2 depletes HSC, implying that “dormant” cancer stem cells may be susceptible to manipulation via an IFN-alpha2 induced “wake up call” with subsequent proliferation and “unmasking” of the malignant cells for the immune system by targeted treatment [14]. All these studies and the impact of IFN-alpha2 upon the immune system in CML [11, 12] created not only the platform for similar studies in patients with MPNs but also the platform for studies in CML patients on combination therapy with imatinib and IFN-alpha2 and later also studies on IFNs with other TKIs in CML [15,16,17,18]. Indeed, these studies have shown that such combination therapy is far more efficacious than singe-agent therapy based upon the fact that the modes of action and biological effects of TKIs and IFN-alpha2 are quite different. These lessons from the IFN-era in CML are of utmost importance, since so many similarities exist between the CML-IFN-landscape and the MPN-IFN-landscape in regard to highly important questions such as “Why to treat with IFN-alpha2?” and “When to treat with IFN-alpha2?”. Accordingly, several of the lessons in the CML-IFN era can be translated and used in the treatment of MPN patients today and in the future. All these questions will be addressed below.
Despite the very prominent “anti-cancer-effects” as described above, and despite initial studies displaying safety and efficacy of IFN-alpha2 in a large number of patients with the classical Philadelphia-negative MPNs—ET, PV, and MF (MPNs) [reviewed in 19–28], IFN-alpha2 disappeared in the dark and only in recent years the interest in using IFN-alpha2 in MPNs has been revived [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50]. This renaissance of IFN-alpha2 in MPNs is mainly attributed to the increasing number of studies within the last 5–10 years, which have shown sustained complete hematological and major molecular remissions after long-term treatment with IFN-alpha2 [19, 20, 27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50], and have even been sustained up to 3 years after discontinuation of IFN-alpha2 [30, 31, 33, 41]. These highly encouraging and intriguing results envisage “minimal residual disease” (MRD) with normalization of peripheral blood cell values and normal bone marrow architecture to be new treatment objectives in MPNs [23,24,25, 51]. Importantly, they may also open a new horizon for patients with MPNs by promoting the next step towards cure by vaccination strategies as described elsewhere in this theme issue [52].
After a description of the history on IFN-alpha2 in MPNs, mechanisms of actions of IFN, and the novel concept of chronic inflammation as the driving force for clonal evolution in MPNs, we will focus on some controversial issues in MPNs and give our answers to key questions in MPNs—based upon decades of clinical experience with IFN-alpha2 in the treatment of MPNs and most recent novel observations. We will put in perspective the rationales for early treatment with IFN-alpha2-monotherapy in MPNs, for combination therapies, including JAK1–2 inhibitor (e.g., ruxolitinib), DNA-hypomethylators and statins, and the perspectives for such therapies to shape a new horizon with cure being an achievable goal together with vaccination strategies [51, 52].
History of IFNs in MPNs
Already in 1985, Linkesch et al. from Austria described that IFN-alpha2 was able to control myeloproliferation in myeloproliferative diseases with severe thrombocytosis [53, 54]. Since then, several studies during the last 30 years have subsequently confirmed that IFN-alpha2 is also able to inhibit myeloproliferation in the Philadelphia-negative MPNs with a reduction or alleviation of the need of phlebotomies in PV, disappearance of pruritus, normalization of elevated leucocyte and platelet counts, and a reduction in spleen size [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50, 55,56,57,58,59]. Although early studies in MPNs suggested that enhancement and modulation of immune cells might be involved in the mechanisms of action of IFN-alpha2 [58] and these aspects have been extensively studied in CML [11, 12], only recently immune cells and their functionality have been similarly studied during treatment with IFN-alpha2 [42, 49, 60,61,62]. Despite all these studies, IFN-alpha2 has not been the first drug of choice in the treatment of patients with MPNs, for many reasons but mainly because of a relatively high drop-out rate (about 20–40%) due to side effects [reviewed [19–27]. With the identification of the JAK2V617F-mutation in 2005 [63,64,65,66], reports on the potential of IFN-alpha2 to induce major molecular remissions in JAK2V617-positive patients [19, 26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50] and later on after the discovery of the CALR-mutations in 2013 [67,68,69], a reduction in the CALR-mutational load as well [39, 43], the interest in treatment of PV and related neoplasms with IFN-alpha2 has been revived as reviewed in several papers during the last 5–10 years [19,20,21,22,23,24,25,26, 55, 56]. Indeed, several studies have shown that long-term treatment with IFN-alpha2 in a subset of patients is accompanied by deep molecular remissions [30,31,32,33,34, 36,37,38, 41], which may be sustained even after discontinuation of IFN-alpha2 for up to 3 years [30, 31, 41]. These observations show that immune therapy with IFN-alpha2 is able to induce MRD (“operational cure”?) in subgroups of patients with MPNs.
Mechanisms of action of IFN-alpha2
One of the major pathways by which IFN-alpha2 exerts its actions is the Janus-activated kinase/signal transducers and activators of transcription (STAT) signal pathway. The type I IFN-dependent signallying pathways are activated by both human type I IFN-a receptor chains 1 and 2 , their intracellular domains being associated with Janus-activated kinases, which accordingly are activated upon IFN-alpha2 binding to its receptors. Janus-activated kinases phosphorylate and activate STATs (pSTAT), which then translocate to the nucleus and activate gene expression [2,3,4, 6, 70].
The mechanisms of action of IFN-alpha2 have been ascribed to its antiproliferative, proapoptotic, antiangiogenic, and immunomodulatory effects [2,3,4, 70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90]. In addition, IFN-alpha2 has also been shown to downregulate telomerase reverse transcriptase and telomerase activity in both human malignant and non-malignant hematopoietic cells [91]. As interferon-alpha2 being a telomerase-inhibitor itself [91], it has been argued that the efficacy of another telomerase-inhibitor-imetelstat which recently has been investigated in ET and myelofibrosis patients [92,93,94] might actually be mediated through IFN-alpha2 [95] by binding of imetelstat to cell-surface receptors such as toll-like receptor 9 (TLR9) [95] with ensuing TLR9-induced production of type I interferons by plasmacytoid dendritic cells [96].
In most recent years, the impact of IFN-alpha2 upon the immune system has been studied extensively in patients with MPNs [42, 60,61,62, 97] and the studies by Riley et al. [60,61,62] have paved the way for vaccination studies in Danish MPN patients [98,99,100,101,102]. These studies of JAK2V617F-positive patients have shown marked changes in circulating immune cells with low levels of NK-cells, that are boosted during treatment with IFN-alpha2 [61, 62] and profoundly changing the NK-phenotype with a significant increase in the proportion of CD56bright NK cells and a decreasing CD56dim population. The findings in this study might indicate that IFN-alpha2 treatment skews the NK cell immunity towards a more immunostimulatory profile [61].
The frequency of circulating regulatory T cells—CD4 + CD25 + Foxp3+ T cells—(Tregs) was found to be significantly increased during IFN-alpha2 treatment in all patients [60, 62]. Myeloid dendritic cells (DCs) (mDCs) and plasmacytoid DCs (pDCs) displayed decreased frequencies during the course of treatment. On both mDCs and pDCs, HLA-ABC expression was upregulated, but decreased expression levels of HLA-DR were detected on mDCs. By whole-blood transcriptional profiling studies, we have previously described significant downregulation of HLA genes and speculated whether these findings might contribute to immune evasion of MPN cells [103] thereby reflecting immunoderegulation in MPNs [104] with deregulation of several immune genes [105,106,107,108] consequently giving rise to a defective tumor immune surveillance and an increased risk of second cancers, which has been demonstrated both before and after the MPN diagnosis [105,106,107,108]. Importantly, during treatment with IFN-alpha2, the downregulated HLA genes are upregulated, indicating that IFN-alpha2 is able to restore this defective component in the impaired immune surveillance [109]. It remains to be established whether long-term treatment may also decrease or eliminate the increased risk of second cancers in MPNs [106,107,108].
Interestingly, PD-L1 expression was reduced on mDC and increased on pDCs during treatment with IFN-alpha2 [62]. Importantly, we and others have most recently found PD-L1 upregulated in MPNs, this being yet another mechanism by which the malignant cells may evade the immune system in MPNs [102, 110]. Highly intriguing, Prestipino et al. show that the JAK2V617F induces the expression of PD-L1 through activation of STAT3, thereby likely mediating the immune escape in JAK2V617F-positive MPNs [110]. Since the JAK2V617F mutation is also a generator of reactive oxygen species (ROS) [111], it is relevant to consider whether the increased PD-L1 expression by JAK2V617F is further enhanced by inflammation.
The reasons for the consistent increase in circulating Tregs after institution of IFN-alpha2 [60, 62] might reflect IFN-alpha2-mediated mobilization of Tregs to the periphery [97]. If so, the migration of Tregs from the bone marrow to the periphery may decrease their immunosuppressive and tumor-promoting influence on the marrow microenvironment. An alternative interpretation might be that this expansion of Tregs reflects a counter-response to an overall activated immune system induced by IFN-alpha2 by unknown mechanisms and, thus, indeed represents a beneficial response to prevent auto-immunity as adverse effects to treatment [62].
