
Abstract
Purpose
To review the developments of recent decades and the current status of PET molecular imaging in Huntington’s disease (HD).
Methods
A systematic review of PET studies in HD was performed. The MEDLINE, Web of Science, Cochrane and Scopus databases were searched for articles in all languages published up to 19 August 2015 using the major medical subject heading “Huntington Disease” combined with text and key words “Huntington Disease”, “Neuroimaging” and “PET”. Only peer-reviewed, primary research studies in HD patients and premanifest HD carriers, and studies in which clinical features were described in association with PET neuroimaging results, were included in this review. Reviews, case reports and nonhuman studies were excluded.
Results
A total of 54 PET studies were identified and analysed in this review. Brain metabolism ([18F]FDG and [15O]H2O), presynaptic ([18F]fluorodopa, [11C]β-CIT and [11C]DTBZ) and postsynaptic ([11C]SCH22390, [11C]FLB457 and [11C]raclopride) dopaminergic function, phosphodiesterases ([18F]JNJ42259152, [18F]MNI-659 and [11C]IMA107), and adenosine ([18F]CPFPX), cannabinoid ([18F]MK-9470), opioid ([11C]diprenorphine) and GABA ([11C]flumazenil) receptors were evaluated as potential biomarkers for monitoring disease progression and for assessing the development and efficacy of novel disease-modifying drugs in premanifest HD carriers and HD patients. PET studies evaluating brain restoration and neuroprotection were also identified and described in detail.
Conclusion
Brain metabolism, postsynaptic dopaminergic function and phosphodiesterase 10A levels were proven to be powerful in assessing disease progression. However, no single technique may be currently considered an optimal biomarker and an integrative multimodal imaging approach combining different techniques should be developed for monitoring potential neuroprotective and preventive treatment in HD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease (HD) is an autosomal dominant monogenic neurodegenerative disease with a prevalence of 0.4 – 5.7 per 100,000 worldwide [1]. HD is caused by an expanded CAG trinucleotide repeat sequence in the huntingtin gene on chromosome 4, which leads to the formation of intranuclear inclusions of mutated huntingtin in the brain, resulting in loss of GABAergic medium spiny neurons (MSNs) in the striatum and in cortical areas [2]. HD is clinically characterized by motor symptoms (chorea and parkinsonism), cognitive symptoms (slowed mentation, attention, mental flexibility, planning and emotion recognition) and psychiatric symptoms (depression, apathy, impulsivity, irritability, disinhibition and psychosis), with a progressive course and a typical onset in adult middle age (40 – 55 years) [3]. The age at onset is inversely correlated with the size of the CAG repeat expansion [4]. This allows us to predict the motor onset [5] and classify the disease in stages such as premanifest HD (before motor onset) and manifest HD (after motor onset) [6] providing a tremendous opportunity to investigate subclinical and pathological changes in asymptomatic HD gene carriers. This offers a therapeutic window for potential preventive treatments aiming to delay the clinical onset of HD.
The mechanisms underlying the progressive neurodegeneration in HD are still unclear and, currently, there is no single proven biomarker that allows us to monitor disease progression and assess the efficacy of novel disease-modifying drugs. The lack of biomarkers may be related to the fact that HD pathology causes only minor brain alterations in early stages [7].
Molecular imaging techniques are able to identify subtle alterations at the nanomolecular level and this is a prerequisite to understanding minimal changes in brain activity [8, 9]. PET molecular probes bind a target, such as a receptor, a transporter or an enzyme, with high specificity and power of resolution [10]. PET molecular imaging has revolutionized the ability to gain insights into human brain biology and beyond this to understand the physiology and pathophysiology of neurological diseases [11]. PET radiotracers have provided invaluable insights into the natural history of HD and have been used to measure brain metabolism, dopaminergic function, neuroinflammation, phosphodiesterases and other targets in HD [12]. They have contributed to the identification of disease characteristics at different stages mainly in cross-sectional studies but also in some longitudinal studies.
This review describes the developments during recent decades and current applications of PET molecular imaging techniques in HD.
Methods
Search strategy
The MEDLINE, Web of Science, Cochrane CENTRAL and Scopus databases were searched for articles in all languages published up to 19 August 2015. Studies were identified and evaluated by two of the authors (G.P. and F.N.) using the major medical subject heading “Huntington Disease” combined with text and key words “Huntington Disease”, “Neuroimaging” and “PET”. Additional eligible studies were identified by screening the reference lists of the studies found.
Inclusion criteria
Studies were excluded if the title and/or abstract was not appropriate for the aim of the review. The full text of eligible studies and of studies whose relevance was uncertain were obtained. Selected studies were eligible if they met the following criteria: (1) peer-reviewed, primary research studies, (2) studies including HD patients, (3) studies including a description of the clinical features of the HD patients in association with neuroimaging results, and (4) studies including PET neuroimaging. Reviews, case reports and nonhuman studies were excluded.
Results
Of 944 articles identified by the initial search, 86 were retrieved for more detailed evaluation, and 54 PET studies were finally identified and analysed in this review. The results of 32 of the main studies are summarized in Table 1.
Brain metabolism
[15O]H2O and [18F]FDG have been used as markers of cerebral blood flow and cerebral glucose metabolism providing an index of neuronal integrity and the functional state of neurons [13]. Changes in motor-related cortical activation have been evaluated using [15O]H2O PET [14, 15]. In manifest HD, impaired activation of the striatum and its frontal motor projection areas during motor tasks, such as paced joystick movements or sequential finger-to-thumb opposition, has been found. There was a parallel increased activity in the parietal areas [14] and insular areas [15]. These findings suggest that the loss of MSNs in the striatum leads to impairment of the basal ganglia-thalamocortical motor output and may induce a compensatory recruitment of additional accessory motor pathways [14, 15]. Moreover, different patterns of brain activation have been shown in HD patients during word generation task [16]. During the word generation task, HD patients show decreased cerebral blood flow in the anterior cingulate and the inferior frontal gyri (related to lexical selection) and a compensatory activation of the left supramarginal gyrus and the right inferior frontal gyrus, suggesting the presence of compensatory language strategies in HD [16].
