
Abstract
Quantum chemical studies using density functional theory were carried out on M(NHBMe)2 and M[Mn(CO)5]2 (M=Zn, Cd, Hg) complexes. The calculations suggest that M(NHBMe)2 and M[Mn(CO)5]2 have D2d and D4d symmetry, respectively, with a 1A1 electronic ground state. The bond dissociation energies of the ligands have the order of Zn > Cd > Hg. A thorough bonding analysis using charge and energy decomposition methods suggests that the title complexes are best represented as NHBMe⇆M0⇄NHBMe and Mn(CO)5⇆M0⇄Mn(CO)5 where the metal atom M in the electronic ground state with an ns2 electron configuration is bonded to the (NHBMe)2 and [Mn(CO)5]2 ligands through donor–acceptor interaction. These experimentally known complexes are the first examples of mononuclear complexes with divalent group 12 metals with zero oxidation state that are stable at ambient condition. These complexes represent the rare situation where the ligands act as a strong acceptor and the metal center acts as strong donor. The relativistic effect of Hg leads to a weaker electron donating strength of the 6s orbital, which explains the trend of the bond dissociation energy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The chemistry of group 12 metals (M=Zn, Cd, Hg) is dominated by their + 2 oxidation state which is caused by the ease of loss of the outermost ns2 valence electrons [1]. A far less common oxidation state is + 1. Cp*Zn–ZnCp* (Cp* = pentamethylcyclopentadiene) [2, 3], Cd2[AlCl4]2 [4, 5] and Hg2Cl2 [6] are prominent examples of the latter category. The examples of molecular complexes having zero-valent oxidation state of the group 12 metals are very scarce. Scant examples of possible zero-valent Zn in complicated systems like Zn-encapsulated MFI-type zeolite [7] and [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] featuring a Mo4Zn6 tetrahedron [8] with Mo0–Zn0–Mo0 edges were reported. A set of comparatively simpler systems containing an M0 (M=Zn, Cd, Hg) center between two ZnI centers in (LZn)2M, where L is a sterically bulky amide, was reported by Jones et al. in 2015 [9]. Transient Zn(CO)n (n = 1, 2) and Cd(CO)2 molecules were also made in solid neon and characterized using matrix-isolation infrared spectroscopy [10, 11].
In 2006, Segawa et al. [12] synthesized {(HCNDipp)2B}Li(THF)2 (Dipp = 2,6-iPr2C6H3; henceforth {(HCNDipp)2B} is abbreviated as NHBDipp), where the anionic boron ligand acts as an excellent nucleophile, and consequently, a variety of stable complexes of NHBDipp ligand with main group and transition metal atoms were reported in the literature [13,14,15,16,17,18,19,20,21]. In 2008, Nozaki and co-workers prepared the Zn(NHBDipp)2 complex [22], and in 2014, Aldridge and co-workers synthesized its Cd and Hg homologues [23]. Given the fact that NHBDipp ligand is available in anionic form as Li salt, they were often treated as M(II) complexes [23, 24]. Very recently, it was theoretically shown that NHBMe facilitates the formation of an M0–M0 single bond in M2(NHBMe)2 (M=Zn, Cd, Hg) complexes [25].
On the other hand, M[Mn(CO)5]2 (M=Zn, Cd, Hg) complexes have been known synthetically for a long time. While Hg[Mn(CO)5]2 was first synthesized in 1960 by Hieber and Schropp [26], its lighter homologues, M[Mn(CO)5]2 (M=Zn, Cd), were prepared in 1968 by Burlitch [27]. Later on, some alternative procedures were also reported to synthesize these complexes [28,29,30,31,32]. They were characterized by IR and/or Raman spectroscopic studies which suggest that these complexes possess highly symmetric D4h or D4d structures. However, an X-ray structure is only available for Hg[Mn(CO)5]2 showing the molecule with an approximate D4h symmetry [33, 34] Since Mn(CO)5− is an 18-electron complex, these complexes are generally described with interaction between M2+ and two Mn(CO)5− ligands, giving the formal oxidation state + 2 for M [30]. But an in-depth bonding analysis for these complexes is missing which leads to the question about the correct oxidation state of the metal center in M(NHBDipp)2 and M[Mn(CO)5]2 (M=Zn, Cd, Hg).
We report in this work a theoretical analysis on M(NHBMe)2 and M[Mn(CO)5]2 (M=Zn, Cd, Hg) using state-of-the-art charge and energy decomposition methods, which shows that the title complexes are best described with dative bonds NHBMe⇆M0⇄NHBMe and Mn(CO)5⇆M0⇄Mn(CO)5, where M is in the electronic ground state with an ns2 electron configuration binding to with (NHBMe)2 and [Mn(CO)5]2 ligands via donor–acceptor interaction. It is suggested that the adducts are the first examples of mononuclear complexes of divalent group 12 metals with zero oxidation state that are stable at ambient condition.
