
Abstract
The randomized phase III ADMYRE trial evaluated plitidepsin plus dexamethasone (DXM) versus DXM alone in patients with relapsed/refractory multiple myeloma after at least three but not more than six prior regimens, including at least bortezomib and lenalidomide or thalidomide. Patients were randomly assigned (2:1) to receive plitidepsin 5 mg/m2 on D1 and D15 plus DXM 40 mg on D1, D8, D15, and D22 (arm A, n = 171) or DXM 40 mg on D1, D8, D15, and D22 (arm B, n = 84) q4wk. The primary endpoint was progression-free survival (PFS). Median PFS without disease progression (PD) confirmation (IRC assessment) was 2.6 months (arm A) and 1.7 months (arm B) (HR = 0.650; p = 0.0054). Median PFS with PD confirmation (investigator’s assessment) was 3.8 months (arm A) and 1.9 months (arm B) (HR = 0.611; p = 0.0040). Median overall survival (OS, intention-to-treat analysis) was 11.6 months (arm A) and 8.9 months (arm B) (HR = 0.797; p = 0.1261). OS improvement favoring arm A was found when discounting a crossover effect (37 patients crossed over from arm B to arm A) (two-stage method; HR = 0.622; p = 0.0015). The most common grade 3/4 treatment-related adverse events (% of patients arm A/arm B) were fatigue (10.8%/1.2%), myalgia (5.4%/0%), and nausea (3.6%/1.2%), being usually transient and reversible. The safety profile does not overlap with the toxicity observed with other agents used in multiple myeloma. In conclusion, efficacy data, the reassuring safety profile, and the novel mechanism of action of plitidepsin suggest that this combination can be an alternative option in patients with relapsed/refractory multiple myeloma after at least three prior therapy lines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The prognosis for patients with multiple myeloma (MM) who are refractory to both proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs) is poor: with further treatment, the median survival is 9 months and 3 months in patients without further treatment [1]. Furthermore, treatment options for MM decrease with each relapse and outcomes with subsequent treatment using standard therapies are characterized by short duration of response and increasing drug resistance [2]. Therefore, there is a need for alternative antimyeloma treatments for patients with advanced illness following refractory/multiply relapsed disease.
Plitidepsin is a cyclic depsipeptide, originally isolated from a Mediterranean marine tunicate, Aplidium albicans, which is currently produced by total synthesis. Plitidepsin effects are related to the induction of early oxidative stress, which induces the sustained activation of c-Jun N-terminal kinase (JNK) and p38MAPK and finally apoptosis [3, 4]. The eukaryotic translation elongation factor 1 alpha 2 (eEF1A2), a protein which is overexpressed in MM, has been identified as the primary target for plitidepsin [5]. In vitro studies showed antiproliferative activity against several human MM cell lines [6, 7]. In vivo studies showed an antitumour effect of plitidepsin in xenograft MM models as a single agent or in combination with dexamethasone (DXM) [6]. Plitidepsin plus DXM showed activity in a phase II clinical trial conducted in relapsed/refractory MM patients [8]. Based on the data from the randomized phase III ADMYRE trial, which compared plitidepsin plus DXM with DXM alone, this plitidepsin combination has been recently approved in Australia for the treatment of patients with relapsed/refractory MM who have received at least three prior treatment regimens, including both a PI and an IMiD. Plitidepsin plus DXM has been also approved for its use after two prior lines of therapy in the case of patients refractory and/or intolerant to both a PI and an IMiD. Efficacy and safety results from the ADMYRE trial are presented here.
Methods
Patients
Patients were recruited worldwide at 61 investigational sites from 17 countries. The study protocol received protocol assistance by the Committee for Medicinal Products for Human Use (CHMP), was approved by the Independent Local Ethics Committee of each participating center, and was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulations on clinical trials. Signed informed consent was obtained from all patients prior to any study-specific procedure (ClinicalTrials.gov identifier: NCT01102426).