[18F]FDG studies in manifest HD patients have shown glucose hypometabolism in the striatum and the cortex [17–19]. Striatal hypometabolism was associated with motor dysfunction [20], and cortical hypometabolism with cognitive dysfunction [19, 21]. Striatal and cortical hypometabolism has also been found to precede neuronal loss in premanifest HD gene carriers [22–24]. However, it has been suggested that a possible hypermetabolism precedes the decrease in these areas [25]. Moreover, [18F]FDG hypometabolism was not localized in a specific area, and an approach using a network of regions with altered metabolism has been used to identify spatial covariance patterns in premanifest HD [25–28]. Following this approach, relative bilateral increases in thalamic, occipital and cerebellar glucose metabolism associated with bilateral decreases in striatal metabolism has been identified as a characteristic pattern of HD, which discriminated well between HD patients and healthy controls [26]. This characteristic pattern has shown good reliability as a marker of disease progression although there was a tendency to be quite stable after an initial impairment (nonlinear trend), thus limiting its clinical utility [26]. Reduction in [18F]FDG uptake in the striatum, and frontal, parietal and temporal cortex indicates an impairment in intracellular processes, such as calcium handling, transcription regulation and mitochondrial energy production, while an increase in [18F]FDG uptake in the thalamus, and occipital and cerebellar cortex might indicate an increased glycolytic response due to mitochondrial dysfunction [25] or due to the lack of inhibitory activity exerted by the damaged basal ganglia on the thalamocortical system [26].
Dopaminergic function
Pathognomonic chorea and the other motor features (parkinsonism and dystonia) of HD are related to impairment in dopaminergic function [29]. The neurodegenerative process mainly affects the striatal MSNs expressing dopaminergic type 1 (D1) and type 2 (D2) receptors [30]. This leads to severe involvement of the postsynaptic dopaminergic system with relatively spared presynaptic dopaminergic nerve terminals. However, presynaptic dopaminergic function in HD has been investigated only in small and older studies, with discordant results. In manifest HD patients, caudate [18F]fluorodopa uptake (targeting dopa decarboxylase) has been found to be reduced [31] or normal [32], [11C]β-CIT levels (targeting striatal dopamine transporter) were 50 % reduced [33] and [11C]DTBZ levels (targeting type-2 vesicular monoamine transporter) showed a gradient from a rostral increase to a caudal decrease in the striatum [34]. However, in these PET studies no correction for partial volume effects was performed and their results may have been affected by regional volume loss, and thus it is unclear whether they measured degeneration of neurons or pure presynaptic dopaminergic terminal dysfunction. Presynaptic dopaminergic function has not been investigated in premanifest HD patients, nor has the association with CAG repeat expansion or other HD-related clinical features.
Postsynaptic dopaminergic dysfunction is a key characteristic of HD. This has been investigated using [11C]SCH22390 as marker of D1 receptors density and using [11C]raclopride and [11C]FLB457 [35] as markers of D2 receptor density. The densities of D1 and D2 receptors were significantly reduced in the striatum, ranging from 40 % [33] to 75 % [36] in HD patients and from 25 % [37] to 50 % [38] in premanifest HD gene carriers. This loss was found to be higher in akinetic-rigid HD patients than in choreic manifest HD patients [39]. Reductions in striatal D1 and D2 receptors were also correlated with worse motor dysfunction, as assessed using the unified Huntington’s disease rating scale (UHDRS) [37, 40]. In extrastriatal areas, a reduction in D1 receptors has been found in the temporal cortex and was associated with cognitive dysfunction in manifest HD patients [33, 41]. In a study using [11C]raclopride including 16 manifest HD patients and 11 premanifest HD gene carriers [42], the cortical D2 receptor levels were reduced in both manifest HD patients and premanifest HD gene carriers and were associated with the degree of cognitive dysfunction (attention and executive functions). In line with these results, another study using [11C]raclopride [40] showed a reduction in D2 receptor levels in the amygdala, and frontal and temporal cortex of 12 manifest HD patients. Levels of both D1 and D2 receptors (but mainly D2 receptors) were strongly correlated with cognitive performance in 17 premanifest HD gene carriers [43]. Hypothalamic D2 receptor levels were also significantly reduced in presymptomatic and premanifest HD gene carriers [44]. The early involvement of the hypothalamus might explain the prodromal nonmotor symptoms and could be a potential early biomarker in HD. The number of CAG repeat lengths has been associated with a reduction in striatal D2 receptor levels in premanifest HD patients [38, 45], suggesting that mutant huntingtin plays a role in the expression of dopamine receptors. These findings support the primary involvement of sensorimotor striatum dysfunction in the motor component of the disease [40] and of associative striatum and temporal cortex dysfunction in the cognitive component of the disease [42], with D2 receptor loss as the common biological substrate.