In the above immune cell studies, no significant correlations were found between the changes in immune cells and hematological or molecular responses, which might be partly explained by a short interval of 9-month IFN-alpha2 treatment only. Similar studies after long-term treatment with IFN-alpha2 (> 12 months) are needed to assess whether the profound changes in circulating immune cells in the initial phase of IFN-alpha2 treatment are consistent and instrumental for the beneficial effects of long-term IFN-a2 treatment in some patients [62].
In the context of immune deregulation and defective immune surveillance as being potentially important mechanisms for clonal expansion in MPNs, it is intriguing to consider that the JAK2V617F mutation has been shown to generate the accumulation of ROS [111], thereby contributing to the chronic inflammatory state in MPNs (see below). In this regard, we have also by transcriptional profiling studies described a marked deregulation of oxidative and antioxidative stress genes [112], supporting the concept of chronic inflammation as the driving force for clonal evolution in MPNs. Most recently, our mathematical modeling studies have also delivered the proof of concept for MPNs as a human inflammation model for cancer development [113].
As previously alluded to, IFN-alpha2 has profound biologic effects on the MPN stem cells [13, 14, 114,115,116]. Pietras et al. elucidated the relationship between the proliferative and suppressive effects of IFN-alpha2 during acute versus chronic drug exposure [117]. These authors showed that the cell cycle entry due to acute exposure to IFN was but transient and that HSCs re-enter into quiescence during chronic IFN-alpha2 exposure [114]. Mullaly also demonstrated in a murine model of polycythemia vera that IFN-alpha2 depletes JAK2V617F myeloproliferative neoplasm-propagating stem cells [115]. Stein et al. have excellently described the biological rationales and use of IFN in MPNs [118].
MPNs as inflammatory diseases
The MPNs are acquired stem cell diseases that include essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF) [119, 120]. A long pre-diagnostic phase with abnormal hematological parameters usually precedes the final diagnosis [121, 122]. The MPNs have a low incidence but a prevalence comparable to lung cancer, since most MPN patients live for decades, although with a huge morbidity/comorbidity burden due to a high risk of cardio- and cerebrovascular complications, an increased risk of autoimmune and chronic inflammatory diseases [120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137], and an increased risk of second cancers (SCs) [105,106,107]. Even patients in the early cancer stages (ET and PV) exhibit shorter survival than the general population [119, 120]. Most recently, these blood cancers have been described as “a human inflammation model for cancer development” [124] reflecting chronic inflammation to be a major driving force for clonal evolution and disease progression [124,125,126,127,128] and accordingly contributing substantially to the symptom burden and an impaired quality of life (QoL) [129]. Chronic inflammation is the common link between highly prevalent diseases such as atherosclerosis, the metabolic syndrome, type II diabetes, and cancer [138,139,140]. Several of the signaling pathways activated in these diseases (e.g., the JAK-STAT pathway) are constitutively activated in MPNs due to driver mutations [63,64,65,66,67,68,69, 136]. Additional mutations are associated with an increased risk of leukemic transformation [69, 141]. Chronic inflammation is also involved in the huge inflammation-mediated disease burden [120,121,122,123,124,125,126,127, 130,131,132,133,134] very similar to that seen in patients with type II diabetes.
As previously noted, chronic inflammation has been suggested to be the driving force for clonal evolution, the development of premature atherosclerosis, and secondary cancers in MPNs [124,125,126], which accordingly have been described as “a human inflammation model” [124]. However, how chronic inflammation elicits MPN is a matter of intense investigation. By generating ROS, the JAK2V617 mutation is considered to be an important inflammatory driver [111]. In MPNs, the chronic inflammatory state per se with elevated levels of several inflammatory cytokines [126], deregulation of immune and inflammation genes [142,143,144], and/or oxidative stress and anti-oxidative defense genes [112] may all contribute to defective tumor immune surveillance, being most severely affected in the advanced myelofibrosis stage, where the deregulation of the above genes is most pronounced [112, 142,143,144]. In the context of the JAK2V617F mutation as a generator of ROS, it is most intriguing to note that the JAK2V617F mutation per se may actually modulate the T cell response by generating excessive ROS through an upregulation of Akt/phosphatidylinositol-3′-kinase, which in turn decreases the amount of the ROS-converting enzyme catalase [111]. Indeed, since ROS has been shown to be a potent inhibitor of T cell function [145, 146], it is tempting to speculate if the excessive ROS might attenuate the specific immune response against the JAK2V617F-clone. The implications of excessive ROS production in MPNs have previously been described and discussed [112, 147].
Most recently, another “inflammatory” mutation has been described in the background population—the TET2-mutation [148]. The Jaiswal paper brings several important pieces to the puzzle that might associate inflammation, atherosclerosis, and second cancer in MPNs. First, the TET2 mutation gives rise to impaired resolution of inflammation by fostering the production of several inflammatory cytokines (e.g., IL-1beta and IL-6) [148] which are elevated in MPNs [126]. Second, TET2 has been shown to exacerbate JAK2V617F-induced disease by eliciting prolonged leukocytosis and extramedullary hematopoiesis with splenomegaly and a shorter survival. It was concluded that the TET2-mutations might be a disease accelerator and disease initiator and sustainer in combination with JAK2V617F in MPNs [149]. Third, TET2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation [150], which might be explained by enhanced and sustained inflammation in the stem cell compartment [151] Fourth, TET2 deficiency elicits monocytosis in mice and both the TET2 mutation and monocytosis associate with inferior prognosis in MPNs [152,153,154]. Indeed, this association may explain the high cardiovascular morbidity and mortality in MPN patients with monocytosis and perhaps also the increased mortality associated with secondary cancers [105,106,107,108, 120,121,122,123,124, 131]. Accordingly, the TET2 mutation may be yet another “inflammatory” mutation, which together with the JAK2V617F and CALR mutations may fuel the inflammatory drive, ultimately founding the soil for the development of overt MPN diseases from clonal hematopoiesis of indeterminate potential (CHIP) in the background population.
After the history on the journey of IFNs during the last 30 years, the mechanisms of action of IFN, and the novel concept of chronic inflammation as the driving force for clonal evolution in MPNs, we will in the following focus upon describing the rationales for IFN to be a successful story in the future treatment of MPNs and the perspectives for its use in MPNs. We do so by addressing some controversial issues in MPNs and provide our answers to some key questions.
Some key questions on IFN-alpha2
Does the efficacy of IFN-alpha2 reflect interference with a reactivated dormant virus—human endogenous retrovirus (HERV)?
Since IFN-alpha2 has highly potent antiviral activity, it is tempting to consider if the efficacy of IFN-alpha2 in MPNs reflects that IFN-alpha2 interferes with replication of a virus that is involved in the pathogenesis of MPNs. In this regard, particular attention has been payed to the potential role of human endogenous retrovirus (HERV), which has recently been revived as a potential causative factor for the development of MPNs [124]. Thus, the story on HERV being involved in MPN pathogenesis is not new. Indeed, HERV-K particles have been reported in megakaryocytes cultured from patients with ET [155, 156]. In the context of chronic inflammation as a potential trigger and driver of clonal evolution [124,125,126], it is intriguing to consider if the marked deregulation of inflammation and immune genes in MPNs [142,143,144]—several of these being deregulated in virus-induced malignancies as well—might be due to chronic inflammation elicited by a virus infection, e.g., reactivation of an endogenous retrovirus [124]. Thus, a chronic HERV infection of myeloid cells might account for activation of immune cells with deregulation of inflammation and immune genes. The immune attack with apoptosis of virus-infected cells might consequently elicit a sustained compensatory myeloproliferation of non-infected cells. However, ultimately, the immune system fails to clear the virus and from an early stage (ET) the disease progresses during the next 10–20 years in concert with a steady increase in bone marrow fibrosis, reflecting sustained reparative processes in an attempt to heal “the wound that won’t heal” [124, 157].
Does interferon-alpha alter the frequency and functionality of immune cells in MPNs restoring a defective tumor immune surveillance?
As alluded to previously, IFN-alpha2 induces marked alterations in both the frequency and functionality of immune cells in MPNs [60,61,62]. Whole-blood gene expression profiling studies have unraveled massive deregulation of inflammation and immune genes with downregulation of several HLA-genes of importance for tumor immune surveillance [142,143,144]. Thus, immune deregulation in MPNs is well established [104]. Importantly, treatment with IFN-alpha2 is associated with upregulation of HLA genes [109] thereby improving the defective tumor immune surveillance. It has been speculated that the defective tumor immune surveillance might not only contribute to MPN disease progression through the biological continuum from the early cancer stages (ET and PV) to the advanced metastatic myelofibrosis stage but actually might also account for the increased risk of second cancers both before and after the MPN diagnosis [108]. The observation that discontinuation of IFN-alpha2 after long-term treatment (e.g., 5 years) may be followed by several years with normal cell counts and low-JAK2V617F burden (MRD) [30, 31, 33, 41] also support the concept that IFN-alpha2—by modulation and enhancement of the immune system and accordingly the defense against cancer development—is actually able to restore a defective tumor immune surveillance in MPNs with a sustained and powerful control of the malignant clone prohibiting clonal evolution. Studies are ongoing to elucidate if IFN-alpha treatment of MPNs may also reduce the increased risk of second cancers in MPNs as recently suggested [108] and most recently preliminarily described [158, 159].
