
Abstract
Purpose
To compare bacteria recovered by standard cultures and metataxonomics, particularly with regard to ventilator-associated pneumonia (VAP) pathogens, and to determine if the presence of particular bacteria or microbiota in tracheal and oropharyngeal secretions during the course of intubation was associated with the development of VAP.
Methods
In this case–control study, oropharyngeal secretions and endotracheal aspirate were collected daily in mechanically ventilated patients. Culture and metataxonomics (16S rRNA gene-based taxonomic profiling of bacterial communities) were performed on serial upper respiratory samples from patients with late-onset definite VAP and their respective controls.
Results
Metataxonomic analyses showed that a low relative abundance of Bacilli at the time of intubation in the oropharyngeal secretions was strongly associated with the subsequent development of VAP. On the day of VAP, the quantity of human and bacterial DNA in both tracheal and oropharyngeal secretions was significantly higher in patients with VAP than in matched controls with similar ventilation times. Molecular techniques identified the pathogen(s) of VAP found by culture, but also many more bacteria, classically difficult to culture, such as Mycoplasma spp. and anaerobes.
Conclusions
Molecular analyses of respiratory specimens identified markers associated with the development of VAP, as well as important differences in the taxa abundance between VAP and controls. Further prospective trials are needed to test the predictive value of these markers, as well as the relevance of uncultured bacteria in the pathogenesis of VAP.
Similar content being viewed by others

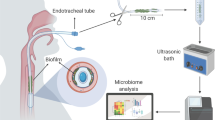
Molecular analyses of respiratory samples showed important taxonomic differences between patients with VAP and matched controls, and permitted the identification of early markers associated with the development of VAP. In the future, this could lead to an improvement in VAP diagnosis and prevention. |
Introduction
Ventilator-associated pneumonia (VAP) is a frequent complication in the intensive care unit (ICU) and associated with prolonged mechanical ventilation (MV), longer ICU stay and outcome [1,2,3,4]. VAP diagnosis relies on a combination of clinical and radiological signs, usually coupled with a quantitative culture of bronchoalveolar lavage (BAL) fluid [5, 6], with the drawback that cultures postpone VAP diagnosis by 48–72 h. Microbiological culture surveillance of endotracheal aspirates (ETA) helps to identify early and with reasonable probability the VAP pathogen and improves empirical treatment. However, an effect on mortality has not been proven [7,8,9,10]. Studies with a clear demonstration of the efficacy and safety of routine culture surveillance are still lacking. In addition, the presence of a pathogen in surveillance cultures, even with a high bacterial burden, is not systematically associated with the development of VAP and clearing of bacteria by the host immune system is possible, even in the absence of antibiotics [11]. Other strategies are, therefore, needed for the early identification of patients at highest risk of VAP and with a high likelihood to benefit from preventive measures, such as aspiration of oropharyngeal secretions (OPS), silver-coated tubes, continuous control of cuff pressure, selective oropharyngeal or digestive decontamination [12, 13], or preemptive antibiotic treatment [14].
There is an unmet need of culture-independent strategies that allow the rapid and extensive identification of bacteria in biological fluids. The 16S rRNA gene-based taxonomic profiling of bacterial communities (metataxonomics) in respiratory specimens allows to study the whole respiratory microbiota and identify many bacteria and potential pathogens that are not routinely cultured [15,16,17]. Only a few studies have evaluated the usefulness of metataxonomics and other molecular approaches for the prediction or diagnosis of VAP [18,19,20,21], but the specificity of the results obtained with such sensitive techniques has been a concern [22, 23].
We aimed to identify metataxonomic VAP markers early in the course of intubation and to compare bacteria recovered by standard cultures and metataxonomics, particularly VAP pathogens.
This work was presented as a poster at the European Congress of Clinical Microbiology and Infectious Diseases in 2018 (available online at https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=63447).
Methods
Study design and patients
This is a case-control study nested in a prospective single-centre cohort of mechanically ventilated (MV) adult patients. The study was conducted at Geneva University Hospitals (Geneva, Switzerland) between October 2012 and March 2014. Patients were recruited in a 36-bed polyvalent ICU treating ~ 2500 patients/year with a mean length of stay of 4 days. Inclusion criteria were antibiotic-naïve patients intubated for less than 24 h, with an expected duration of > 4 days. The study protocol (clinicaltrials.gov # NCT01875692) was approved by the institutional ethics committee. Informed consent was obtained from patients’ next-of-kin and confirmed in patients whenever possible.