Activation of microglia
Activation of microglia is considered one of the mechanisms underlying neurodegeneration [46]. However, it is not clear if it represents a compensatory mechanism for neuronal loss or a trigger of damage [47]. Microglia release cytokines when stimulated by abnormal proteins, such as mutant huntingtin. These cytokines are proinflammatory and further stimulate microglia, resulting in a self-propagating cascade, which may lead to neuronal dysfunction and death [48]. This process is not confined to the brain, but peripheral plasma cytokines are also increased in premanifest and manifest HD gene carriers [47]. Activation of microglia can be measured using 18-kDa translocator protein (TSPO) ligands, such as [11C]PK11195, [11C]GE180, [11C]PBR28, and others [49]. So far only [11C]PK11195 has been used to measure TSPO in HD gene carriers. [11C]PK11195 is a first-generation TSPO PET ligand, whereas the others were developed recently and represent second-generation TSPO PET [50]. Increased activation of striatal and cortical microglia has been found in manifest HD gene carriers [51] and in premanifest HD gene carriers and was associated with loss of striatal D2 receptors ([11C]raclopride binding) [52]. In manifest HD patients, striatal TSPO levels are also associated with motor dysfunction (UHDRS) [52]. In a more recent study, increased TSPO binding has been observed specifically in the associative striatum of premanifest HD patients and correlated with cognitive dysfunction [53]. We have also recently demonstrated that plasma levels of IL-1β, IL-6, IL-8 and TNF-α are correlated with increased [11C]PK11195 binding in the somatosensory cortex of premanifest HD patients [54]. These findings demonstrate for the first time in vivo a link between peripheral and central immune dysfunction and support the role of immune dysfunction in the pathogenesis of HD. In terms of pathology, it is reasonable to assume that the more dorsal part of the striatum (related to motor and cognitive function) is affected earliest in HD, whereas the ventral part of the striatum (related to psychiatric function) is affected later, possibly after the onset of motor symptoms [53].
Phosphodiesterases
Phosphodiesterases are a family of intracellular enzymes hydrolysing cyclic nucleotides (cAMP and cGMP) [55]. Phosphodiesterase 10A (PDE10A) is mainly expressed in striatal MSNs where it regulates the cAMP/PKA/DARPP-32 signalling cascade, and thus plays a key role in the regulation of striatal output and in promoting neuronal survival [55, 56]. PDE10A has received increasing attention after the observation that its pharmacological inhibition in an animal model of HD significantly improves HD symptoms and pathology [57]. Post-mortem studies have confirmed that PDE10A is severely reduced in manifest HD patients [58].
[18F]JNJ42259152, [18F]MNI-659 and [11C]IMA107 have been used to quantify of PDE10A expression in vivo in HD patients [59–61]. In five manifest HD patients, Ahmad et al. [59] found 70.7 % and 62.6 % [18F]JNJ42259152 binding reductions in the caudate and putamen, respectively (Fig. 1). These authors found no correlations between [18F]JNJ42259152 levels and clinical scales. Russell et al. [60], found 47.6. % decreases in striatal and pallidal [18F]MNI-659 binding in eight patients with early manifest HD. Lower striatal [18F]MNI-659 binding was associated with worse UHDRS motor scores, disease burden of pathology and regional atrophy. In three premanifest HD gene carriers, who were a mean of 12 years from predicted onset, striatal PDE10A expression was also found to be decreased although to a lesser degree compared to the group of manifest HD patients [60].

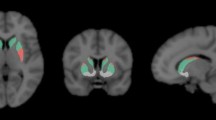
Example PET images in healthy controls (HC) and premanifest Huntington’s disease gene carriers (preHD). Axial [11C]MePPeP, [11C]IMA107, [11C]raclopride images in stereotaxic space overlaid onto the T1-weighted MNI template show decreased cortical CB1 receptor, striatal PDE10A and striatal D2 receptor binding in the preHD subjects compared to the healthy controls . Images from our data
Using [11C]IMA107 PET, our group has recently investigated PDE10A expression in 12 HD gene carriers with early premanifest asymptomatic HD who were a mean of 25 years before the predicted onset of motor symptoms [61]. We found 25 – 33 % reductions in striatal and pallidal [11C]IMA107 binding, and 35 % increases in the motor thalamic nuclei. We then combined PET and diffusor tensor imaging MRI data to perform a connectivity-based striatal parcellation analysis of PDE10A expression according to (a) cortex–striatum connectivity profiles and (b) striatal connections with the globus pallidus externus and substantia nigra/globus pallidus internus, which reflect the major parts of the indirect and direct pathways, respectively. We found that PDE10A decreases were confined to the sensorimotor striatum and to the striatonigral and striatopallidal projecting segments. Moreover, the altered balance of PDE10A expression between the motor thalamic nuclei and the striatopallidal internal projecting segments of the striatum was strongly associated with the predicted risk of symptomatic conversion. Our findings are a major breakthrough in the field of HD in showing the earliest biochemical abnormality identified in HD to date, and showing bidirectional alteration of PDE10A expression within the neuropathological salient networks in HD gene carriers up to 43 years before the development of overt clinical symptoms, which are crucial to prognosis. These findings provide in vivo evidence that PDE10A plays a role in the pathophysiology of HD, although longitudinal studies are needed to confirm the relationship between PDE10A and clinical symptomatology.
Adenosine and cannabinoid receptors
The identification of novel targets is a primary objective of current PET research in HD. Type 1 adenosine (A1) and type 1 cannabinoid (CB1) receptors represent only a few among many potential novel targets that have recently become investigable with PET imaging. In animal studies, adenosine was increased by about 50 % in a mouse model of HD [62], and A1 receptor levels in the hypothalamus were found to be lower in HD rats carrying 51 CAG repeats [63]. [18F]CPFPX has been used as a marker of A1 receptors in HD [64]. Matusch et al. [64] found a 25 % reduction in [18F]CPFPX binding in the caudate of eight manifest HD patients (Fig. 1). They studied premanifest HD patients, dividing them into premanifest gene carriers far from predicted symptom onset (preHD-A; n = 7) and near to predicted symptom onset (preHD-B; n = 6). Interestingly, in preHD-A patients, [18F]CPFPX binding in the thalamus was 31 % higher than in healthy controls, while in preHD-B patients, thalamic [18F]CPFPX binding was similar to the levels in healthy controls. There was a strong correlation between A1 receptor binding and years to onset [65]. These findings suggest that A1 receptors switch from supranormal to subnormal levels during phenoconversion of HD. This differential regulation may play a role in the pathophysiology of altered energy metabolism.