How does the mutational and cytogenetic landscape impact the efficacy of interferon-alpha2 in MPNs?
The mutational landscape in MPNs is complex and highly heterogeneous. Thus, in addition to the driver mutations—JAK2V617F, CALR, and MPL—several mutations outside the JAK-STAT pathway have been comprehensively described during the years [69, 141, 160]. Importantly, disease progression and clonal evolution in the biological continuum from the early cancer stages (ET/PV) along the path towards the advanced myelofibrosis stage have been closely linked to the development of additional subclonal mutations (ASXL1, SRSF2, CBL, IDH1/IDH2, TP53, and SRSF2), being independently associated with leukemic transformation and poor survival [69, 152, 160]. Thus, despite a low mutation rate, it has been shown that the presence of two or more somatic mutations significantly reduces overall survival and increases the risk for leukemic transformation in patients with MPNs [152].
Recent studies have suggested that mutations in the epigenetic modifiers—TET2, DNMT3A, ASXL1, EZH2, and IDH1/2—may lead to alterations in hematopoietic stem cell (HSC) function [150, 161,162,163,164]. Since IFN-alpha2 directly targets the malignant HCS [13, 14, 26], thereby potentially depleting and eliminating the disease-initiating HSC compartment [115], such alterations might negatively affect the response to IFN-alpha2. Indeed, in a small series of JAK2V617F patients, Kiladjian et al. showed that a subset had persistent TET2-positive clones during IFN-alpha2a treatment despite eradication of the JAK2 mutations, indicating that IFN-alpha2 is able to reduce or eliminate the JAK2V617F mutant clone but not the TET2 mutant clone [165]. These preliminary data might imply that patients with concurrent JAK2V617F and TET2 mutations have a less favorable response to treatment with IFN-alpha2 taking into account that the TET2 mutation—as the JAK2V617F mutation—is an “inflammatory mutation,” which gives rise to increased production of IL-6 and thereby an “inflammatory soil” in the bone marrow with potential impairment of IFN signaling and accordingly impaired clinical and molecular response to IFN-alpha2.
Highly interestingly, by serial sequencing of TET2, ASXL1, EZH2, DNMT3A, and IDH1/2 in ET and PV patients treated with pegylated IFN-alpha2a, Quintas-Cardama et al. showed that the frequency of mutations in genes outside of JAK2 was higher in patients failing to achieve a complete molecular remission (CMR) (56%) versus those achieving CMR (30%), although this difference did not reach statistical significance. Furthermore, patients not achieving CMR were more prone to acquire new mutations during therapy [36]. Of note, TET2 mutations at therapy onset had a higher JAK2V617F mutant allele burden and a less significant reduction in JAK2V617F allele burden compared with JAK2 mutant/TET2 wild-type patients [36]. Surprisingly, in this study, TET2 mutant alleles were shown to be eradicated by IFN-alpha2a in a subset of patents. However, all together, TET2 mutant clones most commonly persisted during IFN-alpha2a treatment despite eradication of JAK2V617F mutant clones [36]. The authors speculated if the discovery that mutations in TET2 [150, 162, 163, 166], DNMT3A [161], and IDH1/2 [167] elicit an increased self-renewal might actually negatively influence the ability of IFN-alpha2 to reduce or eliminate mutant MPN disease initiating cells, which harbor these mutations and accordingly conferring acquired resistance to IFN-alpha2 [36]. The authors concluded that IFN-alpha2 induces CMR in a subset of PV or ET patients, and that the molecular signature may impact clinical and molecular responses to IFN-alpha2a [36]. Larger studies are needed to assess whether mutations in TET2 and/or other genes that regulate the HSC compartment (such as DNMT3a and IDH1/2) result in persistence of malignant clones during IFN-alpha2 therapy and if their persistence indeed impact upon the prognosis of ET and PV patients being treated long-term with IFN-alpha2.
In regard to patients with early myelofibrosis, Silver and co-workers have most recently described the impact of the mutational landscape on the response to IFN-alpha2 in a phase 2 study of 30 patients with early myelofibrosis [117, 168], including their initial cohort of 17 patients [169,170,171]. The authors correlated response to IFN-alpha2 treatment with the mutation profile at the time of diagnosis, including both driver mutations (JAK2V617F, CALR, and MPL) and high risk mutations (HRMs), including ASXL1, EZH2, SRSF2, and IDH1/2 [168]. Importantly, patients with these HRM did not respond to IFN-alpha2 therapy, irrespective of spleen size. Of note, the longest surviving patient who was in complete remission for more than 25 years had a molecular profile that included positive CALR and TET2 mutation status. This observation is of utmost importance since it dictates that the TET2 mutation may not consistently imply a poor response to IFN-alpha2 treatment [117, 168]. The findings by Silver et al. suggest that treatment with IFN-alpha2 in patients with early myelofibrosis may offer a survival benefit, putting in perspective the rationales for early therapeutic intervention with IFN-alpha2 in this patient group [22, 23, 51, 120] instead of “watchful waiting,” which is recommended in patients with low-risk MF at most MPN centers. The authors argue for early intervention with IFN-alpha2 before the development of the advanced myelofibrosis stage with large splenomegaly and bone marrow failure. At this stage of increasing genomic instability and subclone formation, IFN-alpha2 has only a minor impact, in part due to the presence of HRMs. The observations by Silver et al. substantiate “The Early IFN Intervention Concept” in MPNs [22,23,24,25, 51, 120], implying treatment with IFN-alpha2 to be initiated as early after the diagnosis as possible, when the tumor burden is at a minimum, because, at this stage, IFN-alpha2 is likely to have the optimal chance of inducing MRD as defined by normalization of the bone marrow and low JAK2V617F allele burden sustained even several years after discontinuation of IFN-alpha2 [22,23,24,25, 51, 120].
Several studies of smaller series of patients have documented cytogenetic remissions during treatment with IFN-alpha2 [reviewed in 24]. In recent years, larger studies, including the above study by Quintas-Cardama et al., have convincingly confirmed that long-term treatment with IFN-alpha2 may be followed by complete cytogenetic remissions [36, 37]. Thus, this highly important observation has also been confirmed by Gisslinger et al. using the new formulation of pegylated interferon alpha (peg-proline-IFNa-2b, AOP2014/P1101) [37]. In addition to high response rates being obtained on both hematologic and molecular levels, (the JAK2V617F mutational load) peg-proline-IFNa-2b treatment also led to cytogenetic remissions in a subset of their PV patients, even in those with complex cytogenetic findings at treatment onset [37]. In a previous study, Gisslinger et al. have reported that chromosomal aberrations emerged at the time of IFN-alpha2 resistance in a patient with primary myelofibrosis [172]. The impact of the mutational and cytogenetic landscape upon the immediate and long-term responses to IFN-alpha2 in MPNs remains to be definitely described in larger studies.
How does the chronic inflammatory state in MPNs impact the efficacy of interferon-alpha2?
Chronic inflammation may impact the efficacy of IFN-alpha2 in MPNs. Thus, it has been shown that inflammatory signaling impedes the effect of IFN-alpha2 [173]. As previously alluded to, all effects of IFN-alpha2 on cells are elicited through interaction with the type I IFN receptor on the cell surface. This receptor consists of IFNAR1 and IFNAR2c chains. Among the potential mechanisms of refractoriness to IFN-alpha2 is downregulation of IFNAR1. Indeed, low levels of IFNAR1 correlate with poor response to IFN-alpha2 in patients with malignant melanoma [174]. Highly intriguing, Huang Fu et al. have shown that inflammatory cytokines interleukin 1-alpha (IL1-alpha) and tumor necrosis factor alpha (TNF-alpha)) stimulate IFNAR1 degradation and attenuate IFN-alpha signaling [173]. In patients with chronic hepatitis C, unresponsiveness to IFN-alpha is common, partly being explained by oxidative stress, impairing IFN-alpha signaling [175]. Since MPNs are associated with elevated levels of several inflammatory cytokines, including IL1-alpha and TNF-alpha, being produced by the malignant clone itself but also by the stroma cells in the bone marrow, and the highest levels have been reported in patients in the advanced myelofibrosis stage [126], these data also support the concept of early intervention with IFN-alpha2 when the inflammatory state is less pronounced. The fact that the effects of IFN-alpha are negatively impacted by inflammation may have several implications. First, one may speculate if smoking—exposing a huge systemic inflammatory load—may actually interfere with IFN signaling in MPN patients [176], implying either a weaker response to IFN-alpha2 or larger doses to be used to obtain adequate IFN responses in terms of inducing CHR. Second, agents with an anti-inflammatory potential in terms of lowering inflammatory cytokines, including IL1-alpha and TNF-alpha, might improve the IFN-alpha2 response. Indeed, the effects of IFN-alpha2 have most recently been shown to be enhanced by combination therapy with the JAK1–2 inhibitor, ruxolitinib, which is potently anti-inflammatory and immunosuppressive as well [177, 178]. Studies are ongoing to elucidate if statins, which have been suggested as potential useful agents in MPNs due to their anti-proliferative, anti-angiogenic, proapoptotic, and not least anti-inflammatory capabilities [179, 180], may also enhance the efficacy of IFN-alpha2 in MPNs. Taking into account that patients with MPNs have a 40% increased risk of second cancers [105], and statins have been shown to reduce cancer-associated mortality by 15% [181], their role in the treatment of MPNs certainly deserves to be investigated in the future [179, 180].