Eligibility criteria included the following: patients ≥ 18 years old with relapsed/refractory MM after at least three, but not more than six, prior therapeutic regimens, including at least bortezomib and lenalidomide or thalidomide; measurable disease; Eastern Cooperative Oncology Group performance status (ECOG PS) ≤ 2; life expectancy ≥ 3 months; and adequate major organ function. Patients were excluded if they had the following: a concomitant unstable or serious medical condition (e.g., myocardial infarction, angina, congestive heart failure, severe dyspnea or oxygen requirement, active uncontrolled infection, immune deficiency, or myopathy), grade > 2 peripheral neuropathy, myelodysplasia and/or post-chemotherapy aplasia, or mood disturbance associated with previous steroid-based therapy.
Treatment
In 2009, at the time of designing the ADMYRE trial, no standard treatment existed for the intended population (MM patients relapsed/refractory to all standard available therapy) that could be considered a gold standard comparator, and treatment options were limited. DXM was an active compound widely used as a single agent as well as part of combination regimens for treatment of MM patients in different settings. For that reason, the European Society of Medical Oncology recommendations included DXM added to these agents as treatment for relapsed/refractory MM [9]. On this basis, the choice of the control arm (low-dose DXM) was agreed with the health authorities (European Medicines Agency and Food and Drug Administration of the USA).
Patients were stratified according to their ECOG PS score (0 and 1 vs. 2) and Durie-Salmon stage (I/II vs. III) and were randomly assigned (with a 2:1 ratio) to receive plitidepsin 5 mg/m2 on D1 and D15 intravenously (i.v.) over 3 h plus DXM 40 mg orally on D1, D8, D15, and D22 (arm A) or DXM 40 mg orally on D1, D8, D15, and D22 (arm B), every 4 weeks (q4wk). Patients in the control arm (DXM alone, arm B) with documented disease progression after a minimum of 8 weeks from randomization could crossover to the combination arm (arm A).
Efficacy assessment
The primary efficacy endpoint was progression-free survival (PFS), which was assessed according to an Independent Review Committee (IRC) per the International Myeloma Working Group (IMWG) criteria current at the time of study protocol design (i.e., without requiring PD confirmation for assigning a PFS event) [2]. Furthermore, a preplanned sensitivity analysis of PFS by an investigator’s assessment was performed following the revised IMWG criteria (i.e., with PD confirmation required for assigning a PFS event) [10]. Secondary efficacy endpoints were objective response rate (ORR; ≥ partial response) according to the IMWG criteria, duration of response, and overall survival (OS).
Safety assessment
Safety was evaluated in all patients who received at least one dose of the study treatment by assessment of adverse events (AEs), clinical laboratory test results, physical examinations, and vital signs. AEs were recorded and coded with the Medical Dictionary for Regulatory Activities (MedDRA) v.16.0. AEs and laboratory values were graded according to the National Cancer Institute-Common Toxicity Criteria for Adverse Events (NCI-CTCAE) v.4. All patients were followed until recovery from any treatment-related AE. Follow-up was longer in arm A partly because it included patients that had crossed over from arm B.
Statistical methods
The number of randomized patients required was calculated on the basis of PFS estimates from the previous phase II study in MM [8]. Approximately 210 progression or death events were required to reject the equality of hazard rates (HR) between both treatment arms, assuming a HR of 0.625 in favor of the combination arm (90% power, 1-sided 2.5% significance level). A futility analysis was done once 40 patients in arm A were evaluable for response. ORR including minor response (MR) was 37.8% (planned threshold was 30%), and therefore patient accrual was resumed. The final PFS analysis was done with data obtained in November 2015.
The unstratified log-rank test was used to compare PFS. Cox regression was used to calculate the risk reduction in PFS. Binomial estimates with exact 95% confidence intervals (CIs) were calculated for the analysis of ORR. Fisher’s exact test was used to compare ORR.
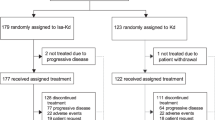
The study was powered for the evaluation of the main endpoint, PFS, and for ascertaining if a trend in OS is observed in favor of the experimental arm. The final OS analysis was done according to the Kaplan-Meier method 2 years after the last patient inclusion (May 2017). Nevertheless, the potential to detect statistically significant differences in OS was hampered by crossover: 44.0% of patients from arm B crossed over to arm A (Fig. 1). Then, a post hoc sensitivity analysis based on the two-stage method proposed by Latimer et al. [11], which was previously reported when evaluating OS in another MM trial [12], was performed.