CB1 receptors are mainly expressed in GABA-ergic striatal MSNs [65] (also expressing D1 and D2 receptors) and are a key modulator of inhibitor neurotransmission [66]. In post-mortem studies in manifest HD patients, a reduction in CB1 receptor levels was demonstrated in the striatum [67] and GABA receptors were decreased in the caudate and putamen and increased in the globus pallidus [68] and substantia nigra [69]. This indicates that output GABAergic projections from the striatum are severely affected in HD patients [70]. This hypothesis has been confirmed in vivo in PET studies conducted to evaluate GABA/benzodiazepine receptors [71, 72] and CB1 receptors [73].
[18F]MK-9470 has been used as a marker of CB1 receptors in PET studies of the cannabinoid system in HD [73]. Van Laere et al. [73] found reduced CB1 receptor binding in the grey matter of the cerebrum and cerebellum, and in the brainstem of 20 patients with manifest HD (Fig. 1). They used a simplified semiquantification model and their findings need to be confirmed in a full quantification model. The widespread CB1 receptor changes observed in HD are in line with the suppressive effect of mutant huntingtin on CB1 transcription [74]. This finding strongly suggests that cannabinoid dysfunction is heavily involved in HD even during the early presymptomatic stages of the disease.
Opioid and GABA receptors
Old targets, such as opioid and GABA, might be reintroduced in combination with novel ones. Post-mortem studies have shown that the opioid system is also affected in HD [75, 76]. A loss of encephalin of striatal projection neurons to the external globus pallidus has been reported in premanifest HD [75] and a reduction in the proencephalin/prodynorphin ratio in the caudate, putamen, and internal and external globus pallidus has been found in manifest HD patients [76]. [11C]Diprenorphine has been used as a marker of opioid receptor in HD [77]. Significant reductions of 31 % and 26 % in [11C]diprenorphine binding in the caudate and putamen, respectively, were demonstrated in five manifest HD patients [77]. These reductions were lower than striatal dopaminergic degeneration (about 60 % in manifest HD [39]), suggesting that opioid receptor sites are less vulnerable to disease processes than dopamine sites, possibly because of preservation of presynaptic opioid sites.
[11C]Flumazenil has been used as marker of GABA/benzodiazepine receptors [71, 72]. Holthoff et al. [71] found a 17 % reduction in [11C]flumazenil binding in the caudate of six manifest HD patients, with normal putamen and thalamic binding. They also found a reduction in [18F]FDG binding of 47 %, 41 %, 18 % in the caudate, putamen and thalamus, respectively. In ten manifest HD patients, Künig et al. [72] found the same results, showing reduced [11C]flumazenil binding in the caudate (with normal values in the putamen) and reduced [18F]FDG binding in the caudate and putamen. The metabolic impairment of the putamen and thalamus, without detectable loss of benzodiazepine receptor density, confirms that the reduction in [18F]FDG uptake happens before the changes in [11C]flumazenil binding. Künig et al. also compared [11C]flumazenil and [18F]FDG binding with [11C]raclopride binding in 13 premanifest HD patients. [11C]Raclopride binding in the caudate and putamen was reduced in all manifest HD patients and in eight premanifest patients. As previously reported, a reduction in D2 receptor levels indicates neuronal cell loss. Thus, they divided premanifest gene carriers into two groups: those with and those without a decrease in [11C]raclopride. [11C]Flumazenil binding in the caudate was significantly lower in manifest HD patients compared with premanifest patients without a decrease in [11C]raclopride, but not compared with those with a decrease in [11C]raclopride who displayed [11C]flumazenil values between those of the control subjects and the manifest HD patients. These findings indicate that the loss in pallidocaudate GABA projections was marked only in manifest HD patients. [11C]Flumazenil binding in the putamen was not different among healthy controls, and premanifest and manifest HD gene carriers. The authors suggest that the normal [11C]flumazenil binding in the putamen of manifest HD patients might be a compensatory mechanism in the pallidal projection areas of striatal GABAergic MSNs. However, these PET studies were not corrected for partial volume effects and the results may have been affected by regional volume loss.
No studies investigating the cannabinoid system in premanifest HD gene carriers have been published. Interestingly, CB1 receptors and PDE10A transcription is modulated by huntingtin and, in HD animal models expressing a mutant huntingtin, CB1 receptors and PDE10A were strongly reduced [78]. This reduction was present not only in the symptomatic stage but also in the asymptomatic stage, even before decreases in glucose uptake. Thus, these proteins may not only be excellent biomarkers of HD progression but may also represent novel therapeutic targets for HD.
Progression monitoring
Several longitudinal studies have been conducted in HD patients to identity biomarkers, monitor disease progression and evaluate novel treatments. Brain metabolism and dopaminergic function have been evaluated as potential PET biomarkers. In a longitudinal study, Feigin et al. [26] observed a progressive decline in [18F]FDG uptake across three time-points over 4 years. A recent [18F]FDG PET study has shown that premanifest HD gene carriers who become symptomatic after 5 years following a PET scan have a mean glucose uptake in the caudate significantly lower than those who do not convert, and this difference is independent of CAG repeats [24]. These findings suggest that brain metabolic changes may be predictors of HD symptomatic conversion. Decreased cortical metabolism in the early stage of HD is considered indicative of rapid progression [79]. Indeed, cortical metabolism in the frontotemporal and parietal cortices is significantly lower in patients with early HD with faster progression of the disease as measured using the Independence Scale of the UHDRS [79].