Daily clinical and microbiological data were collected and entered into a secure online database (SecuTrial®). Patient charts were reviewed by two independent ICU physicians who allocated patients to the following groups: “definite VAP”; “probable VAP”; or “absence of VAP”. In the case of disagreement, a third senior investigator from the Division of Infectious Diseases was consulted to achieve consensus (needed for two patients only). “Definite VAP” was diagnosed when ventilated patients presented clinical and radiological signs of VAP with a clinical pulmonary infection score (CPIS) > 6 [24] and ≥ 103 bacteria/mL in protected catheter mini-BAL fluid (Combicath®, Kol Bio-Medical, Fairfax, VA) or ≥ 104 bacteria/mL in bronchoscopic BAL fluid. “Probable VAP” was defined as patients with clinical and radiological signs of VAP, a CPIS > 6 and the presence of a bacterial pathogen in ETA irrespective of the abundance of the pathogen. Of note, BAL sampling was not available 24/24 h and some “probable VAP” would have probably been classified as “definite VAP” if BAL sampling had been performed at the time of clinical suspicion.
Early-onset VAP was defined as VAP occurring after 48 h of MV, but before day five; late-onset VAP was defined as VAP occurring from day five onwards. Daily OPS collected before chlorhexidine mouthwashing were sent together with paired ETA to the bacteriology laboratory for culture. Aliquots were frozen at − 80 °C for further batched metataxonomics in selected patients. Clinicians were blinded to the bacteriological results linked to the study. Bacterial cultures of ETAs ordered by clinicians were processed independently from the study.
VAP patients and matched controls were identified in our prospective cohort. Matching was performed essentially on MV duration. Matched controls had a mandatory MV duration of at least 2 days longer than the period preceding the VAP onset of their case, allowing to study the same time points in cases and matched controls. Other criteria used for matching were: APACHE II score at time of admission, diagnosis, age and the use of antibiotics. The matching algorithm used is described in the online data supplement. Three to four matched controls were defined for each patient diagnosed with late-onset VAP for the “culture” study. Given the costs of molecular analyses and to study the most relevant VAP patient population, metataxonomic analyses were limited to patients with late-onset “definite VAP” (N = 18) and compared with their two best-matched controls (N = 36). Whereas OPS and ETA were cultured daily during intubation, metataxonomics was only performed at five time points during MV: on D0 (day of intubation); D3 (3 days after intubation); DVAP-3 (3 days before VAP); DVAP (day of VAP diagnosis); and DVAP + 3 (3 days after VAP). If a sample was not available for a given time point, the sample of the following day was analyzed. The study section related to serial microbiological ETA and OPS cultures, methods and results is presented in the online supplement.
Sample size calculation
Sample size was determined by a power calculation based on hypotheses done with serial cultures (see online supplement). For metataxonomics, we analyzed the subset of late-onset “definite VAP” (N = 18) and their two matched controls (N = 36), at multiple time points.
Metataxonomics
A detailed description of the metataxonomic analyses of study specimens is provided in the online data supplement. Briefly, DNA was extracted by the NucleoSpin Soil Kit (Macherey–Nagel, Düren, Germany) using 400 μL of sample. 16S rRNA gene amplification consisted in a first round of polymerase chain reaction (PCR) targeting the V1-6 region of the bacterial 16S rRNA gene, followed by a nested PCR of the V3-4 region. Sequencing (2 × 300) was performed on an Illumina MiSeq instrument using a MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA) at LGC Genomics (Berlin, Germany). Merged and quality filtered sequence reads were denoized by mapping to Operational Taxonomic Units (OTUs) with the Greengenes reference OTU database [25] pre-clustered at 99% using USEARCH [26]. The taxonomy was assigned to sequence reads using the Mothur’s RDP reference database [27, 28]. To remove putative contaminant OTUs, we first calculated the R-OTU value for each identified OTU [29]. The dataset was decontaminated by removing the reads with R-OTU > 0.1. This arbitrary selected R-OTU value performed well in removing known contaminant taxa, while not affecting taxa typical of oral/respiratory microbiota (Table S1).
Statistical analysis
To assess differences in microbiota composition, we used permutational multivariate analysis of variance (PERMANOVA, PRIMER, Primer-E Ltd., Plymouth, UK) of the square-root-transformed Bray–Curtis similarity matrix with 9999 permutations. Microbiota composition was compared pairwise at each time point: (1) VAP vs. controls samples; and (2) ETA vs. OPS samples.