2 Computational details
The geometry optimizations followed by the harmonic frequency calculations for all the systems presented here were carried out at the BP86-D3(BJ)/def2-TZVPP level [35,36,37,38,39] using the Gaussian 16 suit of program [40]. Scalar-relativistic effective core potentials were used for the 28 and 60 core electrons of Cd and Hg, respectively. Superfine integration grid is considered for all cases. QTAIM (Quantum Theory of Atoms in Molecules) analysis [41] was performed at the BP86-D3(BJ)/def2-TZVPP/x2C-TZVPall//BP86-D3(BJ)/def2-TZVPP level where all-electron x2C-TZVPall [42] basis set is used from Cd and Hg.
The energy decomposition analysis (EDA) [43] in combination with natural orbital for chemical valence (NOCV) [44] method was performed at the BP86-D3(BJ)-ZORA/TZ2P+//BP86-D3(BJ)/def2-TZVPP level using the ADF (2018.105) program package [45, 46]. The zeroth-order regular approximation (ZORA) [47] was used to include scalar relativistic effects for the metals. All electrons were considered in the computations.
In the EDA method, the interaction energy (ΔΕint) between two prepared fragments is divided into three energy terms, viz. the electrostatic interaction energy (ΔEelstat), which represents the quasiclassical electrostatic interaction between the unperturbed charge distributions of the prepared atoms, the Pauli repulsion (ΔEPauli), which is the energy change associated with the transformation from the superposition of the unperturbed electron densities of the isolated fragments to the wavefunction that properly obeys the Pauli principle through explicit antisymmetrization and renormalization of the product wavefunction, and the orbital interaction energy (ΔEorb), which is originated from the mixing of orbitals, charge transfer and polarization between the isolated fragments. Use of D3(BJ) gives additional dispersion interaction energy (ΔEdisp) between two interacting fragments. Therefore, the interaction energy (ΔΕint) between two fragments can be defined as:
The orbital term may be further divided into contributions from each irreducible representation of the point group of the interacting system as follows:
The EDA–NOCV combination allows the partition of ΔEorb into pairwise contributions of the orbital interactions, which gives important information about bonding. The charge deformation Δρk(r) which is originated from the mixing of the orbital pairs ψk(r) and ψ-k(r) of the interacting fragments gives the size and the shape of the charge flow because of the orbital interactions (Eq. 3), and the corresponding ΔEorb reflects the amount of orbital interaction energy coming from such interaction (Eq. 4).
This method is found to be excellent to analyze the intriguing bonding situation in several interesting complexes [48,49,50,51,52,53,54,55,56,57]. For further information about this method and its application, readers are referred to the related reviews [58,59,60,61].
3 Results and discussion
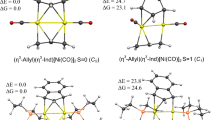
We calculated the model compounds M(NHBMe)2 (M=Zn, Cd, Hg) in place of M(NHBDipp)2 where the larger Dipp group linked to N centers is replaced by the Me group. The minimum energy geometries of the resulting complexes have a D2d symmetry and 1A1 electronic state (Fig. 1). The M–B bond lengths in M(NHBMe)2 match excellently with those of the experimental ones in M(NHBDipp)2. The B–M–B moiety in M(NHBMe)2 is perfectly linear, while due to the unsymmetrical structure of M(NHBDipp)2, the B–M–B angle in the experimental geometries ranges from 177.4° (Cd) to 179.1° (Hg). The only larger difference between the geometries of the model compounds and the experimental structures concerns the torsional angle t(N1B1B2N2) which is 90° in the model systems while it ranges from 41.8 (Cd) to 46.3 (Hg) in M(NHBDipp)2. This is most likely caused by the different steric interactions of the larger Dipp group compared with Me group. But the excellent matching in bond distances and B–M–B angles between the calculated and experimental values indicates that the model systems can be safely used to reflect the bonding situation in the experimental complexes.