Study flow chart. AE, adverse event; DXM, dexamethasone
Results
Patient characteristics
A total of 255 patients were randomized (2:1) between June 2010 and May 2015: 171 in arm A and 84 in arm B. Of these, 167 patients were treated in arm A and 83 in arm B (Fig. 1). Baseline characteristics were well balanced between treatment arms (Table 1). The median number of lines of previous therapy was 4 in both treatment arms. In both arms, around 74% of patients had refractory or relapsed and refractory disease to the last line (38–39% of them relapsed and refractory to lenalidomide/thalidomide and bortezomib).
Efficacy
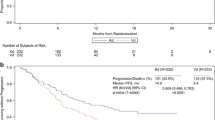
The primary efficacy analysis, with blinded IRC assessment of all randomized patients done without PD confirmation, showed statistically significant longer PFS for patients treated with plitidepsin plus DXM. Median PFS was 2.6 months (95% CI, 1.9–3.0 months) in arm A (plitidepsin plus DXM) and 1.7 months (95% CI, 1.1–2.0 months) in arm B (DXM) (log-rank p = 0.0054) (Fig. 2). The relative risk of progression or death was reduced by 35.0% in patients treated with plitidepsin plus DXM (HR = 0.650; 95% CI 0.477–0.885, p = 0.0062).

Kaplan-Meier curve for progression-free survival by the Independent Review Committee. A, arm A (plitidepsin plus DXM); B, arm B (DXM); C, censored; DXM; dexamethasone; N, number of patients
PFS analysis requiring PD confirmation by an investigator’s assessment showed a median PFS of 3.8 months (95% CI, 2.9–5.6 months) in arm A (plitidepsin plus DXM) and 1.9 months (95% CI, 1.1–2.7 months) in arm B (DXM), with a relative risk of progression or death reduced by 38.9% in patients treated with plitidepsin plus DXM (HR = 0.611; p = 0.0048).
ORR according to the IRC in evaluable patients was 13.8% (95% CI, 8.3–21.2%) in arm A (plitidepsin plus DXM; n = 123), which included two very good partial responses and 15 partial responses (median duration of response was 12.0 months), and 1.7% (95% CI, 0.04–9.1%) in arm B (DXM; n = 59) (one patient with partial response; duration of 1.8 months) (p < 0.0080).
The clinical benefit rate, defined as patients with response (including MR) or stable disease (SD), was 48.0% (95% CI, 40.3–55.7%) in arm A (plitidepsin plus DXM) and 28.6% (95% CI, 19.2–39.5%) in arm B (DXM) (p < 0.0044).
The final intention-to-treat OS analysis was based on 195 death events (76.5% of the 255 randomized patients). Median OS was 11.6 months (95% CI, 9.2–16.1 months) in arm A (plitidepsin plus DXM) and 8.9 months (95% CI, 6.0–15.4 months) in arm B (DXM) (log-rank p = 0.1261) (Fig. 3a). Despite crossover, relative risk of death was reduced by 20.3% in patients treated with plitidepsin plus DXM (HR = 0.797; p = 0.1273). Median OS for patients with response or clinical benefit in arm A (plitidepsin plus DXM) was 37.8 months and 30.3 months, respectively (Fig. 4).

Kaplan-Meier curves for overall survival in all randomized patients (a) and all randomized patients (two-stage method) (b). A, arm A (plitidepsin plus DXM); B, arm B (DXM); C, censored; DXM, dexamethasone; N, number of patients

Progression-free survival, post-progression survival, and overall survival according to the response observed. IRC, Independent Review Committee; MR, minor response; PFS, progression-free survival; PR, partial response; SD, stable disease
A two-stage OS analysis, which mitigates the effect of crossover, showed a statistically significant difference in favor of arm A (plitidepsin plus DXM). Median OS remained as 11.6 months (95% CI, 9.2–16.1 months) in arm A (plitidepsin plus DXM) and was estimated as 6.4 months (95% CI, 5.1–8.3 months) in arm B (DXM) (log-rank p = 0.0015) (Fig. 3b). Relative risk of death was reduced by 37.8% in patients treated with plitidepsin plus DXM (HR = 0.622; p = 0.0016).