Do we have predictors of IFN response in MPNs?
As earlier addressed, the mutational landscape may influence the response to IFN-alpha2. Highly interestingly, Andreassson et al. have recently shown that variation in IL28B genotype influences hematologic response in IFN-alpha2-treated MPN patients [50] similar to the response to IFN-alpha2 treatment of chronic hepatitis C, which has been shown to be strongly influenced by several related single nucleotide polymorphisms (SNP) in a region adjacent to the IL28B gene [182]. These observations are of utmost importance, and if confirmed in larger studies, they may help in identifying those patients who might benefit from IFN-alpha2 treatment.
Rationales for treatment with IFN-alpha2 in MPNs
Why to treat with IFN-alpha2?
As previously addressed, IFN-alpha2 is increasingly being recognized as the treatment of choice in the early disease stages (ET, PV) and in early myelofibrosis [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50, 56] based upon safety and efficacy data on > 1000 patients being enrolled in single-arm studies during the last 30 years.
These studies have convincingly shown that complete hematological remissions (CHR) are achieved in the large majority with normalization of elevated cell counts within the first 6 months [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50, 56] being accompanied by molecular remissions with a reduction in the JAK2V617F allele burden, in many patients already within the first few months and a subset of ET and PV patients achieving major molecular remissions after about 5-year IFN-alpha2 treatment. In a subset of patients, long-term treatment with IFN-alpha2 (approximately 5 years) is associated with normalization of the bone marrow, reflecting that IFN-alpha2 is a disease-modifying agent [22,23,24,25, 30, 31, 33, 41]. Since thrombocytosis is associated with an inferior prognosis in several cancers and MPNs are associated with an increased risk of second cancers [105, 106] that also have an inferior prognosis as compared to the background population [106], it seems highly relevant to normalize elevated platelet counts in patients [108]. Importantly, elevated platelet counts may attribute to the inferior survival of second cancers in MPNs, since platelets enhance cancer invasiveness in solid tumors and accordingly their metastatic potential [183]. In addition, platelets surround tumor cells during their journey to metastatic sites, thereby protecting them from being attacked and killed by NK-cells. In this perspective, it seems most rational to normalize elevated platelet counts by IFN-alpha2, which concomitantly strongly enhance and boost the number and functionality of several immune cells, including NK-cells. These important aspects have recently been described as “The Platelet-Cancer –Loop in MPNs” [183]. Another heavy-weight rationale includes the fact that both leukocytes and platelets are deeply involved in the atherosclerotic process and leukocytosis is a risk factor for thrombosis—both in the background population and in patients with MPNs [125]. Accordingly, sustained leukocytosis and thrombocytosis are likely key players in the development of premature atherosclerosis in MPNs, being also substantiated by the association between the occurrence of the JAK2V617F-mutation and ischemic heart disease in a large epidemiological study [184]. In this study, the JAK2V617-mutation was also linked to the emergence of second cancers [184], raising the possibility that the JAK2V617F mutation actually is a “tumor promoter” not only eliciting genomic instability in blood cells but also increasing the risk of other cancers—perhaps by generating ROS and chronic inflammation in several organs other than the bone marrow compartment [108].
When to start treatment with IFN-alpha2?
All untreated cancers progress from an early stage to the advanced metastatic stage due to increasing genomic instability, subclone formation, and ultimately metastasis. As cancers, the MPNs are no exception to this general rule on cancer biology. Accordingly, institution of IFN at the earliest time point possible in MPNs may offer the best chance of a successful outcome [22,23,24,25]. The “Early IFN Intervention Concept” is based upon Danish studies, which have demonstrated that long-term treatment with IFN may induce a state of MRD as defined by deep molecular remissions (< 1% mutated JAK2V617F alleles) in concert with a normalization of the bone marrow—even being sustained in a subset of patients after discontinuation of IFN for several years [30, 31, 33, 41]. Since chronic inflammation may be a highly important driving force for clonal evolution in MPNs, combination therapy with the JAK1–2 inhibitor ruxolitinib and IFN (COMBI) has recently been suggested to be a rational treatment modality [51] being based upon the first clinical observation in a Danish PV-patient treated with COMBI [177] and the highly encouraging results in the Danish COMBI trial [178].
Side effects of IFN-alpha2
IFN-alpha2 treatment is associated with side effects that account for drop-out rates of about 20–30% in most studies, even when using low-dose pegylated IFN-alpha2 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48]. Many patients only experience the initial flu-like symptoms and afterwards they tolerate Peg-IFN-alpha2 exceedingly well. However, in some patients, chronic fatigue and/or musculoskeletal pain may persist, ultimately necessitating withdrawal of the treatment. A minority of patients develop depression which necessitates pausing or discontinuation of IFN-alpha2. In patients with previous or present psychiatric disease, IFN-alpha2 should be administered cautiously. Some patients may develop symptoms and signs of autoimmune disease. Thyroid dysfunction—thyroiditis with ensuing hypothyroidism—may develop in a subset of patients and accordingly it is recommended to test thyroid function before and during treatment. Other rare autoimmune diseases include polyarthritis, dermatomyositis, immune hemolytic anemia, immune thrombocytopenia, and glomerulonephritis.
In previous studies in patients with CML and in patients with malignant melanoma, the development of autoimmune phenomena/diseases during IFN-alpha2 treatment has been linked to an enhanced anti-leukemia or antitumor effect reflecting a very efficient immune attack on the malignant cells.
Whether similar associations exist in patients with MPNs has never been investigated. A comprehensive description of side effects to IFN-alpha2 in patients with MPNs has been given in several reviews during recent years [22,23,24,25,26].
Conclusion and perspectives
The MPNs are inflammatory cancers, in which the malignant clone per se generates inflammatory products that in a self-perpetuating vicious circle sustain the inflammatory drive and accordingly disease progression in the biological continuum from the early cancer stages (ET/PV) to the advanced “burnt-out” myelofibrosis stage and imminent leukemic transformation [120,121,122,123,124,125,126,127,128]. During this evolution, additional mutations, other than the driver mutations, emerge. The MPNs are associated with several “inflammatory” co-morbidities, including an increased risk of second cancers [105,106,107,108], which are likely due to a defective tumor immune surveillance system being partly attributed to the chronic inflammatory state [108].
The cornerstone treatment of MPNs in the future is foreseen to be IFN-alpha2, which as monotherapy in several studies during the last three decades has demonstrated safety and efficacy and as the only agent within MPNs is able to induce MRD and accordingly being disease modifying [30, 31, 33, 41]. Thus, recently, the apparent disease-modifying potential of IFN-alpha2 in PV and ET as evidenced by the progressive reduction of the JAK2V617 tumor burden during prolonged therapy has elicited renewed efforts to evaluate its clinical efficacy as front-line therapy for early stage disease in terms of reducing thrombo-hemorrhagic events, normalization of biochemical, hematologic, and molecular variables, and, ultimately, altering the natural history of these diseases.
The perspectives for the future treatment of MPNs with the goal of inducing MRD and hopefully cure in a subset of MPN patients are combination therapies, in which IFN-alpha2—primarily and directly targeting the malignant clone [26]—is being combined with agents targeting the concurrent inflammatory state (JAK1–2 inhibitors and statins), that are driving clonal expansion and disease progression [124,125,126]. The rationales for these combinations have been thoroughly described and discussed in most recent reviews [51, 124,125,126,127,128], and preliminary results from the first Danish studies are indeed very promising [177, 178]. In patients in the accelerated phase towards leukemic transformation and in patients having transformed to acute myeloid leukemia, the prognosis is dismal [185]. However, even in these stages, IFN-alpha2 may be an option [186] with the potential as monotherapy to revert imminent or overt leukemic transformation [186]. Importantly, recent studies have shown that monotherapy with the DNA-hypomethylator azacytidine [187] may be efficacious in these patients, and combination therapy with a DNA-hypomethylator and ruxolitinib may be even more efficacious [188]. Based upon the above studies of monotherapy with IFN-alpha2 and combination therapy with DNA-hypomethylating agents and ruxolitinib in patients towards or with leukemic transformation, it is intriguing to consider if “triple therapy” (IFN-alpha2 + DNA-hypomethylator + ruxolitinib) may be even more efficacious. The rationales for this “triple therapy” are several. First, such a combination directly targets the malignant clone (IFN-alpha2 + DNA-methylator) and dampens the fire—the inflammation—that fuels the malignant clone. Second, as noted above, hypomethylators have shown efficacy as monotherapy in MPN patients in the accelerated phase [187] and combination therapy (aza and ruxolitinib) seems even more efficacious [188]. Third, Aza stimulates the expression of retroviral proteins, and this expression of retroviral proteins activates immune signaling through the viral defense pathway causing a type I interferon response and apoptosis [189]. Fourth, the type I interferon response is accompanied by upregulation of hypermethylated endogenous retrovirus (ERV) genes and ERV overexpression which activates the response [190]. Fifth, by stimulating the expression of retrovirus (virus mimicry) [190], aza may render MPN cells more immunogenic and thus more susceptible to attack by immune cells. Sixth, by enhancing immune cell function, IFN may—in combination with aza—further accelerate MPN cell killing.