The Wilcoxon rank-sum test was used to compare VAP patients vs. controls in terms of the absolute abundance (see Supplementary material) of individual bacterial taxa in samples of the same type (ETA, OPS) taken at the same time point. The Wilcoxon signed-rank test was used to assess the statistical significance of differences between bacterial taxa in samples taken from the same patient either at two time points from the same sample type (ETA, OPS) or at the same time point from different sample types. Wilcoxon rank-sum test was used to compare intra-individual vs. inter-individual Bray–Curtis similarities and DNA loads of VAP patients vs. controls at different time points. ROC curves were computed to assess the performance of biomarkers (MedCalc), following the methodology of DeLong et al. [30] and the binomial exact test.
Temporal changes of OPS and ETA microbiota centroids were visualized using Principal Coordinates Analysis plots in PRIMER. The analysis was based on Bray–Curtis similarity matrix of the square-root-transformed relative abundance of bacterial genera. Group average clustering of the Bray–Curtis similarity matrix was performed in PRIMER, based on the square-root transformed relative abundance of bacterial families. Canonical Analysis of Principal Coordinates (CAP) [31] was performed on the Euclidean distance matrix of the relative abundance of genera or OTUs using PRIMER. CAP maximizes the group differences in the multivariate cloud of points [31] to separate samples into a priori defined groups (VAP, controls) and the proportion of correct classifications can then be computed.
Statistical significance was set at the 95% confidence level (p < 0.05). The Benjamini–Hochberg procedure was used to correct p values for multiple testing using a false discovery rate of 0.05.
Results
Among 1154 MV patients screened, 286 were included and 263 could be analyzed (Fig. 1). Of these, 213 were extubated and 50 died before extubation. Sixty-seven VAP episodes (40 “definite”; 27 “probable”) were diagnosed in 62 patients (24%) of the 263 patients ventilated for more than 48 h. Only the first episode of VAP was studied in each patient. VAP incidence density was not available at the beginning of the study, but was 24 VAP/1000 ventilation days ≥ 48 h during the last 3 months of the study [13]. Table 1 shows the characteristics of the population studied by metataxonomics, i.e., late-onset “definite VAP” patients (N = 18) and their controls (N = 36). Male patients were predominant and proportionally more VAP developed in men compared with women (42.4% vs. 19%, respectively; p = 0.139). Patients with late-onset “definite VAP” required more days of MV than controls and were twice as likely to die (mortality 27.8% vs. 13.9%; p = 0.199), despite similar APACHE II scores. However, these differences did not reach statistical significance. Characteristics of all late-onset VAP patients are shown in Table S2.

Study flow chart. ICU intensive care unit, MV mechanical ventilation, VAP ventilator-associated pneumonia
Metataxonomics
16S rRNA gene amplicon libraries were sequenced from 240 OPS, 235 ETA and 24 BAL fluids obtained from 18 patients with late-onset “definite VAP” and 36 matched controls. Whenever available, 5 OPS and 5 ETA samples were selected from each patient so as to correspond as far as possible to D0, D3, DVAP-3, DVAP and DVAP + 3. Processing of 88 negative controls (Fig. S1) allowed the removal of sequence reads putatively derived from reagent contaminants using a method combining next-generation sequencing and qPCR data [29]. After this decontamination step, 233 OPS, 218 ETA and 22 BAL samples were normalized to 1000 reads per sample and analyzed.
Changes in respiratory microbiota during mechanical ventilation
OPS and ETA microbiota from VAP patients and controls showed a similar overall trend of changes over time (Fig. 2a), represented by a progressive increase in phyla Proteobacteria (which includes Gram-negative VAP pathogens) and Tenericutes (cell-wall free bacteria including Mycoplasma), and a decrease in the proportion of other major phyla, including Firmicutes. These changes were associated with a decrease in bacterial diversity (Fig. 2a and S2). At the genus level, the significant differences corresponded to a decrease in the abundance over time, except for Mycoplasma which increased (OPS samples; Fig. S3). Temporal changes of both OPS and ETA microbiota centroids were negatively related to the first Principal Coordinate (PCo) axis (Fig. 2b) and, to a lesser extent, positively correlated to the second PCo. Although this general trend was found in most patients, variations between individuals were substantial (Fig. 2c, d), as shown in a previous report [21]. For example, for OPS samples, the intra-individual D0–DVAP Bray–Curtis similarity was in the 2.5–65.7 range (median 50.4) in VAP patients and between 3.9 and 63 (median 39.4) in controls. Intra-individual Bray–Curtis similarities between baseline and later time points (i.e., D0–D3, D0–DVAP-3, D0–DVAP, D0–DVAP + 3) were not significantly different (p > 0.05, Wilcoxon rank-sum test) when VAP cases were compared to controls. However, differences between VAP and control patients reached statistical significance for D0–DVAP changes along the PCo2 axis for OPS samples (Fig. 2d).