Safety
All treated patients were evaluable for safety. The median (range) of cycles received was 3 (1–33) in arm A (plitidepsin plus DXM; total cycles = 842) and 2 (1–21) in arm B (DXM; total cycles = 251). Median time on treatment was 12.3 weeks (1.3–137.1 weeks) in arm A and 8.3 weeks (1.4–85.3 weeks) in arm B. Fifteen patients (9.0%) discontinued treatment because of treatment-related AEs in arm A and eight patients (9.6%) in arm B.
In arm A (plitidepsin plus DXM), the most common AEs (all grades) related to the study treatment (or with unknown causality) were nausea (37.1% of patients), fatigue (36.5%), vomiting (16.8%), diarrhea (14.4%), myalgia (14.4%), peripheral edema (12.0%), decreased appetite (12.6%), and muscular weakness (9.6%). The most common grade 3/4 treatment-related (or with unknown causality) AEs were fatigue (10.8%), myalgia (5.4%), muscular weakness (3.6%), and nausea (3.6%) (Table 2). Other grade 3/4 AEs of specific interest were as follows: creatine phosphokinase (CPK) increase (20.0%), alanine aminotransferase (ALT) increase (14.5%), peripheral sensory neuropathy (0.6%), and infection/pneumonia (2.4%). One patient died following a treatment-related AE (grade 4 myopathy) after having received one cycle (n = 1/167 patients; 0.6%).
In arm B (DXM), the most common AEs (all grades) related to the study treatment (or with unknown causality) were nausea (10.8%), fatigue (8.4%), and insomnia (9.6%). All grade 3/4 AEs occurred in one (1.2% of patients) or two patients (2.4%) each (Table 2). One patient died following a treatment-related AE (grade 4 respiratory tract infection) after having received two cycles (n = 1/83 patients; 1.2%).
Discussion
The ADMYRE study met its primary endpoint, demonstrating a 35% reduction in the relative risk of progression or death for the combination of plitidepsin plus DXM compared with DXM alone in relapsed/refractory MM patients pretreated with at least three regimens, including bortezomib and either lenalidomide or thalidomide. Short PFS values reported in the primary PFS analysis (2.6 months in arm A; 1.7 months in arm B) can be explained by the conservative adjudication of PD time points, which were calculated according to the IMWG criteria version current when the study was planned and designed [2], but also to the advanced stage of the heavily pretreated population, who generally had exhausted most available therapies. A preplanned sensitivity analysis of PFS in line with updated IMWG criteria [10], which require confirmation of PD in two consecutive assessments, showed a longer PFS value for the combination (3.8 months), with the difference between treatments reaching almost 2 months.
The reduction in the relative risk of death observed with mature survival data (23.5% of patients censored) was 20.3% despite the substantial crossover rate (44.0% of patients). The finding of a similar outcome in survival, a secondary but robust endpoint, when discounting the effect of crossover in the two-stage method (statistically significant 37.8% reduction in the risk of death relative to the control arm) supports the benefit demonstrated in PFS, the primary study endpoint (statistically significant 35.0% reduction in the relative risk of progression or death). Survival results for patients with response (median OS of 37.8 months, with a median duration of response of 12.0 months) or clinical benefit (median OS of 30.3 months) are of interest in the setting of a pretreated MM patient population (expected median survival is about 9 months).
Several drugs (carfilzomib, ixazomib, pomalidomide, daratumumab, elotuzumab, and panobinostat) have been more recently approved for the treatment of patients in whom the use of bortezomib and lenalidomide has been exhausted. Beyond these, there are few options for salvage treatment, which are limited to re-challenge with a previously used treatment (alone or in combination with corticosteroids or other novel agents), use of older drugs (e.g., thalidomide, melphalan, vincristine, doxorubicin, etoposide, bendamustine, and carmustine), or enrolment in a clinical trial [1].