Most recently, the JAK2V617F and the CALR mutations, found in > 90% of patients, were shown to be highly immunogenic neo-antigens [98,99,100,101]. Additionally, patients with MPN display frequent and strong T cell responses against the immunoregulatory proteins programmed death ligand-1 (PD-L1) and arginase-1 [102, 191]. Accordingly, peptide vaccination with either JAK2-mutant or CALR-mutant epitopes in combination with vaccination against PD-L1 and/or arginase-1 may be a new and potentially curable treatment modality for MPN [98,99,100,101] as also reviewed by Holmström and Hassselbalch elsewhere in this theme issue [52].
By early detection of MPNs at the earliest time point in target populations in combination with early intervention with IFN and in subsets of patients COMBI, it is envisaged that MRD may be induced in a substantial proportion of patients along the path towards ultimate cure being obtained by novel vaccination strategies. The IFN story in MPNs will never end.
References
Isaacs A, Lindenmann J, Virus interference I (1957) The interferon. Proc R Soc Lond B Biol Sci 147:258–267
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32
Krause CD, Pestka S (2005) Evolution of the class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther 106:299–346
Platanias LC (2005) Mechanisms of type-I- and type-II-interferon mediated signalling. Nat Rev Immunol 5:375–386
de Weerd NA, Samarajiwa SA, Hertzog PJ (2007) Type I interferon receptors: biochemistry and biological functions. J Biol Chem 282: 20053–20057
Pestka S (2007) The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem 282:20047–20051
Tough DF, Sun S, Zhang X, Sprent J (1999) Stimulation of naive and memory T cells by cytokines. Immunol Rev 170:39–47
Ortaldo JR, Mason A, Rehberg E et al (1983) Effects of recombinant and hybrid recombinant human leukocyte interferons on cytotoxic activity of natural killer cells. J Biol Chem 258:15011–15015
Cantell K, Hirvonen S, Kauppinen HL, Myllyla G (1981) Production of interferon in human leukocytes from normal donors with the use of Sendai virus. Methods Enzymol 78(Part A):29–38
Mellstedt H, Bjorkholm M, Johansson B, Ahre A, Holm G, Strander H (1979) Interferon therapy in myelomatosis. Lancet 313:245–247
Kujawski LA, Talpaz M (2007) The role of interferon-alpha in the treatment of chronic myeloid leukemia. Cytokine Growth Factor Rev 18(5–6):459–471
Guilhot F, Roy L, Saulnier PJ et al (2008) Immunotherapy of chronic myelogenous leukemia. Leuk Lymphoma 49(4):629–634
Essers MAG, Offner S, Blanco-Bose WE et al (2009) IFNa activates dormant haematopoietic stem cells in vivo. Nature 458:904–908
Trumpp A, Essers M, Wilson A (2010) Awakening dormant haematopoietic stem cells. Nat Rev Immunol 10(3):201–209
Simonsson B, Gedde-Dahl T, Markevärn B, Remes K, Stentoft J, Nordic CML Study Group et al (2011) Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 118(12):3228–3235
Simonsson B, Hjorth-Hansen H, Bjerrum OW, Porkka K (2011) Interferon alpha for treatment of chronic myeloid leukemia. Curr Drug Targets 12(3):420–428
Talpaz M, Mercer J, Hehlmann R (2015) The interferon-alpha revival in CML. Ann Hematol 94(Suppl 2):S195–S207
Cayssials E, Guilhot F (2016) Beyond tyrosine kinase inhibitors: combinations and other agents. Best Pract Res Clin Haematol 29(3):271–283
Kiladjian JJ, Cassinat B, Turlure P et al (2006) High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood 108:2037–2040
Kiladjian JJ, Chomienne C, Fenaux P (2008) Interferon-alpha therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia 22(11):1990–1998
Kiladjian JJ, Mesa RA, Hoffman R (2011) The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 117(18):4706–4715
Hasselbalch HC, Larsen TS, Riley CH, Jensen MK, Kiladjian JJ (2011) Interferon-alpha in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Status and perspectives. Curr Drug Targets 12(3):392–419
Hasselbalch HC (2011) A new era of interferon-alpha2 in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol 4(6):637–655
Silver RT, Kiladjian JJ, Hasselbalch HC (2013) Interferon in the treatment of essential thrombocythemia, polycythemia vera and myelofibrosis. Expert Rev. Hematology 6(1):49–58
Hasselbalch HC, Silver RT (2015) Interferon in polycythemia vera and related neoplasms. Can it become the treatment of choice without a randomized trial? Expert Rev Hematol 8(4):439–445
Kiladjian JJ, Giraudier S, Cassinat B (2016) Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the malignant clone. Leukemia 30(4):776–781
Samuelsson J, Hasselbalch H, Bruserud O et al (2006) A phase II trial of pegylated interferon alpha- 2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 106:2397–2405
Steimle C, Lehmann U, Temerinac S et al (2007) Biomarker analysis in polycythemia vera under interferon-alpha treatment: clonality, EEC, PRV-1, and JAK2 V617F. Ann Hematol 86(4):239–244
Kiladjian JJ, Cassinat B, Chevret S et al (2008) Pegylated interferon-alfa-2a induces complete haematological and molecular responses with low toxicity in polycythemia vera. Blood 112(8):3065–3072
Larsen TS, Pallisgaard N, Moller MB, Hasselbalch HC (2008) Complete molecular remission of polycythemia vera during long-term treatment with pegylated interferon alpha-2b. Ann Hematol 87:847–850
Larsen TS, Møller MB, de Stricker K et al (2009) Minimal residual disease and normalization of the bone marrow after long-term treatment with alphainterferon2b in polycythemia vera. A report on molecular response patterns in seven patients in sustained complete haematological remission. Hematology 14(6):331–334
Quintás-Cardama A, Kantarjian H, Manshouri T et al (2009) Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 27(32):5418–5424
Larsen TS, Iversen KF, Hansen E, Mathiasen AB, Marcher C, Frederiksen M, Larsen H, Helleberg I, Riley CH, Bjerrum OW, Rønnov-Jessen D, Møller MB, de Stricker K, Vestergaard H, Hasselbalch HC (2013) Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res;37(9):1041–1045
Kuriakose E, Vandris K, Wang YL, Chow W, Jones AV, Christos P, Cross NC, Silver RT (2012) Decrease in JAK2 V617F allele burden is not a prerequisite to clinical response in patients with polycythemia vera. Haematologica 97(4):538–542
Huang BT, Zeng QC, Zhao WH, Li BS, Chen RL (2014) Interferon-alpha2b gains high sustained response therapy for advanced essential thrombocythemia and polycythemia vera with JAK2V617F positive mutation. Leuk Res 38(10):1177–1183
Quintás-Cardama A, Abdel-Wahab O, Manshouri T et al (2013) Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon α-2a. Blood 122(6):893–901
Them NC, Bagienski K, Berg T, Gisslinger B, Schalling M, Chen D, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Zagrijtschuk O, Klade C, Greil R, Gisslinger H, Kralovics R (2015) Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 90(4):288–294
Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R (2015) Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 126(15):1762–1769
Verger E, Cassinat B, Chauveau A, Dosquet C, Giraudier S, Schlageter MH, Ianotto JC, Yassin MA, Al-Dewik N, Carillo S, Legouffe E, Ugo V, Chomienne C, Kiladjian JJ (2015) Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 126(24):2585–2591
King KY, Matatall KA, Shen CC, Goodell MA, Swierczek SI, Prchal JT (2015) Comparative long-term effects of interferon α and hydroxyurea on human hematopoietic progenitor cells. Exp Hematol 43(10):912–918
Utke Rank C, Weis Bjerrum O, Larsen TS, Kjær L, de Stricker K, Riley CH, Hasselbalch HC (2015) Minimal residual disease after long-term interferon-alpha2 treatment: a report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk Lymphoma; 1–7
Kovacsovics-Bankowski M, Kelley TW, Efimova O, Kim SJ, Wilson A, Swierczek S, Prchal J (2016) Changes in peripheral blood lymphocytes in polycythemia vera and essential thrombocythemia patients treated with pegylated-interferon alpha and correlation with JAK2V617F allelic burden. Exp Hematol Oncol 5:28 eCollection 2015
Kjær L, Cordua S, Holmström MO, Thomassen M, Kruse TA, Pallisgaard N, Larsen TS, de Stricker K, Skov V, Hasselbalch HC. Differential dynamics of CALR mutant allele burden in myeloproliferative neoplasms during interferon alfa treatment. PLoS One 2016;11(10):e0165336. doi: https://doi.org/10.1371/journal.pone.0165336. eCollection 2016
O'Neill C, Siddiqi I, Brynes RK, Vergara-Lluri M, Moschiano E, O'Connell C (2016) Pegylated interferon for the treatment of early myelofibrosis: correlation of serial laboratory studies with response to therapy. Ann Hematol 95(5):733–738
Gowin K, Jain T, Kosiorek H, Tibes R, Camoriano J, Palmer J, Mesa R (2017) Pegylated interferon alpha - 2a is clinically effective and tolerable in myeloproliferative neoplasm patients treated off clinical trial. Leuk Res 54:73–77