A distinct spatial covariance pattern has been identified by Tang et al. [28] as associated with disease progression using longitudinal metabolic imaging data in premanifest HD patients. This pattern showed a progressive impairment in striatothalamic and cortical metabolic activity over 7 years and was not influenced by symptomatic conversion. Moreover, premanifest HD patients at higher risk of symptomatic conversion were those with higher metabolic network activity at baseline [28]. Thus, metabolic network may be a sensitive biomarker for disease progression during presymptomatic stages.
In a combined [18F]FDG and [11C]raclopride longitudinal study, premanifest HD gene carriers showed an annual loss of 2.3 % in striatal glucose metabolism and 6.3 % in D2 receptor binding [17]. These findings suggest that glucose metabolism is a less sensitive marker of disease progression than [11C]raclopride [17]. In another longitudinal study, the decline in D2 receptor levels was constant (around 5 % per year) [40], although no correlation between changes in UHDRS motor scores and reductions in striatal binding were observed [40]. This may be explained by the sigmoid model, in which the motor symptoms reach a plateau (threshold of caudate degeneration/hypometabolism) and further degeneration is not associated with worsening of symptoms. In premanifest HD patients, longitudinal [11C]raclopride studies have shown rates of decline from 4 % [37] up to 6.3 % [17]. In a 40-month longitudinal study [37], nine premanifest HD patients and four manifest HD patients showed a mean annual D1 receptor binding reductions of 2 % and 5 %, respectively, and D2 receptor binding reductions of 4 % and 3 %, respectively [37]. Among them, premanifest HD patients with annual losses ≥5 % demonstrated faster disease progression. Thus, striatal D1 and D2 receptor binding may be used to identify asymptomatic HD subjects with a higher risk of conversion [37]. However, in another study [80] reduction in putaminal dopamine D2 receptor binding was weakly correlated with the probability of symptomatic conversion within 5 years, as calculated using a model based on age and CAG repeats, and the rate of change of putaminal D2 receptor binding was not increased around the time of HD symptom onset [80]. Striatal degeneration might proceed in a non-linear fashion in HD. A correlation between CAG repeat length and the estimated percentage loss of striatal D2 receptor binding were found in a cross-sectional study among premanifest and manifest HD subjects [45]. However, while CAG repeat length influenced the rate of disease progression, the slopes of the correlation for premanifest and manifest HD population were significantly different, suggesting that the rate of disease progression is faster during the earlier asymptomatic stages of the disease [45]. Therefore, striatal D2 receptor measures should be used mainly in patients during premanifest HD stages, when they are more sensitive.
Recently, novel biomarkers have been used to monitor disease progression. Russell et al. [81] found reduced striatal [18F]MNI-659 uptake of about 50 % at baseline in eight manifest HD patients compared with seven healthy controls. At 1 year, [18F]MNI-659 uptake had declined in the basal ganglia in all eight manifest HD patients with mean annualized rates of reduction in of 16.6 % in the caudate, 6.9 % in the putamen, and 5.8 % in the globus pallidus. A decline in clinical status as assessed using UHDRS was also observed in this cohort. To date, three subjects had completed imaging assessment at 2 years and they showed a mean annualized reduction in [18F]MNI-659 uptake at similar rate to that found at 1 year (caudate 15.5 %, putamen 7.2 %, and globus pallidus 9.4 %). Despite these promising results, head-to-head studies need to be performed comparing [18F]MNI-659 with [18F]FDG and [11C]raclopride to finally determine which functional PET markers are most sensitive for monitoring HD progression.
Differences found between HD patients and healthy subjects in the uptake of PET tracers in various brain regions are summarized in Table 2.
Unfortunately, current biomarkers did not show consistent results in terms of changes over time, thus limiting their utility as markers of disease progression. A novel model (sigmoid acceleration–deceleration) recently applied in Alzheimer’s disease [82] might also be useful in HD. This model has been suggested to identify biomarkers with a nonlinear reduction over time and assumes an initial acceleration of volume loss up to an inflection point following which deceleration of atrophy occurs [82]. The pathophysiological changes in HD extend far beyond the striatum, cortical changes occur even at the earliest stages of the disease. Using structural MRI, cortical thinning has been found in early manifest HD patients [83] and cortical degeneration of premotor and supplementary motor areas has been found to be associated with bradykinesia [84]. Functional MRI studies have shown reorganization of cortical circuitry at the early stage of HD [85]. Using an “interference” task that activates multiple brain regions including the anterior cingulate, inferior parietal, inferior temporal, sensorimotor, premotor and inferior frontal cortices, different patterns of brain activation have been found in premanifest HD gene carriers further from disease onset (increased activation) compared with premanifest HD gene carriers closer to disease onset (reduced activation) [85].
Brain restoration
Brain metabolism and dopamine receptors levels have been used to assess the efficacy of restorative therapy in HD patients. A multicentre open-label pilot study was designed to evaluate the safety and efficacy of bilateral fetal striatal transplantation [86]. Transplantation was performed in five HD patients who were followed clinically and with PET over 3 – 10 years [87, 88]. No significant differences were found over time using the UHDRS between patients who had and had not received the transplant and striatal D1 and D2 receptor binding indicated that there was no obvious surviving striatal graft tissue [87, 88]. A 2-year follow-up study with [18F]FDG in five HD patients who underwent bilateral striatal transplantation [89, 90] was performed to assess the restoration of striatocortical function [90]. Three patients showed an increase in striatal/cortical glucose metabolic rate, associated with clinical improvement or stabilization suggesting restoration of striatocortical function. However, a 2-year follow-up multicentre study failed to show significant this change in [18F]FDG uptake [87]. Therefore, whether bilateral striatal transplantation may restore striatocortical connections remains a matter of debate [91, 92].