Carfilzomib (a new irreversible PI) was evaluated in a phase II trial in patients pretreated with five lines of therapy (95% of whom had disease refractory to the last therapy) [13]. Median PFS was 3.7 months, and median OS in patients refractory to both bortezomib and lenalidomide was 15.6 months. The data from the ADMYRE study was obtained in a population less heavily pretreated (four prior lines; 74% of patients with refractory disease). Nevertheless, a median PFS of 3.8 months (updated IMWG criteria) [10] and a median OS of 11.6 months observed with plitidepsin plus DXM confirm a level of activity close to that observed for carfilzomib.
Daratumumab, a human monoclonal IgG1 antibody that binds to the CD38 myelomatous cell surface antigen, was evaluated in a phase II study (SIRIUS) in patients who had received at least three prior lines of therapy or were refractory to the most recent PI and IMiD combination [14]. The median duration of response was 7.4 months and median PFS was 3.7 months. In the ADMYRE study, conducted in a heavily pretreated and largely refractory population, the median duration of response was longer (12.0 months) in patients achieving VGPR or PR, and median PFS (updated IMWG criteria) was similar (3.8 months).
Pomalidomide plus low-dose DXM was evaluated in a randomized phase III study (MM-003) in MM patients pretreated with a median of five previous regimens (82% of them with refractory disease status) [15]. The median PFS (4.0 months) and median OS (12.7 months) were quite similar to the ADMYRE results.
Plitidepsin plus DXM showed a low incidence of toxicities that are common with available agents used in the treatment of relapsed/refractory MM, such as venous thromboembolism, neurotoxicity, neutropenia and associated infections, thrombocytopenia and associated bleeding, or cardiac events [13,14,15,16,17,18,19], which represents a favorable safety profile in a disease setting of heavily pretreated patients. Most of the toxicities observed with plitidepsin plus DXM were transient, non-cumulative laboratory abnormalities that usually occur in the first two cycles of treatment and are controlled by dose adjustment (cycle delay, dose omission, and in ultimate instance, dose reduction). eEF1A2 is overexpressed in MM [20] and has been identified as the primary intracellular target of plitidepsin [5, 21]. eEF1A2 is responsible for the enzymatic delivery of aminoacyl-tRNAs to the ribosome, but also has pro-oncogenic activities including regulation of oxidative stress [22], inhibition of apoptosis [23], or control of unfolded protein degradation by the proteasome [24]. All recently introduced new myeloma drugs have mechanisms of activity not targeting eEF1A2; this fact, together with the favorable safety profile and the lack of overlapping toxicities with commonly used agents, places plitidepsin as an alternative option for designing combinations or even for its administration after relapse in patients treated with immunotherapy.
In conclusion, the combination of plitidepsin and DXM has shown antimyeloma activity compared with DXM alone, introduces a new agent with a novel mechanism of action into the MM therapeutic armamentarium, has an acceptable safety profile different from that of PIs, IMiDs, or histone deacetylase inhibitors, and could thus be considered an alternative treatment option for patients with relapsed/refractory MM.