Masarova L, Patel KP, Newberry KJ, Cortes J, Borthakur G, Konopleva M, Estrov Z, Kantarjian H, Verstovsek S (2017) Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase2 trial. Lancet Haematol 4(4):e165–e175. https://doi.org/10.1016/S2352-3026(17)30030-3 Epub 2017 Mar 11
Masarova L, Yin CC, Cortes JE, Konopleva M, Borthakur G, Newberry KJ, Kantarjian HM, Bueso-Ramos CE, Verstovsek S (2017) Histomorphological responses after therapy with pegylated interferon α-2a in patients with essential thrombocythemia (ET) and polycythemia vera (PV). Exp Hematol Oncol 6:30. https://doi.org/10.1186/s40164-017-0090-5 eCollection 2017
Crisà E, Cerrano M, Beggiato E, Benevolo G, Lanzarone G, Manzini PM, Borchiellini A, Riera L, Boccadoro M, Ferrero D (2017) Can pegylated interferon improve the outcome of polycythemia vera patients? J Hematol Oncol 10(1):15. https://doi.org/10.1186/s13045-017-0395-1
Tashi T, Swierczek S, Kim SJ, Salama ME, Song J, Heikal N, King KY, Hickman K, Litton S, Prchal JT (2018, 2018) Pegylated interferon alfa-2a and hydroxyurea in polycythemia vera and essential thrombocythemia: differential cellular and molecular responses. Leukemia. https://doi.org/10.1038/s41375-018-0080-6 [Epub ahead of print]
Lindgren M, Samuelsson J, Nilsson L, Knutsen H, Ghanima W, Westin J, Johansson PL, Andréasson B (2018) Genetic variation in IL28B (IFNL3) and response to interferon-alpha treatment in myeloproliferative neoplasms. Eur J Haematol 100(5):419–425
Bjørn ME, Hasselbalch HC (2017) Minimal residual disease or cure in MPNs? Rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev Hematol 10(5):393–404
Holmström MO, Hasselbalch HC (2018) Cancer immune therapy for myeloid malignancies – present and future. Sem Immunopathol Submitted
Linkesch W, Gisslinger H, Ludwig H, Flener R, Sinzinger H (1985) Therapy with interferon (recombinant IFN-alpha-2C) in myeloproliferative diseases with severe thrombocytosis. Acta Med Austriaca 12(5):123–127
Ludwig H, Linkesch W, Gisslinger H et al (1987) Interferon alfa corrects thrombocytosis in patients with myeloproliferative disorders. Cancer Immunol Immunother 25:266–273
Lengfelder E, Griesshammer M, Hehlmann R (1996) Interferon-alpha in the treatment of essential thrombocythemia. Leuk Lymphoma 22(Suppl 1):135–142
Lengfelder E, Berger U, Hehlmann R (2000) Interferon alpha in the treatment of polycythemia vera. Ann Hematol 79(3):103–109
Silver RT (1988) Recombinant interferon-alpha for treatment of polycythaemia vera. Lancet 2:403
Gilbert HS (1998) Long term treatment of myeloproliferative disease with interferon-alpha-2b: feasibility and efficacy. Cancer 83:1205–1213
Silver RT (2006) Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 107:451–458
Riley CH, Jensen MK, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, Svane IM (2011) Increase in circulating CD4(+) CD25(+) Foxp3(+) T cells in patients with Philadelphia-negative chronic myeloproliferative neo- plasms during treatment with IFN-alpha. Blood 118(8):2170–2173
Riley CH, Hansen M, Brimnes MK, Hasselbalch HC, Bjerrum OW, Svane IM, Jensen MK (2015) Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur J Haematol 94(3):227–234
Riley CH, Brimnes MK, Hansen M, Jensen MK, Hasselbalch HC, Kjaer L, Svane IM (2016) Interferon-alpha induces marked alterations in circulating regulatory T cells, NK cell subsets and dendritic cells in patients with JAK2 -positive essential thrombocythemia and polycythemia vera. Eur J Haematol 97(1):83–92
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR, Cancer genome project (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365(9464):1054–1061
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature;434(7037):1144–1148
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC (2005) A gain-of-function mutation of JAK2 in myeloprolif- erative disorders. N Engl J Med 352(17):1779–1790
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7(4):387–397
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD et al (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369(25):2379–2390
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC et al (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 369(25):2391–2405
Cazzola M, Kralovics R (2014) From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood 123(24):3714–3719
Billiau A (2006) Interferon: the pathways of discovery 1. Molecular and cellular aspects. Cytokine Growth Factor Rev 17:381–409
Dukes PP, Izadi P, Ortega JA, Shora NA, Gomperts E (1980) Inhibitory effects of interferon on mouse megakaryocytic progenitor cells in culture. Exp Hematol 8:1048
Lotzova E, Savary CA, Gutterman JU, Hersch EM (1982) Modulation of natural killer cell- mediated cytotoxicity by partially purified and cloned interferon-alpha. Cancer Res 42:2480–2488
Neumann HA, Fauser AA (1982) Effect of interferon on pluripotent hematopoietic progenitors (CFU-GEMM) derived from human bone marrow. Exp Hematol 10:587–590
Broxmeyer HE, Lu L, Platzer E, Feit C, Juliano L, Rubin BY (1983) Comparative analysis of the influences of human gamma, alpha and beta interferons on human multipotent (CFU-GEMM), erytrhoid (BFU-E) and granulocyte-macrophage (CFU-GM) progenitor cells. J Immunol 131:1300–1305
Ganser A, Carlo-Stella C, Greher J, Volkers B, Holzer D (1987) Effect of interferons alpha and gamma on human bone marrow-derived megakaryocytic progenitor cells. Blood 70:1173–1179
Nissen MH, Larsen JK, Plesner T, Olesen BK, Ernst P (1987) Alpha interferon induces enhanced expression of HLA-ABC antigens and beta2-microglobulin in vivo and in vitro in various subsets of human lymphoid cells. Clin Exp Immunol 69:632–638
Carlo Stella C, Cazolla M, Ganser A et al (1987) Effects of recombinant alpha and gamma interferons on the in vitro growth of circulating hematopoietic progenitor cells (CFU-GEMM, CFU-Mk, BFU-E, and CFU-GM) from patients with myelofibrosis with myeloid metaplasia. Blood 70:1014–1019
Carlo Stella C, Cazzola M (1988) Interferons as biologic modulators of hematopoietic cell proliferation and differentiation. Hematologica 1988(73):225
Gugliotta L, Bagnara GP, Catani L, Gaggiol L, Guarni A, Zauli G (1989) In vivo and in vitro inhibitory effect of interferon on megakaryocyte colony growth in essential thrombocythemia. Br J Hematol 71:177–181
Chott A, Gisslinger H, Thiele J et al (1990) Interferon-alpha-induced morphological changes of megakaryocytes: a histomorphometrical study on bone marrow biopsies in chronic myeloproliferative disorders with excessive thrombocytosis. Br J Hematol 74:10–16
Muller CA, Walz J, Zinser R, Buhring HJ, Steinke B, Schmidt H (1991) In vivo induction of HLA molecules in patients with myeloproliferative syndrome during IFN-alfa treatment. Ann Hematol 63:259–263
Wadenvik H, Kutti J, Ridelli B et al (1991) The effect of alpha-interferon on bone marrow megakaryocytes and platelet production rate in essential thrombocythemia. Blood 77:2103–2108
Franco V, Florena AM, Aragona F, Campesi G (1993) Morphometric study of the bone marrow in polycythemia vera following interferon therapy. Pathol Res Pract 189:52–57
Castello G, Lerza R, Cerruti A et al (1994) The in vitro effect of recombinant interferon-alpha-2a on circulating hematopoietic progenitors in polycythemia vera. Br J Hematol 87:621–623
Peschel C, Aulitzky WE, Huber C (1996) Influence of interferon –alpha on cytokine expression by bone marrow microenvironment – impact on treatment of myeloproliferative disorders. Leukemia Lymphoma 22(Suppl 1):129–134
Grander D, Sangfelt O, Erickson S (1997) How does interferon exert its cell growth inhibitory effect? Eur J Hematol 59:129–135
Wang O, Miyakawa Y, Fox N et al (2000) Interferon-alfa directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 96:2093–2097
Brassard DL, Grace MJ, Bordens RW (2002) Interferon-alpha as an immunotherapeutic protein. J Leucocyte Biol 71:565–581
Ferrantini M, Capone I, Belardelli F (2007) Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie 89:884–893
Bracci L, Proietti E, Belardelli F (2007) IFN-alpha and novel strategies of combination therapy for cancer. Ann N Y Acad Sci 1112:256–268
Xu D, Erickson S, Szeps M, Gruber A, Sangfelt O, Einhorn S, Pisa P, Grandér D (2000) Interferon alpha down-regulates telomerase reverse transcriptase and telomerase activity in human malignant and nonmalignant hematopoietic cells. Blood 96(13):4313–4318
Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, McDevitt MA et al (2015) Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med 373(10):920–928
Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM et al (2015) A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med 373(10):908–919
Armanios M, Greider CW (2015) Treating myeloproliferation — on target or off? N. Engl. J Med 373(10):965–966