The minimum energy geometries of M(NHBMe)2 and M[Mn(CO)5]2, M=Zn {Cd} [Hg] complexes at the BP86-D3(BJ)/def2-TZVPP level. The bond distances and angles are given in Å and in degree, respectively. The experimental parameters are given in italics. The computed BDE values at 298 K for ML2 → M + 2L are given in kcal/mol. Experimental bond distances correspond to M(NHBDipp)2 complexes which have two nonequivalent M–B bonds differed by only 0.001–0.002 Å
The equilibrium geometries of M[Mn(CO)5]2 (M=Zn, Cd, Hg) have D4d symmetry at the BP86-D3(BJ)/def2-TZVPP level which is in contrast to the X-ray structure of Hg[Mn(CO)5]2 which has approximately a D4h symmetry. The calculated D4h symmetric structure has a small imaginary frequency which corresponds to the internal rotation of two Mn(CO)5 units with respect to each other (see Table S1). Very soft modes of rotation and small relative energies indicate very flat potential energy surface with respect to internal rotation, and the complexes are very floppy. Therefore, the D4h symmetry in Hg[Mn(CO)5]2 is more likely because of solid state effect in the crystal structure.
Figure 1 also shows the computed bond dissociation energy (BDE) for the most favorable dissociation pathway, ML2 → M + 2L, while the complete set of BDE values for three possible dissociations via homolytic bond cleavage (ML2 → M + 2L), heterolytic bond cleavage (ML2 → M2+ + 2L−) and a mixture of them (ML2 → M+ + L + L−) is given in Table S2, where L is NHBMe or Mn(CO)5. The BDE value at 298 K ranges from 69.4 kcal/mol to 81.6 kcal/mol for M(NHBMe)2 and from 31.7 kcal/mol to 51.9 kcal/mol for M[Mn(CO)5]2 showing the order Zn > Cd > Hg. The same order was earlier found for the methyl and phenyl compounds M(Me)2 and M(Ph)2 [62]. The calculations suggest that NHBMe is a much stronger ligand than Mn(CO)5. Note that the stability order contradicts the usual trend for transition metals which usually shows the heaviest (sixth row) element having the maximum BDE value [63]. The reason can be understood from the EDA–NOCV results (vide infra).
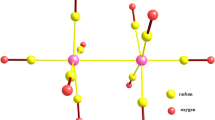
We employed the QTAIM method to analyze the electronic structure of the complexes. Figure 2 shows the contour plots of the Laplacian of electron density (∇2ρ(r)) at the Zn–B–N and Zn–Mn–C planes of Zn(NHBMe)2 and Zn[Mn(CO)5]2 complexes, respectively. The Laplacian distributions of the complete set of the systems are displayed in Figure S1. For a given type of systems, the distribution of ∇2ρ(r) looks very similar. However, there are significant differences in the Laplacian distribution between M(NHBMe)2 and M[Mn(CO)5]2 complexes. In the former systems, there is local electron density accumulation (indicated by a red dotted region) between B and M centers. Note that the bond critical point (BCP) is lying outside of the red region because of the polar nature of the bond. On the other hand, in the latter cases, there is no area of charge accumulation between Mn and M centers. There are only spherical areas of relative charge depletion (indicated by blue solid lines) around the metal atoms. Figure 2 also shows the values of ∇2ρ(rc) and local energy density (H(rc)) at the BCP of M–B and M–Mn bonds. ∇2ρ(rc) value is positive in all cases. But it does not mean that the interaction is noncovalent in nature. In fact, for such bonds where heavier elements are involved H(rc) is a more reliable descriptor to describe the nature of a bond than ∇2ρ(rc) [64]. For the M–B bonds, the polarized nature of the bond is also responsible for the positive ∇2ρ(rc) value. The H(rc) values at the BCP of M–B and M–Mn bonds are negative, indicating the covalent nature of the bond. The larger negative value in the former bond indicates larger covalent character in the M–B bond than in M–Mn bond. Note that there are no bond paths between M and CO groups. A similar situation was observed previously in the isoelectronic [M{Fe(CO)5}2]+ (M = Cu, Ag, Au) complexes [65,66,67]. This is in contrast to the recently reported hexacarbonyl cations [Ag{M(CO)6}2]+ (M = Cr, Mo, W) and the isoelectronic anions [Ag{M(CO)6}2]− (M = V, Nb, Ta) where bond paths and BCPs exist between Ag and tilted CO groups [68]. Figure 2 gives also the partial charges of the central atoms q(M) in the two sets of complexes. The group-12 metals carry a positive charge, which has the order Zn < Cd < Hg. The partial charges suggest that the [Mn(CO)5]2 ligands are stronger donor than (NHBMe)2.