References
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, Haessler J, Feather J, Hoering A, Moreau P, LeLeu X, Hulin C, Klein SK, Sonneveld P, Siegel D, Blade J, Goldschmidt H, Jagannath S, Miguel JS, Orlowski R, Palumbo A, Sezer O, Rajkumar SV, Durie BG, International Myeloma Working G (2012) Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 26(1):149–157. https://doi.org/10.1038/leu.2011.196
Anderson KC, Kyle RA, Rajkumar SV, Stewart AK, Weber D, Richardson P, Myeloma AFPCEM (2008) Clinically relevant end points and new drug approvals for myeloma. Leukemia 22(2):231–239. https://doi.org/10.1038/sj.leu.2405016
Cuadrado A, Garcia-Fernandez LF, Gonzalez L, Suarez Y, Losada A, Alcaide V, Martinez T, Fernandez-Sousa JM, Sanchez-Puelles JM, Munoz A (2003) Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. J Biol Chem 278(1):241–250. https://doi.org/10.1074/jbc.M201010200
Cuadrado A, Gonzalez L, Suarez Y, Martinez T, Munoz A (2004) JNK activation is critical for Aplidin-induced apoptosis. Oncogene 23(27):4673–4680. https://doi.org/10.1038/sj.onc.1207636
Losada A, Munoz-Alonso MJ, Garcia C, Sanchez-Murcia PA, Martinez-Leal JF, Dominguez JM, Lillo MP, Gago F, Galmarini CM (2016) Translation elongation factor eEF1A2 is a novel anticancer target for the marine natural product plitidepsin. Sci Rep 6:35100. https://doi.org/10.1038/srep35100
Mitsiades CS, Ocio EM, Pandiella A, Maiso P, Gajate C, Garayoa M, Vilanova D, Montero JC, Mitsiades N, McMullan CJ, Munshi NC, Hideshima T, Chauhan D, Aviles P, Otero G, Faircloth G, Mateos MV, Richardson PG, Mollinedo F, San-Miguel JF, Anderson KC (2008) Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res 68(13):5216–5225. https://doi.org/10.1158/0008-5472.CAN-07-5725
Depenbrock H, Peter R, Faircloth GT, Manzanares I, Jimeno J, Hanauske AR (1998) In vitro activity of aplidine, a new marine-derived anti-cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. Br J Cancer 78(6):739–744
Mateos MV, Cibeira MT, Richardson PG, Prosper F, Oriol A, de la Rubia J, Lahuerta JJ, Garcia-Sanz R, Extremera S, Szyldergemajn S, Corrado C, Singer H, Mitsiades CS, Anderson KC, Blade J, San Miguel J (2010) Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin Cancer Res 16(12):3260–3269. https://doi.org/10.1158/1078-0432.CCR-10-0469
Moreau P, San Miguel J, Ludwig H, Schouten H, Mohty M, Dimopoulos M, Dreyling M, Group EGW (2013) Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 24(Suppl 6):vi133–vi137. https://doi.org/10.1093/annonc/mdt297
Rajkumar SV, Harousseau JL, Durie B, Anderson KC, Dimopoulos M, Kyle R, Blade J, Richardson P, Orlowski R, Siegel D, Jagannath S, Facon T, Avet-Loiseau H, Lonial S, Palumbo A, Zonder J, Ludwig H, Vesole D, Sezer O, Munshi NC, San Miguel J, International Myeloma Workshop Consensus P (2011) Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 117(18):4691–4695. https://doi.org/10.1182/blood-2010-10-299487
Latimer NR, Abrams KR, Lambert PC, Crowther MJ, Wailoo AJ, Morden JP, Akehurst RL, Campbell MJ (2014) Adjusting survival time estimates to account for treatment switching in randomized controlled trials--an economic evaluation context: methods, limitations, and recommendations. Med Decis Mak 34(3):387–402. https://doi.org/10.1177/0272989X13520192
Morgan G, Palumbo A, Dhanasiri S, Lee D, Weisel K, Facon T, Delforge M, Oriol A, Zaki M, Yu X, Sternas L, Jacques C, Akehurst R, Offner F, Dimopoulos MA (2015) Overall survival of relapsed and refractory multiple myeloma patients after adjusting for crossover in the MM-003 trial for pomalidomide plus low-dose dexamethasone. Br J Haematol 168(6):820–823. https://doi.org/10.1111/bjh.13227
Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, Chanan-Khan A, Buadi F, Reu FJ, Somlo G, Zonder J, Song K, Stewart AK, Stadtmauer E, Kunkel L, Wear S, Wong AF, Orlowski RZ, Jagannath S (2012) A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 120(14):2817–2825. https://doi.org/10.1182/blood-2012-05-425934