Bjørn ME, Nielsen CH, Hasselbalch HC (2015) Telomerase inhibitor imetelstat in essential thrombocythemia and myelofibrosis. N Engl J Med 373(26):2579–2580
Frazier KS (2015) Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol Pathol 43(1):78–89
Swierczek S, Kelley TW, King KY, Ching-Chieh S, Hickman K, Kim SJ, et al (2012) Salutary effect of pegylated interferon in PV and ET as evaluated by quantitation of Pre-JAK2V617F and JAK2V617F-bearing stem cells and granulocytes and correlation with circulating regulatory T cells and HSC cell cycle status. Blood 2012 (ASH Annu Meet Abstr 2012 120 Abstr 807)
Holmström MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH (2016) The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia 30(12):2413–2416
Holmström MO, Hjortsø MD, Ahmad SM, Met Ö, Martinenaite E, Riley C et al (2017) The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 31(2):495–498
Holmström MO, Hasselbalch HC, Andersen MH (2017) The JAK2V617F and CALR exon 9 mutations are shared immunogenic neoantigens in hematological malignancy. Oncoimmunology 6(11):e1358334. https://doi.org/10.1080/2162402X.2017.1358334 eCollection 2017
Holmström MO, Martinenaite E, Ahmad SM, Met Ö, Friese C, Kjær L (2018) The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 32(2):429–437
Holmström MO, Riley CH, Skov V, Svane IM, Hasselbalch HC, Andersen MH (2018) Spontaneous T-cell responses against the immune check point programmed-death-ligand 1 (PD-L1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. Oncoimmunology 7(6):e1433521. https://doi.org/10.1080/2162402X.2018.1433521 eCollection 2018
Skov V, Riley CH, Thomassen M, Larsen TS, Jensen MK, Bjerrum OW et al (2013) Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leuk Lymphoma 54(10):2269–2273
Barosi G (2014) An immune dysregulation in MPN. Curr Hematol Malig Rep 9(4):331–339
Frederiksen H, Farkas DK, Christiansen CF et al (2011) Chronic myeloproliferative neoplasms and subsequent cancer risk: a Danish population-based cohort study. Blood 118:6515–6520
Frederiksen H, Farkas DK, Christiansen CF et al (2015) Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: a population-based cohort study. Lancet. Haematol 2:e289–e296
Pettersson H, Knutsen H, Holmberg E, Andréasson B (2015) Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur J Haematol 94(2):152–156
Hasselbalch HC (2015) Perspectives on the increased risk of second cancer in patients with essential thrombocythemia, polycythemia vera and myelofibrosis. Eur J Haematol 94:96–98
Skov V, Riley CH, Thomassen M, Kjær L, Stauffer Larsen T, Bjerrum OW, Kruse TA, Hasselbalch HC (2017) The impact of interferon-alpha2 on HLA genes in patients with polycythemia vera and related neoplasms. Leuk Lymphoma 58(8):1914–1921
Prestipino A, Emhardt AJ, Aumann K, O'Sullivan D, Gorantla SP et al (2018) Oncogenic JAK2V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med 10(429):eaam7729. https://doi.org/10.1126/scitranslmed.aam7729
Marty C, Lacout C, Droin N, Le Couédic JP, Ribrag V, Solary E, Vainchenker W, Villeval JL, Plo I (2013) A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 27(11):2187–2195
Hasselbalch HC, Thomassen M, Riley CH, Kjær L, Larsen TS, Jensen MK, Bjerrum OW, Kruse TA, Skov V (2014) Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS One 9(11):e112786. https://doi.org/10.1371/journal.pone.0112786 eCollection 2014
Andersen M, Sajid Z, Pedersen RK, Gudmand-Hoeyer J, Ellervik C, Skov V (2017) Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS One 12(8):e0183620. https://doi.org/10.1371/journal.pone.0183620 eCollection 2017
Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, Passegué E (2014) Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 211(2):245–262
Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T et al (2013) Depletion of Jak2V617F myeloproliferative neoplasm- propagating stem cells by interferon-a in a murine model of polycythemia vera. Blood 121(18):3692–36702
Hasan S, Lacout C, Marty C, Cuingnet M, Solary E, Vainchenker W et al (2013) JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNa. Blood 122(8):1464–1477. https://doi.org/10.1182/blood-2013-04-498956
Hasselbalch HC (2017) Molecular profiling as a novel tool to predict response to interferon-α2 in MPNs: the proof of concept in early myelofibrosis. Cancer 123(14):2600–2603
Stein BL, Tiu RV (2013) Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interf Cytokine Res 33(4):145–153
Campbell PJ, Green AR (2006) The myeloproliferative disorders. N Engl J Med 355(23):2452–2466
Hasselbalch HC, Bjørn ME (2015) MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediat Inflamm 102476:2015. https://doi.org/10.1155/2015/102476
Marchioli R, Finazzi G, Landolfi R et al (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23(10):2224–2232
Sørensen AL, Hasselbalch HC (2016) Antecedent cardiovascular disease and autoimmunity in Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Res 41:27–35
Enblom-Larsson A, Girodon F, Bak M, Hersby D, Jooste V, Hasselbalch HC, Johansson P, Andreasson B (2017) A retrospective analysis of the impact of treatments and blood counts on survival and the risk of vascular events during the course of polycythaemia vera. Br J Haematol. https://doi.org/10.1111/bjh.14625 [Epub ahead of print]
Hasselbalch HC (2013) Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res 37(2):214–220
Hasselbalch HC (2012) Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 119:3219–3225
Hasselbalch HC (2013) The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev 24(2):133–145
Hermouet S, Bigot-Corbel E, Gardie B (2015) Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediat Inflamm 2015:145293. https://doi.org/10.1155/2015/14529
Koschmieder S, Mughal TI, Hasselbalch HC et al (2016) Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia 30(5):1018–1024
Geyer HL, Dueck AC, Scherber RM, Mesa R (2015) Impact of inflammation on myeloproliferative neoplasm symptom burden. Mediat Inflamm 2015:284706
Bak M, Sørensen TL, Flachs EM, Zwisler AD, Juel K, Frederiksen H, Hasselbalch HC (2017) Age-related macular degeneration in patients with chronic myeloproliferative neoplasms. JAMA Ophthalmol 135(8):835–843
Christensen AS, Møller JB, Hasselbalch HC (2014) Chronic kidney disease in patients with the Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Res 38(4):490–495
Farmer S, Horváth-Puhó E, Vestergaard H, Hermann AP, Frederiksen H (2013) Chronic myeloproliferative neoplasms and risk of osteoporotic fractures; a nationwide population-based cohort study. Br J Haematol 163(5):603–610
Farmer S, Ocias LF, Vestergaard H, Broesby-Olsen S, Hermann AP, Frederiksen H (2015) Bone morbidity in chronic myeloproliferative neoplasms. Expert Rev Hematol 8(4):447–456
Farmer S, Shanbhogue VV, Hansen S, Stahlberg CI, Vestergaard H, Hermann AP, Frederiksen H (2017) Bone mineral density and microarchitecture in patients with essential thrombocythemia and polycythemia vera. Osteoporos Int 28(2):677–668
Lussana F, Rambaldi A (2017) Inflammation and myeloproliferative neoplasms. Autoimmun 85:58–63. https://doi.org/10.1016/j.jaut.2017.06.010 Epub 2017 Jun 30
Lussana F, Carobbio A, Salmoiraghi S, Guglielmelli P, Vannucchi AM, Bottazzi B et al (2017) Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J Hematol Oncol 22(10(1)):1054
Craver BM, El Alaoui K, Scherber RM, Fleischman AG (2018) The critical role of inflammation in the pathogenesis and progression of myeloid malignancies. Cancers 10(4). https://doi.org/10.3390/cancers10040104
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Pikarsky E, Porat RM, Stein I et al (2004) NF-kappaB functions as a tumour promoter in inflammation associated cancer. Nature 431(7007):461–466
Pittet MJ, Swirski FK (2011) Monocytes link atherosclerosis and cancer. Eur J Immunol 41(9):2519–2522
Mughal TI, Gotlib J, Mesa R, Koschmieder S, Khoury HJ, Cortes JE et al (2018) Recent advances in the genomics and therapy of BCR/ABL1-positive and -negative chronic myeloproliferative neoplasms. Leuk Res 67:67–74
Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse TA, Hasselbalch HC (2011) Whole-blood transcriptional profiling of interferon-inducible genes identifies highly upregulated IFI27 in primary myelofibrosis. Eur J Haematol 87(1):54–60
Skov V, Larsen TS, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse TA, Hasselbalch HC (2012) Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: identification of deregulated genes of significance for inflammation and immune surveillance. Leuk Res;36(11):1387–1392
Skov V, Thomassen M, Riley CH, Jensen MK, Bjerrum OW, Kruse TA, Hasselbalch HC, Larsen TS (2012) Gene expression profiling with principal component analysis depicts the biological continuum from essential thrombocythemia over polycythemia vera to myelofibrosis. Exp Hematol 40(9):771–780