The plot of Laplacian of electron density, ∇2ρ(r) at the Zn–B–N and Zn–Mn–C plane of Zn(NHBMe)2 and Zn[Mn(CO)5]2 complexes at the BP86-D3(BJ)/def2-TZVPP/x2C-TZVPall//BP86-D3(BJ)/def2-TZVPP level. Values of ∇2ρ(rc) in e/Å5 and H(rc) in Hartree/Å3 are given at BCP of M–B and M–Mn bonds. The values q(M) give the partial charge of the atom M. The blue solid lines indicate area of ∇2ρ(r) > 0, and red dotted lines represent the area of ∇2ρ(r) < 0. Blue spheres show the bond critical point
More details about the nature of chemical bonding between M and NHBMe or Mn(CO)5 groups can be gained from the results of EDA–NOCV method. To get a reliable bonding situation in the complexes in the EDA–NOCV method, the selection of proper charge and electronic state of the interacting fragments is very crucial. One of the strengths of this method is that if there are more than one partitioning scheme available, one can choose the most suitable scheme to describe the bonding by using the size of ΔEorb as a probe. For a given interaction, those fragments, which give the lowest ΔEorb value, are the best one to describe the bonding situation as it indicates that the chosen fragments are most nearly prepared as those in the complex [69,70,71,72,73,74]. For both M(NHBMe)2 and M[Mn(CO)5]2 complexes, we have carried out EDA by considering M and (L)2 with different charges and electronic states as interacting fragments. Details about the results are given in Tables S3–S8. A comparison of the relative size of ΔEorb value indicates in all cases that the best description is provided using M in the ground state with (ns)2(np)0 valence electronic configuration and (L)2 in singlet spin state interacting through donor–acceptor type of bonding. Therefore, the complexes should be represented as NHBMe⇆M0⇄NHBMe and Mn(CO)5⇆M0⇄Mn(CO)5, where M is in the zero oxidation state.
Detailed numerical results of EDA–NOCV for the most favorable scheme are provided in Tables 1 and 2. Similar to the BDE values, the intrinsic interaction between M and (L)2 is the strongest for M=Zn followed by Cd and Hg, and for a given M, it is weaker for L = Mn(CO)5 than for NHBMe. Except for Zn(NHBMe)2, the M–L interaction is somewhat more electrostatic than covalent. Dispersion interaction is only responsible for 3–6% of total attraction. There are differences in the origin of obtained order in ΔEint between M(NHBMe)2 and M[Mn(CO)5]2 complexes. In the former case, increased Pauli repulsion and weakened ΔEorb values in going from Zn to Cd to Hg are responsible for the observed trend, whereas in the latter one, both weakened ΔEorb and ΔEelstat are accountable for this.
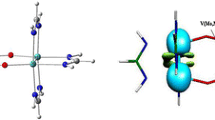
The breakdown of the ΔEorb into pairwise orbital interaction provides the most important information regarding the bonding between M and ligands. We have tabulated seven distinct pairwise contributions of ΔEorb for M(NHBMe)2 (Table 1) and nine such orbital terms for M[Mn(CO)5]2 (Table 2). Corresponding deformation densities Δρ for Zn complex are provided in Fig. 3 which help to understand the involved orbitals in the interaction. The Δρ plots for Cd and Hg homologues are very similar to the Zn complex. The results show that the strongest orbital contribution, ∆Eorb(1), is originated from the in-phase L←M(s)→L σ backdonation which accounts for 64–72% of total ΔEorb value. Note that because of relativistic contraction of 6s orbital in Hg, L←Hg(s)→L σ backdonation is the weakest one. The next strongest interaction, ∆Eorb(2), comes from the out-of-phase L→M(pσ)←L σ donation which is responsible for 17–22% of total ΔEorb. These two interactions together make the 81–94% of covalent interaction. There are two weak degenerate L→M(pπ)←L π donations ∆Eorb(3) and ∆Eorb(4), which only contribute 4–7% to ΔEorb. There is also some d orbital participation in the L←M(d)→L backdonation, albeit even weaker than the participation of the pπ orbitals. In case of M(NHBMe)2, the participation of only three d orbitals is found, whereas in M[Mn(CO)5]2, all five d orbitals are involved in the bonding where the CO groups also participate in the orbital interactions. Nevertheless, the combined effect of d orbitals provides only 3–5% to the covalent interaction. Interestingly, the present cases represent a rather rare scenario where L = NHBMe, Mn(CO)5 act as dominant acceptor and M=Zn, Cd, Hg act as donor centers. We have recently carried out EDA–NOCV calculations on [M{Fe(CO)5}2]+ (M=Cu, Ag, Au), the isoelectronic complexes of M[Mn(CO)5]2 (M=Zn, Cd, Hg), taking M+ as one fragment and (Fe(CO)5)2 as another [65]. The intrinsic interaction between coinage metal cation and (Fe(CO)5)2 is much stronger than that in the latter complex where both enhanced orbital and electrostatic interaction in the cationic complexes are responsible for this. Notably, despite cationic charge, [Fe(CO)5]←M+(d)→[Fe(CO)5] backdonation is much stronger (responsible for 13–24% of ΔEorb) than Mn(CO)5←M(d)→Mn(CO)5 backdonation.