Neuroprotective drugs
Brain metabolism has been used to assess the efficacy of potential neuroprotective drugs in HD. A randomized-controlled trial using riluzole was performed in 23 HD patients (11 treated with riluzole and 12 with placebo) [93]. After a mean follow-up of 24 months placebo-treated patients showed a significantly greater proportional volume loss of grey matter and decrease in metabolic [18F]FDG uptake than patients treated with riluzole in all cortical areas. The decreased rate of metabolic [18F]FDG uptake correlated with worsening clinical scores in placebo-treated patients [93]. A small open-label trial using pridopidine was performed in eight HD patients [94] and increased metabolic activity in several brain regions including the precuneus and mediodorsal thalamic nucleus was found after treatment [94]. Memantine was investigated in an open-label pilot trial in four HD patients and no differences in cortical or striatal metabolism were found between before and after treatment [95].
Conclusion
There are no treatments currently available to stop disease progression in HD. Brain metabolism (relative bilateral increases in thalamic, occipital and cerebellar glucose metabolism associated with bilateral decreases in striatal [18F]FDG binding), postsynaptic dopaminergic function (reduction in striatal and cortical D1 receptor [11C]SCH22390 binding and D2 receptor [11C]raclopride binding) and PDE10A levels (reduction in striatal and pallidal [11C]IMA107 binding and increase in motor thalamic nuclei [11C]IMA107 binding) have been shown to be powerful biomarkers for assessing progression in patients with manifest HD and also during premanifest stages. However, no single biomarker may currently be considered optimal due to specific limitations of each ligand. An integrated multimodal imaging approach, combining different techniques, should be developed for evaluating potential neuroprotective and preventive treatments in HD.
References
Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord. 2012;27:1083–91.
The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–83.
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16.
Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, et al. Self-assembly of polyglutamine containing huntingtin fragments into amyloid-like fibrils: implications for Huntington disease pathology. Proc Natl Acad Sci U S A. 1999;96:4604–9.
Langbehn DR, Hayden MR, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. CAG repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:397–408.
Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington’s disease based on natural history. Mov Disord. 2014;29:1335–41.
Rüb U, Vonsattel JP, Heinsen H, Korf HW. The neuropathology of Huntington´s disease: classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol. 2015;217:1–146.
Politis M, Piccini P. Positron emission tomography imaging in neurological disorders. J Neurol. 2012;259(9):1769–80.
Politis M. Neuroimaging in Parkinson disease: from research setting to clinical practice. Nat Rev Neurol. 2014;10(12):708–22.
Loane C, Politis M. Positron emission tomography neuroimaging in Parkinson’s disease. Am J Transl Res. 2011;3(4):323–41.
Rocchi L, Niccolini F, Politis M. Recent imaging advances in neurology. J Neurol. 2015;262(9):2182–94.
Niccolini F, Politis M. Neuroimaging in Huntington’s disease. World J Radiol. 2014;6(6):301–12.
Sokoloff L. Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J Cereb Blood Flow Metab. 1981;1:7–36.
Bartenstein P, Weindl A, Spiegel S, Boecker H, Wenzel R, Ceballos-Baumann AO, et al. Central motor processing in Huntington’s disease. A PET study. Brain. 1997;120(Pt 9):1553–67.
Weeks RA, Ceballos-Baumann A, Piccini P, Boecker H, Harding AE, Brooks DJ. Cortical control of movement in Huntington’s disease. A PET activation study. Brain. 1997;120(Pt 9):1569–78.
Lepron E, Péran P, Cardebat D, Démonet JF. A PET study of word generation in Huntington’s disease: effects of lexical competition and verb/noun category. Brain Lang. 2009;110:49–60.
Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;119(Pt 6):2085–95.
Mazziotta JC, Phelps ME, Pahl JJ, Huang SC, Baxter LR, Riege WH, et al. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington’s disease. N Engl J Med. 1987;316:357–62.
Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain. 1990;113(Pt 5):1405–23.
Young AB, Penney JB, Starosta-Rubinstein S, Markel DS, Berent S, Giordani B, et al. PET scan investigations of Huntington’s disease: cerebral metabolic correlates of neurological features and functional decline. Ann Neurol. 1986;20:296–303.
Berent S, Giordani B, Lehtinen S, Markel D, Penney JB, Buchtel HA, et al. Positron emission tomographic scan investigations of Huntington’s disease: cerebral metabolic correlates of cognitive function. Ann Neurol. 1988;23:541–6.
Hayden MR, Martin WR, Stoessl AJ, Clark C, Hollenberg S, Adam MJ, et al. Positron emission tomography in the early diagnosis of Huntington’s disease. Neurology. 1986;36:888–94.
Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47:215–22.
Ciarmiello A, Giovacchini G, Orobello S, Bruselli L, Elifani F, Squitieri F. 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging. 2012;39:1030–6.
Feigin A, Leenders KL, Moeller JR, Missimer J, Kuenig G, Spetsieris P, et al. Metabolic network abnormalities in early Huntington’s disease: an [18F]FDG PET study. J Nucl Med. 2001;42:1591–5.
Feigin A, Tang C, Ma Y, Mattis P, Zgaljardic D, Guttman M, et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain. 2007;130:2858–67.
Eidelberg D, Surmeier DJ. Brain networks in Huntington disease. J Clin Invest. 2011;121:484–92.
Tang CC, Feigin A, Ma Y, Habeck C, Paulsen JS, Leenders KL, et al. Metabolic network as a progression biomarker of premanifest Huntington’s disease. J Clin Invest. 2013;123:4076–88.
Schrag A, Quinn N. Disorders of the basal ganglia and their modern management. J R Coll Physicians Lond. 1999;33(4):323–7.
Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28(45):11603–14.
Martin WR, Hayden MR. Cerebral glucose and dopa metabolism in movement disorders. Can J Neurol Sci. 1987;14(3 Suppl):448–51.