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, Belch A, Krishnan A, Vescio RA, Mateos MV, Mazumder A, Orlowski RZ, Sutherland HJ, Blade J, Scott EC, Oriol A, Berdeja J, Gharibo M, Stevens DA, LeBlanc R, Sebag M, Callander N, Jakubowiak A, White D, de la Rubia J, Richardson PG, Lisby S, Feng H, Uhlar CM, Khan I, Ahmadi T, Voorhees PM (2016) Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 387(10027):1551–1560. https://doi.org/10.1016/S0140-6736(15)01120-4
San Miguel J, Weisel K, Moreau P, Lacy M, Song K, Delforge M, Karlin L, Goldschmidt H, Banos A, Oriol A, Alegre A, Chen C, Cavo M, Garderet L, Ivanova V, Martinez-Lopez J, Belch A, Palumbo A, Schey S, Sonneveld P, Yu X, Sternas L, Jacques C, Zaki M, Dimopoulos M (2013) Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol 14(11):1055–1066. https://doi.org/10.1016/S1470-2045(13)70380-2
Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, San Miguel J, Hellmann A, Facon T, Foa R, Corso A, Masliak Z, Olesnyckyj M, Yu Z, Patin J, Zeldis JB, Knight RD, Multiple Myeloma Study I (2007) Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 357(21):2123–2132. https://doi.org/10.1056/NEJMoa070594
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352(24):2487–2498. https://doi.org/10.1056/NEJMoa043445
Richardson PG, Schlossman RL, Alsina M, Weber DM, Coutre SE, Gasparetto C, Mukhopadhyay S, Ondovik MS, Khan M, Paley CS, Lonial S (2013) PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 122(14):2331–2337. https://doi.org/10.1182/blood-2013-01-481325
Richardson PG, Hungria VTM, Yoon S-S, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Guenther A, Nakorn TN, Siritanaratkul N, Schlossman RL, Hou J, Moreau P, Lonial S, Lee JH, Einsele H, Sopala M, Bengoudifa B-R, Corrado C, Binlich F, San-Miguel JF (2016) Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by prior treatment. Blood 127(6):713–721. https://doi.org/10.1182/blood-2015-09-665018
Li Z, Qi CF, Shin DM, Zingone A, Newbery HJ, Kovalchuk AL, Abbott CM, Morse HC 3rd (2010) Eef1a2 promotes cell growth, inhibits apoptosis and activates JAK/STAT and AKT signaling in mouse plasmacytomas. PLoS One 5(5):e10755. https://doi.org/10.1371/journal.pone.0010755
Leisch M, Egle A, Greil R (2018) Plitidepsin: a potential new treatment for relapsed/refractory multiple myeloma. Future Oncol 15:109–120. https://doi.org/10.2217/fon-2018-0492
Chang R, Wang E (2007) Mouse translation elongation factor eEF1A-2 interacts with Prdx-I to protect cells against apoptotic death induced by oxidative stress. J Cell Biochem 100(2):267–278. https://doi.org/10.1002/jcb.20969
Sun Y, Du C, Wang B, Zhang Y, Liu X, Ren G (2014) Up-regulation of eEF1A2 promotes proliferation and inhibits apoptosis in prostate cancer. Biochem Biophys Res Commun 450(1):1–6. https://doi.org/10.1016/j.bbrc.2014.05.045
Hotokezaka Y, Tobben U, Hotokezaka H, Van Leyen K, Beatrix B, Smith DH, Nakamura T, Wiedmann M (2002) Interaction of the eukaryotic elongation factor 1A with newly synthesized polypeptides. J Biol Chem 277(21):18545–18551. https://doi.org/10.1074/jbc.M201022200
Acknowledgments
The authors wish to thank the patients, their families, and the clinical research teams for their time and trust and for enabling this clinical trial and associated research.
Author information
Authors and Affiliations
Contributions
IS and MAD designed the study. IS, EMO, HEO, RG, RHB, SYH, JMDR, MAD, SM, AMC, NM, CKM, and PC collected and assemble the data. IS, EMO, RHB, MAD, SM, SE, CK, VA, NM, RH, AS, HL, PS, and MVM developed the analytical plan and/or interpreted the data. MVM and VA co-wrote the first draft of the paper; all authors contributed to the further writing of the paper and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
IS has honoraria (Amgen, Takeda, Janssen-Cilag, BMS, Celgene); consulting or advisory role (Amgen, Takeda, Janssen-Cilag, Celgene); speakers’ bureau (Amgen, BMS Celgene, Jansen-Cilag); and travel, accommodation, or expenses (Celgene, Amgen, Jansen-Cilag) to disclose.