The shape of the deformation densities, Δρ(n), which are associated with ∆Eorb(n) for Zn(NHBMe)2 and Zn[Mn(CO)5]2 complexes at the BP86-D3(BJ)-ZORA/TZ2P+//BP86-D3(BJ)/def2-TZVPP level. The isovalue is 0.0001 au. The direction of the charge flow of the deformation densities is red → blue
The dominant orbital interaction ∆Eorb(1) coming from the L←M(s)→L σ backdonation is in agreement with the calculated partial charges q(M) given by the AIM method (Fig. 2). But the order of the donor strength of the group-12 atoms Zn > Cd > Hg given by ∆Eorb(1) is opposite to the order of the partial charges. Also, the [Mn(CO)5]2 ligands are more weakly bonded to the group-12 atoms than the (NHBMe)2 groups, although the charge donation of the former is larger compared with the latter. This shows that the size of the charge migration is not always a measure of the associated stabilization energy. This is a warning against premature correlations between charge migration and energy changes.
4 Summary and conclusions
Quantum chemical calculations are performed taking two sets of experimentally known complex of group 12 elements, M(NHBMe)2 and M[Mn(CO)5]2 (M=Zn, Cd, Hg), where the formal oxidation state of M was previously considered to be + 2 because of the anionic nature of the ligands. M(NHBMe)2 and M[Mn(CO)5]2 have D2d and D4d symmetry, respectively, with 1A1 electronic ground state. The bond dissociation energies of the ligands have the order of Zn > Cd > Hg. A thorough bonding analysis using charge and energy decomposition methods suggests that the title complexes are best represented as NHBMe⇆M0⇄NHBMe and Mn(CO)5⇆M0⇄Mn(CO)5, where M is in the electronic ground state with ns2 electron configuration binding to (NHBMe)2 and [Mn(CO)5]2 ligands through donor–acceptor interaction. The complexes are the first examples of mononuclear adducts with divalent group 12 metals with zero oxidation state that are stable at ambient condition. The complexes also represent a rare situation where the ligand acts as a strong acceptor and the metal center acts as strong donor. The relativistic effect of Hg leads to a weaker electron donor strength of the 6s orbital, which explains the trend of the bond dissociation energy. The present study shows that a thorough bonding analysis is needed to understand the actual bonding situation in the complex and the correct oxidation state of the metal.
References
Liu K, Shi W, Cheng P (2011) The coordination chemistry of Zn (II), Cd (II) and Hg (II) complexes with 1, 2, 4-triazole derivatives. Dalton Trans 40:8475–8490
Resa I, Carmona E, Gutierrez-Puebla E, Monge A (2004) Decamethyldizincocene, a stable compound of Zn (I) with a Zn–Zn bond. Science 305:1136–1138
Li T, Schulz S, Roesky PW (2012) Synthesis, reactivity and applications of zinc–zinc bonded complexes. Chem Soc Rev 41:3759–3771
Corbett JD, Burkhard WJ, Druding LF (1961) Stabilization of the cadmium(I) oxidation state. The system Cd–Cd2I(AlCl4)2–Cd2(AlCl4)2. J Am Chem Soc 83:76–80
Staffel T, Meyer G (1987) Synthesis and crystal structures of Cd[AlCl4]2 and Cd2[AlCl4]2. Z Anorg Allg Chem 548:45–54
Roseveare WE (1930) The X-ray photochemical reaction between potassium oxalate and mercuric chloride. J Am Chem Soc 52:2612–2619
Oda A, Torigoe H, Itadani A, Ohkubo T, Yumura T, Kobayashi H, Kuroda Y (2013) Success in making Zn+ from atomic Zn0 encapsulated in an MFI-type zeolite with UV light irradiation. J Am Chem Soc 135:18481–18489
Banh H, Dilchert K, Schulz C, Gemel C, Seidel RW, Gautier R, Kahlal S, Saillard JY, Fischer RA (2016) Atom-precise organometallic zinc clusters. Angew Chem Int Ed 55:3285–3289
Hicks J, Underhill EJ, Kefalidis CE, Maron L, Jones C (2015) A mixed-valence tri-zinc complex, [LZnZnZnL] (L = Bulky Amide), bearing a linear chain of two-coordinate zinc atoms. Angew Chem Int Ed 54:10000–10004
Jiang L, Teng YL, Xu Q (2006) Reactions of laser-ablated zinc and cadmium atoms with CO: infrared spectra of the Zn(CO)x (x + 1–3), CdCO−, and Cd(CO)2 molecules in solid neon. J Phys Chem A 110:7092–7096
Jin L, Fu L-j, Ding Y-h (2010) Zinc (0) chemistry: Does the missing 18-electron zinc tricarbonyl really exist? Phys Chem Chem Phys 12:10956–10962