Leenders KL, Frackowiak RS, Quinn N, Marsden CD. Brain energy metabolism and dopaminergic function in Huntington’s disease measured in vivo using positron emission tomography. Mov Disord. 1986;1:69–77.
Ginovart N, Lundin A, Farde L, Halldin C, Backman L, Swahn CG, et al. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain. 1997;120(Pt 3):503–14.
Bohnen NI, Koeppe RA, Meyer P, Ficaro E, Wernette K, Kilbourn MR, et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54(9):1753–9.
Esmaeilzadeh M, Farde L, Karlsson P, Varrone A, Halldin C, Waters S, et al. Extrastriatal dopamine D(2) receptor binding in Huntington’s disease. Hum Brain Mapp. 2011;32:1626–36.
Sedvall G, Karlsson P, Lundin A, Anvret M, Suhara T, Halldin C, et al. Dopamine D1 receptor number – a sensitive PET marker for early brain degeneration in Huntington’s disease. Eur Arch Psychiatry Clin Neurosci. 1994;243:249–55.
Andrews TC, Weeks RA, Turjanski N, Gunn RN, Watkins LH, Sahakian B, et al. Huntington’s disease progression. PET and clinical observations. Brain. 1999;122(Pt 12):2353–63.
van Oostrom JC, Maguire RP, Verschuuren-Bemelmans CC, Veenma-van der Duin L, Pruim J, Roos RA, et al. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology. 2005;65:941–3.
Turjanski N, Weeks R, Dolan R, Harding AE, Brooks DJ. Striatal D1 and D2 receptor binding in patients with Huntington’s disease and other choreas. A PET study. Brain. 1995;118(Pt 3):689–96.
Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: a PET study. Brain. 2003;126:1127–35.
Bäckman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington’s disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120(Pt 12):2207–17.
Pavese N, Politis M, Tai YF, Barker RA, Tabrizi SJ, Mason SL, et al. Cortical dopamine dysfunction in symptomatic and premanifest Huntington’s disease gene carriers. Neurobiol Dis. 2010;37:356–61.
Lawrence AD, Weeks RA, Brooks DJ, Andrews TC, Watkins LH, Harding AE, et al. The relationship between striatal dopamine receptor binding and cognitive performance in Huntington’s disease. Brain. 1998;121(Pt 7):1343–55.
Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P. Hypothalamic involvement in Huntington’s disease: an in vivo PET study. Brain. 2008;131:2860–9.
Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington’s disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253–5.
van Dijk G, van Heijningen S, Reijne AC, Nyakas C, van der Zee EA, Eisel UL. Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front Neurosci. 2015;9:173.
Politis M, Su P, Piccini P. Imaging of microglia in patients with neurodegenerative disorders. Front Pharmacol. 2012;3:96.
Nakanishi H. Microglial functions and proteases. Mol Neurobiol. 2003;27:163–76.
Hatano K, Sekimata K, Yamada T, Abe J, Ito K, Ogawa M, et al. Radiosynthesis and in vivo evaluation of two imidazopyridineacetamides, [(11)C]CB184 and [(11)C]CB190, as a PET tracer for 18 kDa translocator protein: direct comparison with [(11)C](R)-PK11195. Ann Nucl Med. 2015;29(4):325–35.
Guo Q, Owen DR, Rabiner EA, Turkheimer FE, Gunn RN. Identifying improved TSPO PET imaging probes through biomathematics: the impact of multiple TSPO binding sites in vivo. Neuroimage. 2012;60(2):902–10. doi:10.1016/j.neuroimage.2011.12.078.
Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–43.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66.
Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. 2011;32:258–70.
Politis M, Lahiri N, Niccolini F, Su P, Wu K, Giannetti P, et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol Dis. 2015;83:115–21. doi:10.1016/j.nbd.2015.08.011.
Nishi A, Kuroiwa M, Miller DB, O'Callaghan JP, Bateup HS, Shuto T, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460–71.
Girault JA. Integrating neurotransmission in striatal medium spiny neurons. Adv Exp Med Biol. 2012;970:407–29.
Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5(10):e13417.
Hebb AL, Robertson HA, Denovan-Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123:967–81.
Ahmad R, Bourgeois S, Postnov A, Schmidt ME, Bormans G, Van Laere K, et al. PET imaging shows loss of striatal PDE10A in patients with Huntington disease. Neurology. 2014;82(3):279–81.
Russell DS, Barret O, Jennings DL, Friedman JH, Tamagnan GD, Thomae D, et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early Huntington disease. JAMA Neurol. 2014;71(12):1520–8.
Niccolini F, Haider S, Marques T, Muhlert N, Tziortzi A, Searle G, et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington’s disease. Brain. 2015;138:3016–29. doi:10.1093/brain/awv214.
Gianfriddo M, Melani A, Turchi D, Giovannini MG, Pedata F. Adenosine and glutamate extracellular concentrations and mitogen activated protein kinases in the striatum of Huntington transgenic mice. Selective antagonism of adenosine A2A receptors reduces transmitter outflow. Neurobiol Dis. 2004;17(1):77–88.
Bauer A, Zilles K, Matusch A, Holzmann C, Riess O, von Horsten S. Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington’s disease mutation. J Neurochem. 2005;94(3):639–50.
Matusch A, Saft C, Elmenhorst D, Kraus PH, Gold R, Hartung HP, et al. Cross sectional PET study of cerebral adenosine A1 receptors in premanifest and manifest Huntington’s disease. Eur J Nucl Med Mol Imaging. 2014;41(6):1210–20.
Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–74.
Glass M, Brotchie JM, Maneuf YP. Modulation of neurotransmission by cannabinoids in the basal ganglia. Eur J Neurosci. 1997;9:199–203.
Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience. 2000;97:505–19.