EMO has honoraria (Novartis, Takeda, Celgene, Amgen, BMS, Janssen); consulting or advisory role (Novartis, Takeda, Celgene, AbbVie, Pharma Mar, Seattle Genetics, Amgen); speakers’ bureau (Takeda); research funding to his institution (Celgene, Amgen, Array, Mundipharma, Sanofi, MSD); patents, royalties, or other intellectual properties (Pharma Mar, Mundipharma, IDP); and travel, accommodation, or expenses (Celgene, Janssen, Takeda) to disclose.
HEO has a leadership role (Vertex, Astenas); stock ownership (Vertex); research funding (BMS, Pharma Mar); and travel, accommodation, or expenses (Novartis) to disclose.
RG has honoraria (Celgene, Roche, Merck, Takeda, Astra Zeneca, Novartis, Amgen, BMS, MSD, Sandoz); consulting or advisory role (Celgene, Novartis, Roche, BMS, Takeda, AbbVie, Astra Zeneca, Janssen); research funding (Celgene, Roche, Merck, Takeda, Astra Zeneca, Novartis, Amgen, BMS, MSD, Sandoz); and travel, accommodation, or expenses (Roche, Amgen, Janssen, Astra Zeneca) to disclose.
RHB has a consulting or advisory role (Specialised Therapeutics, Pharma Mar, BMS); research funding (Acerta, Pharma Mar, Celgene, Gilead, Novotech, Pharmacyclics, Roche); and travel, accommodation, or expenses (Roche, Janssen) to disclose.
JMDR has honoraria (Alexion, Roche); consulting or advisory role (Alexion, Roche); and travel, accommodation, or expenses (Pharma Mar, Alexion) to disclose.
Meletios A. Dimopoulos has honoraria (Amgen, Takeda, Janssen, Celgene, BMS) to disclose.
SM, SE, CK, and VA are employees of Pharma Mar S.A.
SE, CK, and VA have stock ownerships (Pharma Mar).
RH has honoraria (Amgen, BMS, Takeda, Celgene, Janssen); consulting or advisory role (Amgen, BMS, Takeda, Celgene, Janssen); research funding to his institution (Takeda, Novartis, Amgen, Janssen, Celgene); travel, accommodation, or expenses (Amgen, BMS, Celgene, Janssen, Takeda) to disclose.
AS has honoraria (AbbVie, Amgen, Celgene, Janssen, Gilead, MSD, Novartis, Pfizer, Roche, Sanofi, Takeda, WinMedica); consulting or advisory role (AbbVie, Amgen, Celgene, Janssen, Gilead, MSD, Novartis, Pfizer, Roche, Sanofi, Takeda); speakers’ bureau (AbbVie, Amgen, Celgene, Janssen, Gilead, MSD, Novartis, Pfizer, Roche, Sanofi, Takeda, WinMedica); research funding to his institution (AbbVie, Actelion, Alexion, Amgen, Astellas, Bayer, Bristol, Celgene, Demo/ApoPharma, Enorasis, Faran, Gilead, Janssen, MSD, Novartis, Pharmaten, Pfizer, Rafarm, Roche, Sanofi, Takeda, Teva, Vianex, WinMedica); and travel, accommodation, or expenses (AbbVie, Amgen, Celgene, Janssen, Gilead, Roche, Sanofi, Takeda) to disclose.
PC has travel, accommodations, or expenses (Amgen, Roche, BMS—conference attendance sponsorship) to disclose.
HL has a consulting or advisory role (Pharma Mar); speakers’ bureau (Takeda, Amgen, Celgene, Janssen, BMS); and research funding (Takeda, Amgen) to disclose.
PS has honoraria (Janssen, Celgene, Amgen, Takeda); expert testimony (Pharma Mar); and research funding (Janssen, Celgene, Amgen, Takeda) to disclose.
MVM has honoraria to disclose from Pharma Mar because of the involvement in advisory boards.
All other authors declare that they have no competing interests.
Research involving human participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Spicka, I., Ocio, E.M., Oakervee, H.E. et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann Hematol 98, 2139–2150 (2019). https://doi.org/10.1007/s00277-019-03739-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-019-03739-2