Segawa Y, Yamashita M, Nozaki K (2006) Boryllithium: isolation, characterization, and reactivity as a boryl anion. Science 314:113–115
Arnold T, Braunschweig H, Ewing WC, Kramer T, Mies J, Schuster JK (2015) Beryllium bis (diazaborolyl): old neighbors finally shake hands. Chem Commun 51:737–740
Hayashi Y, Segawa Y, Yamashita M, Nozaki K (2011) Syntheses and properties of triborane (5) s possessing bulky diamino substituents on terminal boron atoms. Chem Commun 47:5888–5890
Nozaki K, Aramaki Y, Yamashita M, Ueng S-H, Malacria M, Lacôte E, Curran DP (2010) Boryltrihydroborate: synthesis, structure, and reactivity as a reductant in ionic, organometallic, and radical reactions. J Am Chem Soc 132:11449–11451
Dettenrieder N, Schädle C, Maichle-Mössmer C, Sirsch P, Anwander R (2014) A dimethylgallium boryl complex and its methyllithium addition compound. J Am Chem Soc 136:886–889
Yamashita M, Suzuki Y, Segawa Y, Nozaki K (2007) Synthesis, structure of borylmagnesium, and its reaction with benzaldehyde to form benzoylborane. J Am Chem Soc 129:9570–9571
Dettenrieder N, Dietrich HM, Schädle C, Maichle-Mössmer C, Törnroos KW, Anwander R (2012) Organoaluminum boryl complexes. Angew Chem Int Ed 51:4461–4465
Protchenko AV, Dange D, Harmer JR, Tang CY, Schwartz AD, Kelly MJ, Phillips N, Tirfoin R, Birjkumar KH, Jones C, Kaltsoyannis N, Mountford P, Aldridge S (2014) Stable GaX2, InX2 and TlX2 radicals. Nat Chem 6:315–319
Protchenko AV, Birjkumarm KH, Dange D, Schwartz AD, Vidovic D, Jones C, Kaltsoyannis N, Mountford P, Aldridge S (2012) A stable two-coordinate acyclic silylene. J Am Chem Soc 134:6500–6503
Saha R, Pan S, Merino G, Chattaraj PK (2019) Unprecedented bonding situation in viable E2(NHBMe)2 (E=Be, Mg; NHBMe=(HCNMe)2B) complexes: neutral E2 forms a single E−E covalent bond. Angew Chem Int Ed 58:8372–8377
Kajiwara T, Terabayashi T, Yamashita M, Nozaki K (2008) Syntheses, structures, and reactivities of borylcopper and-zinc compounds: 1, 4-silaboration of an α, β-unsaturated ketone to form a γ-siloxyallylborane. Angew Chem Int Ed 47:6606–6610
Protchenko AV, Dange D, Schwarz AD, Tang CY, Phillips N, Mountford P, Jones C, Aldridge S (2014) Heavy metal boryl chemistry: complexes of cadmium, mercury and lead. Chem Commun 50:3841–3844
Wang SR, Arrowsmith M, Braunschweig H, Dewhurst RD, Dömling M, Mattock JD, Pranckevicius C, Vargas A (2017) Monomeric 16-electron π-diborene complexes of Zn(II) and Cd(II). J Am Chem Soc 139:10661–10664
Saha R, Pan S, Chattaraj PK, Merino G (2020) Filling the void: controlled donor–acceptor interaction facilitates the formation of an M–M single bond in the zero oxidation state of M (M=Zn, Cd, Hg). Dalton Trans 49:1056–1064
Hieber W, Schropp W Jr (1960) Via pentacarbonyl manganese (—I) compounds. Chem Ber 93:455
Burtlich JM (1968) Metal insertion into the Mn–Mn bond of Mn2(CO)10. Chem Commun 887
King RB (1963) The formation of mercury derivatives in reactions involving sodium derivatives of metal carbonyls prepared from sodium amalgam. J Inorg Nucl Chem 25:1296
Carev NA, Noltos JG (1968) ibid. 1471
Louwen JN, Andréa RR, Stufkens DJ, Oskam A, (1982) He (I) and He (II) photoelectron spectra of M[Co(CO)4]2 and M[Mn(CO)5]2 complexes (M=Zn, Cd, and Hg). Z Naturforsch 37b:711–717
Burtlich JM, Ferrari A (1970) Chemistry of bonds between metals. I. synthesis of transition metal carbonyl derivatives of zinc and cadmium by a metal-exchange reaction. Inorg Chem 9:563–569
Adams DM, Squire A (1968) Vibrational spectra of decacarbonyldimanganese and its halogen and mercury derivatives. J Chem Soc A 2817–2819
Katcher ML, Simon GL (1972) Stereochemistry and bonding in bis[pentacarbonylmanganese]mercury, Hg[Mn(CO)5]2. Linear, mercury-bridged derivative of dimanganese decacarbonyl with an eclipsed conformation. Inorg Chem 11:1651
Clegg W, PWheatley PJ (1971) Crystal and molecular structure of mercuriobis(pentacarbonylmanganese), Hg[Mn(CO)5]2. J Chem Soc (A) 3572–3574
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys Rev A 38:3098
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J Chem Phys 132:154104
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297
Weigend F (2006) Accurate Coulomb-fitting basis sets for H to Rn. Phys Chem Chem Phys 8:1057
Gaussian 16, Revision A.03, M. J. Frisch, et al. Gaussian, Inc., Wallingford CT. 2016.