Whitehouse PJ, Trifiletti RR, Jones BE, Folstein S, Price DL, Snyder SH, et al. Neurotransmitter receptor alterations in Huntington’s disease: autoradiographic and homogenate studies with special reference to benzodiazepine receptor complexes. Ann Neurol. 1985;18(2):202–10.
Reisine TD, Wastek GJ, Speth RC, Bird ED, Yamamura HI. Alterations in the benzodiazepine receptor of Huntington’s diseased human brain. Brain Res. 1979;165(1):183–7.
Ribac CE, Vaughn JE, Roberts E. The GABA neurons and their axon terminals in rat corpus striatum as demonstrated by GAD immunocytochemistry. J Comp Neurol. 1979;187:261–84.
Holthoff VA, Koeppe RA, Frey KA, Penney JB, Markel DS, Kuhl DE, et al. Positron emission tomography measures of benzodiazepine receptors in Huntington’s disease. Ann Neurol. 1993;34(1):76–81.
Künig G, Leenders KL, Sanchez-Pernaute R, Antonini A, Vontobel P, Verhagen A, et al. Benzodiazepine receptor binding in Huntington’s disease: [11C]flumazenil uptake measured using positron emission tomography. Ann Neurol. 2000;47(5):644–8.
Van Laere K, Casteels C, Dhollander I, Goffin K, Grachev I, Bormans G, et al. Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J Nucl Med. 2010;51:1413–7.
Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–61.
Albin RL, Reiner A, Anderson KD, Dure 4th LS, Handelin B, Balfour R, et al. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol. 1992;31(4):425–30.
Seizinger BR, Liebisch DC, Kish SJ, Arendt RM, Hornykiewicz O, Herz A. Opioid peptides in Huntington's disease: alterations in prodynorphin and proenkephalin system. Brain Res. 1986;378:405–8.
Weeks RA, Cunningham VJ, Piccini P, Waters S, Harding AE, Brooks DJ. 11C-diprenorphine binding in Huntington’s disease: a comparison of region of interest analysis with statistical parametric mapping. J Cereb Blood Flow Metab. 1997;17:943–9.
Ooms M, Rietjens R, Rangarajan JR, Vunckx K, Valdeolivas S, Maes F, et al. Early decrease of type 1 cannabinoid receptor binding and phosphodiesterase 10A activity in vivo in R6/2 Huntington mice. Neurobiol Aging. 2014;35(12):2858–69.
Shin H, Kim MH, Lee SJ, Lee KH, Kim MJ, Kim JS, et al. Decreased metabolism in the cerebral cortex in early-stage Huntington’s disease: a possible biomarker of disease progression? Int J Clin Neuropsychol. 2013;9:21–5.
Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–7.
Russell D, Jennings D, Barret O, Tamagnan G, Carroll V, Alagille D, et al. Longitudinal assessment of PDE10 in Huntington disease (HD) using [18F]MNI-659 PET imaging. J Nucl Med. 2015;56 Suppl 3:87.
Sabuncu MR, Desikan RS, Sepulcre J, Yeo BT, Liu H, Schmansky NJ, et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68:1040–8.
Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, et al. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology. 2002;58:695–701.
Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131:1057–68.
Reading SA, Dziorny AC, Peroutka LA, Schreiber M, Gourley LM, Yallapragada V, et al. Functional brain changes in presymptomatic Huntington’s disease. Ann Neurol. 2004;55:879–83.
Rosser AE, Barker RA, Harrower T, Watts C, Farrington M, Ho AK, et al. Unilateral transplantation of human primary fetal tissue in four patients with Huntington’s disease: NESTUK safety report ISRCTN no 36485475. J Neurol Neurosurg Psychiatry. 2002;73:678–85.
Furtado S, Sossi V, Hauser RA, Samii A, Schulzer M, Murphy CB, et al. Positron emission tomography after fetal transplantation in Huntington’s disease. Ann Neurol. 2005;58:331–7.
Barker RA, Mason SL, Harrower TP, Swain RA, Ho AK, Sahakian BJ, et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J Neurol Neurosurg Psychiatry. 2013;84:657–65.
Bachoud-Lévi AC, Rémy P, Nguyen JP, Brugières P, Lefaucheur JP, Bourdet C, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356:1975–9.
Gaura V, Bachoud-Lévi AC, Ribeiro MJ, Nguyen JP, Frouin V, Baudic S, et al. Striatal neural grafting improves cortical metabolism in Huntington’s disease patients. Brain. 2004;127:65–72.
Politis M, Piccini P. Brain imaging after neural transplantation. Prog Brain Res. 2010;184:193–203.
Politis M. Optimizing functional imaging protocols for assessing the outcome of fetal cell transplantation in Parkinson’s disease. BMC Med. 2011;9:50.
Squitieri F, Orobello S, Cannella M, Martino T, Romanelli P, Giovacchini G, et al. Riluzole protects Huntington disease patients from brain glucose hypometabolism and grey matter volume loss and increases production of neurotrophins. Eur J Nucl Med Mol Imaging. 2009;36(7):1113–20.
Esmaeilzadeh M, Kullingsjö J, Ullman H, Varrone A, Tedroff J. Regional cerebral glucose metabolism after pridopidine (ACR16) treatment in patients with Huntington disease. Clin Neuropharmacol. 2011;34(3):95–100.
Hjermind LE, Law I, Jønch A, Stokholm J, Nielsen JE. Huntington’s disease: effect of memantine on FDG-PET brain metabolism? J Neuropsychiatry Clin Neurosci. 2011;23(2):206–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was not funded.
Conflicts of Interest
None.
Ethical approval
This article does not describe any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pagano, G., Niccolini, F. & Politis, M. Current status of PET imaging in Huntington’s disease. Eur J Nucl Med Mol Imaging 43, 1171–1182 (2016). https://doi.org/10.1007/s00259-016-3324-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-016-3324-6