Belikov AV, Schraven B, Simeoni L (2015) T cells and reactive oxygen species. J Biomed Sci 22:1–11
Chen X, Song M, Zhang B, Zhang Y (2016) Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev; 11–16
Bjørn ME, Hasselbalch HC (2015) The role of reactive oxygen species in myelofibrosis and related neoplasms. Mediat Inflamm 2015:648090. https://doi.org/10.1155/2015/648090
Jaiswal S, Natarajan P, Silver AJ et al (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377:111–121
Kameda T, Shide K, Yamaja T et al (2015) Loss of TET2 has dual roles in murine myeloproliferative neoplasms: disease sustainer and disease accelerator. Blood 125(2):304–315
Moran-Crusio K, Reavie L, Shih A et al (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20:11–24
Takizawa H, Boettcher S, Manz MG (2012) Demand –adapted regulation of early hematopoiesis in infection and inflammation. Blood 119(13):2991–3002
Lundberg P, Karow A, Nienhold R et al (2014) Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 123(14):2220–2228
Ortmann CA, Kent DA, Nangalia J et al (2015) Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 372:601–612
Barraco D, Cerquozzi S, Gangat N et al (2017) Monocytosis in polycythemia vera: clinical and molecular correlates. Am J Hematol 92(7):640–645
Boyd MT, Maclean N, Oscier DG (1989) Detection of retrovirus in patients with myeloproliferative disease. Lancet 1(8642):814–817
Morgan D, Brodsky I (2004) Human endogenous retrovirus (HERV-K) particles in megakaryocytes cultured from essential thrombocythemia peripheral blood stem cells. Exp Hematol 32(6):520–525
Dvorak HF (1986) Tumors: wounds that do not heal, Similarities between tumor stroma generation and wound healing. N Engl J Med 315(26):1650–1659
Kissova J, Ovesna P, Penka M, Bulikova A, Kiss I (2014) Second malignancies in Philadelphia-negative myeloproliferative neoplasms- single-center experience. Anticancer Res 34:2489–2496
Hansen IO, Sørensen AL, Hasselbalch HC (2017) Second malignancies in hydroxyurea and interferon-treated Philadelphia-negative myeloproliferative neoplasms. Eur J Haematol 98(1):75–84
Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA (2011) New mutations and pathogenesis of myeloproliferative neoplasms. Blood 118(7):1723–1735
Challen GA, Sun D, Jeong M et al (2012) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44(1):23–31
Quivoron C, Couronn’e L, Della Valle V et al (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20(1):25–38
Li Z, Cai X, Cai CL et al (2011) Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118(17):4509–4518
Figueroa ME, Abdel-Wahab O, Lu C et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18(6):553–567
Kiladjian JJ, Masse A, Cassinat B et al (2010) French intergroup of myeloproliferative neoplasms (FIM). Clinical analysis of erythroid progenitors suggests that pegylated interferon alpha-2a treatment targets JAK2V617F clones without affecting TET2 mutant cells. Leukemia 24(8):1519–1523
Ko M, Bandukwala HS, An J et al (2011) Ten-eleventranslocation2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 108(35):14566–14571
Sasaki M, Knobbe CB, Munger JC et al (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488(7413):656–659
Silver RT, Barela A, Lascu E et al (2017) The effect of initial molecular profile on response to recombinant interferon alfa (rIFNa) treatment in early myelofibrosis. Cancer 123:2680–2687
Silver RT, Vandris K (2009) Recombinant interferon alpha (rIFN alpha-2b) may retard progression of early myelofibrosis. Leukemia 23:1366–1369
Silver RT, Vandris K, Goldman JJ (2011) Recombinant interferon alpha may retard progression of early primary myelofibrosis: a preliminary report. Blood 117:6669–6672
Pizzi M, Silver RT, Barel A, Orazi A Recombinant interferon-a in myelofibrosis reduces bone marrow fibrosis, improves its morphology and is associated with clinical response. Mod Pathol 28:1315–1323
Buxhofer-Ausch V, Gisslinger H, Berg T et al (2009) Acquired resistance to interferon alpha therapy associated with homozygous MPL-W515L mutation and chromosome 20q deletion in primary myelofibrosis. Eur J Haematol 82:161–163
Wei-Chun HF, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, Fuchs SY (2012) Inflammatory signaling compromises cell responses to interferon. Oncogene 31(2):161–172
Messina JL, Yu H, Riker AI, Munster PN, Jove RI, Daud AI (2008) Activated STAT-3 in melanoma. Cancer Control 15:196–201
Bona DD, Cippitelli M, Fionda C, Camma C, Licata A, Santoni A, Craxi A (2006) Oxidative stress inhibits IFN-alpha2-induced antiviral gene expression by blocking the JAK-STAT pathway. J Hepatol 45:271–279
Hasselbalch HC (2015) Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk Res pii: S0145-2126(15)30373–8. doi: https://doi.org/10.1016/j.leukres.2015.09.002. [Epub ahead of print]
Bjørn ME, de Stricker K, Kjær L, Ellemann K, Hasselbalch HC (2014) Combination therapy with interferon and JAK1-2 inhibitor is feasible: proof of concept with rapid reduction in JAK2V617F-allele burden in polycythemia vera. Leuk Res Rep 3(2):73–75
Mikkelsen SU, Kjaer L, Bjørn ME, Knudsen TA, Sørensen AL, Andersen CBL et al (2018) Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med 2018 Jun 22. doi: https://doi.org/10.1002/cam4.1619. [Epub ahead of print]
Hasselbalch HC, Riley CH (2006) Statins in the treatment of polycythaemia vera and allied disorders: an antithrombotic and cytoreductive potential? Leuk Res 30(10):1217–1225
Sørensen AL, Kallenbach K, Hasselbalch HC (2016) A remarkable hematological and molecular response pattern in a patient with polycythemia vera during combination therapy with simvastatin and alendronate. Leuk Res Rep 6:20–33
Nielsen SF, Nordestgaard BG, Bojesen SE (2012) Statin use and reduced cancer-related mortality. N Engl J Med 367(19):1792–1802
Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ et al (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401
Hasselbalch HC (2014) The platelet-cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk Res 38(10):1230–1236
Nielsen C, Birgens HS, Nordestgaard BG, Bojesen SE (2013) Diagnostic value of JAK2 V617F somatic mutation for myeloproliferative cancer in 49 488 individuals from the general population. Br J Haematol 160(1):70–79
Mollard LM, Chauveau A, Boyer-Perrard F, Douet-Guilbert N, Houot R, Quintin-Roué I et al (2018) Outcome of Ph negative myeloproliferative neoplasms transforming to accelerated or leukemic phase. Leuk Lymphoma. 2018 :1-9. doi: https://doi.org/10.1080/10428194.2018.1441408. [Epub ahead of print]
Anguille S, Lion E, Willemen Y, Van Tendeloo VF, Berneman ZN, Smits EL (2011). Interferon-α in acute myeloid leukemia: an old drug revisited. Leukemia;25(5):739–748
Thepot S, Itzykson R, Seegers V, Raffoux E, Quesnel B, Chait Y, Groupe Francophone des Myelodysplasies (GFM) et al (2010) treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 116(19):3735–3742
Assi R, Kantarjian HM, Garcia-Manero G, Cortes JE, Pemmaraju N, Wang X et al (2018) A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am J Hematol 93(2):277–285
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B et al (2015) Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162(5):974–986
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY et al (2015) DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162(5):961–973
Aaboe-Jørgensen M, Holmstrøm MO, Martinenaite E, Riley CH, Hasselbalch HC, Andersen MH (2018) Spontaneous T-cell responses against Arginase-1 in chronic myeloproliferative neoplasms relative to disease stage and type of driver mutation. OncoImmunology doi: 10.1080/2162402X.2018.1468957
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No authors have conflicts of interest to disclose. However, it should be noted that Morten Orebo Holmström and Hans Carl Hasselbalch together with Mads Hald Andersen have filed a patent regarding the CALR exon 9 mutations and JAK2V61F-mutation as a target for cancer immune therapy. The patent has been transferred to University Hospital Zealand, Zealand Region, and Copenhagen University Hospital at Herlev, Capital Region, according to Danish Law concerning inventions made at public research institutions.
Additional information
This article is a contribution to the special issue on Anti-cancer immunotherapy: Breakthroughs and Future strategies - Guest Editor: Mads Hald Andersen
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hasselbalch, H.C., Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure?. Semin Immunopathol 41, 5–19 (2019). https://doi.org/10.1007/s00281-018-0700-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-018-0700-2