Bader RFW (1990) Atoms in molecules, a quantum theory. Oxford University Press
Pollak P, Weigend F (2017) Segmented contracted error-consistent basis sets of double-and triple-ζ valence quality for one-and two-component relativistic all-electron calculations. J Chem Theory Comput 13:3696–3705
Ziegler T, Rauk A (1977) On the calculation of bonding energies by the Hartree Fock Slater method. Theor Chim Acta 46:1
Mitoraj M, Michalak A (2007) Donor-acceptor properties of ligands from the natural orbitals for chemical valence. Organometallics 26:6576
ADF2018, SCM, Theoretical chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
te Velde G, Bickelhaupt FM, Baerends EJ, Guerra CF, van Gisbergen SJA, Snijders JG, Ziegler T (2001) Chemistry with ADF. J Comput Chem 22:931
van Lenthe E, Ehlers AE, Baerends EJ (1999) Geometry optimization in the zero order regular approximation for relativistic effects. J Chem Phys 110:8943
Yang W, Krantz KE, Freeman LA, Dickie D, Molino A, Frenking G, Pan S, Wilson DJD, Gilliard RJ Jr (2020) Persistent borafluorene radicals. Angew Chem Int Ed 59:3850
Wang Q, Pan S, Wu Y, Deng G, Wang G, Zhao L, Zhou M, Frenking G (2019) Transition-metal chemistry of alkaline-earth elements: the trisbenzene complexes M(Bz)3 (M=Sr, Ba). Angew Chem Int Ed 58:17365
Wang Q, Pan S, Lei S, Jin J, Deng G, Wang G, Zhao L, Zhou M, Frenking G (2019) Octa-coordinated alkaline earth metal–dinitrogen complexes M(N2)8 (M=Ca, Sr, Ba). Nat Commun 10:3375
Chi C, Pan S, Meng L, Luo M, Zhao L, Zhou M, Frenking G (2019) Alkali metal covalent bonding in nickel carbonyl complexes ENi(CO)3−. Angew Chem Int Ed 58:1732
Wu X, Zhao L, Jin J, Pan S, Li W, Jin X, Wang G, Zhou M, Frenking G (2018) Observation of alkaline earth complexes M(CO)8 (M=Ca, Sr, or Ba) that mimic transition metals. Science 361:912–916
Pan S, Zhao L, Dias HVR, Frenking G (2018) Bonding in binuclear carbonyl complexes M2(CO)9 (M=Fe, Ru, Os). Inorg Chem 57:7780
Pecher L, Pan S, Frenking G (2019) Chemical bonding in the hexamethylbenzene–SO2+ dication. Theor Chem Acc 138:47
Pan S, Frenking G (2020) Comment on “Realization of Lewis Basic Sodium Anion in the NaBH3− Cluster.” Angew Chem Int Ed 59:8756
Zhao L, Pan S, Zhou M, Frenking G (2019) Response to Comment on “Observation of alkaline earth complexes M(CO)8 (M=Ca, Sr, or Ba) that mimic transition metals”. Science 365:eaay5021
Chi C, Pan S, Jin J, Meng L, Luo M, Zhao L, Zhou M, Frenking G (2019) Octacarbonyl ion complexes of actinides [An(CO)8]+/− (An=Th, U) and the role of f orbitals in metal-ligand bonding. Chem Eur J 25:11772
Frenking G, Bickelhaupt FM (2014) The chemical bond 1. Fundamental aspects of chemical bonding, chap. The EDA perspective of chemical bonding, 121. Wiley: Weinheim,.
Zhao L, von Hopffgarten M, Andrada DM, Frenking G (2018) Energy decomposition analysis. WIREs Comput Mol Sci 8:1345
Zhao L, Hermann M, Schwarz WHE, Frenking G (2019) The Lewis electron-pair bonding model: modern energy decomposition analysis. Nat Rev Chem 3:48
Zhao L, Pan S, Holzmann N, Schwerdtfeger P, Frenking G (2019) Chemical bonding and bonding models of main-group compounds. Chem Rev 119:8781
Antes I, Frenking G (1995) Structure and bonding of the transition metal methyl and phenyl compounds MCH3, MC6H5 (M=Cu, Ag, Au) and M(CH3)2, M(C6H5)2 (M=Zn, Cd, Hg). Organometallics 14:4263
Frenking G, Fröhlich N (2000) The nature of the bonding in transition-metal compounds. Chem Rev 100:717–774
Cremer D, Kraka E (1984) Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond? Angew Chem Int Ed 23:627–628
Pan S, Gorantla SMNVT, Parasar D, Dias HVR, Frenking G (2021) Chemical bonding in homoleptic carbonyl cations [M{Fe(CO)5}2]+ (M=Cu, Ag, Au). Chem Eur J 27:6936. https://doi.org/10.1002/chem.202004041
Poater J, Solà M, Bickelhaupt FM (2006) A model of the chemical bond must be rooted in quantum mechanics, provide insight, and possess predictive power. Chem Eur J 12:2902–2905
Poater J, Solà M, Bickelhaupt FM (2006) Hydrogen–hydrogen bonding in planar biphenyl, predicted by atoms-in-molecules theory, does not exist. Chem Eur J 12:2889–2895
Bohnenberger J, Kratzert D, Gorantla SMNVT, Pan S, Frenking G, Krossing I (2020) Group 6 hexacarbonyls as ligands for the silver cation: syntheses, characterization, and analysis of the bonding compared with the isoelectronic group 5 hexacarbonylates. Chem Eur J 26:17203
Fernandez I, Holzmann N, Frenking G (2020) The valence orbitals of the alkaline earth atoms. Chem Eur J 26:14194
Buchner MR, Pan S, Poggel C, Spang N, Müller M, Frenking G, Sundermeyer J (2020) Di-ortho-beryllated carbodiphosphorane: a Compound with a metal-carbon double bond to an element of the s-block. Organometallics 39:3224–3231
Manoj S, Pan S, Mondal KC, Frenking G (2020) Stabilization of linear C3 by two donor ligands. a theoretical study of L–C3–L (L=PPh3, NHCMe, cAACMe). Chem Eur J 26:14211–14220
Deng G, Pan S, Wang G, Zhao L, Zhou M, Frenking G (2020) Side-on bonded beryllium dinitrogen complexes. Angew Chem Int Ed 59:10603–10609
Su W, Pan S, Sun X, Zhao L, Frenking G, Zhu C (2019) Cerium-carbon double dative bonds supported by carbodiphosphorane. Dalton Trans 48:16108–16114
Wang L, Pan S, Lu B, Dong X, Li H, Deng F, Zeng X, Zhou M, Frenking G (2021) Generation and characterization of the C3O2− anion with an unexpected unsymmetrical structure. Angew Chem Int Ed 60:4518. https://doi.org/10.1002/anie.202013921
Acknowledgements
LZ and GF acknowledge the financial support from National Natural Science Foundation of China (Grant No. 21973044), Nanjing Tech University (Grant Nos. 39837123 and 39837132), the State Key Laboratory of Materials-oriented Chemical Engineering (project No. KL19-11), and the High Performance Centre of Nanjing Tech University for supporting the computational resources. SP thanks Nanjing Tech University for a postdoctoral fellowship. SP and GF acknowledge financial support by the Deutsche Forschungsgemeinschaft (Grant FR 641/34-1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts or competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published as part of the special collection of articles “Festschrift in honour of Prof. Ramon Carbó-Dorca.”
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, S., Zhao, L. & Frenking, G. Bonding in M(NHBMe)2 and M[Mn(CO)5]2 complexes (M=Zn, Cd, Hg; NHBMe=(HCNMe)2B): divalent group 12 metals with zero oxidation state. Theor Chem Acc 140, 69 (2021). https://doi.org/10.1007/s00214-021-02751-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-021-02751-y