Microbiota composition and diversity change during intubation. The analysis was based on the dataset normalized to 1000 reads per sample. Twenty groups compared were defined by three factors: development of VAP (VAP, control), sample type (OPS, ETA) and sampling point (D0, D3, DVAP-3, DVAP and DVAP + 3). a Mean relative abundances for seven major bacterial phyla (top panel) and Shannon diversity index (bottom panel). b Principal coordinates analysis of the Bray–Curtis similarity matrix based on the square-root transformed relative abundance of bacterial genera. Bacterial communities defined by the same combination of the three factors were grouped to the centroid. Dotted lines connect neighboring time point centroids. Vectors of Pearson correlation (with length > 0.4) between genera and PCo axes are shown above the plot. In control patients, baseline (D0) ETA and OPS microbial communities were significantly different (PERMANOVA p < 0.05) from those of DVAP-3, DVAP and DVAP + 3. In patients who developed VAP, OPS microbiota was significantly different from those of DVAP and DVAP + 3. c–d Changes per patient between D0 and DVAP bacterial communities along PCo1 and PCo2 axes. Changes are represented by dashed lines, in red for patients who developed VAP and in blue for control patients. The change along PCo2 was significantly lower for OPS of patients who developed VAP (P = 0.02, Wilcoxon rank-sum test). Day 0 samples of all patients were arbitrarily placed at the origin of both axes. OPS oropharyngeal secretions, ETA endotracheal aspirate, VAP ventilator-associated pneumonia, D0 intubation day, D3 3 days after intubation, DVAP-3 3 days before VAP, DVAP day of VAP diagnosis, DVAP + 3 3 days after VAP
Microbiota similarities between different respiratory samples
OPS and ETA microbiota profiles were globally different at each time point (PERMANOVA p < 0.05). However, intra-individual microbiota similarity between sample type pairs (OPS–ETA, OPS–BAL, ETA–BAL) was greater than inter-individual similarities for any given sample type (Figs. S4 and S5). Therefore, microbiota of OPS, ETA and BAL fluid sampled from the same individual at the same day clustered well together (Fig. 3).

Microbial communities from different airway sites of the same patient are closely related. The three sample types (OPS, ETA and BAL) were compared whenever available at exactly the same day for each patient. The group average hierarchical clustering of samples (represented at the top) was based on the Bray–Curtis similarity of square-root-transformed relative abundance of bacterial families. Samples from the same patients are represented on the dendrogram by the patient number and color. Two sets of samples from patient 239 were available for the analysis; they were collected at days 3 and 7 of intubation (indicated in parenthesis). Sample types are indicated by gray symbols below patient numbers. The heat plot (bottom panel) shows the square-root-transformed proportions of bacterial families according to the intensity gradient on the right. OPS oropharyngeal secretions, BAL bronchoalveolar lavage fluid, ETA endotracheal aspirate, VAP ventilator-associated pneumonia
Differences in respiratory microbiota between VAP patients and controls
No significant changes of ETA or OPS microbiota between VAP patients and controls were observed at any time point (PERMANOVA, p > 0.05). However, when looking for VAP-discriminatory taxa that could potentially provide an early distinction between patients who will/will not develop VAP, we found that the quantity of Gammaproteobacteria (Gram-negative bacteria) at DVAP-3 was significantly more abundant in ETA and OPS from VAP patients (Fig. S3). By contrast, the absolute abundance of the class Bacilli (Gram-positive bacteria notably represented by Streptococcus, Enterococcus, Lactobacillus and Staphylococcus) was significantly higher in ETA from controls at D0 (Fig. S3). Similarly, at D0, class Bacilli had a relative abundance of > 11.9% in 29/35 (82.8%) of controls, but only in 3/16 (18.8%) of VAP patients (Fig. 4). CAP performed on OPS microbiota (at genus level) at D0 allowed to allocate patients to VAP or controls with 81% and 69% success, respectively (Fig. 5).

VAP is associated with taxonomic composition of the microbiota and DNA load in respiratory samples. a Dot plots of the relative abundance of 16S rRNA reads assigned to the class Bacilli in OPS samples at D0. b ROC curve for associations between VAP and the relative abundance of class Bacilli in OPS at D0. c Dot plots depicting human DNA load in extracts of ETA at DVAP. d ROC curve for associations between VAP and bacterial or human DNA load at DVAP in ETA or OPS. The area under the curve, specificity, sensitivity and optimal cut-off are shown for each model in the same color as the ROC curve. ROC receiver operating characteristic, OPS oropharyngeal secretions, ETA endotracheal aspirate, VAP ventilator-associated pneumonia, D0 intubation day, DVAP day of VAP diagnosis

CAP performed on OPS microbiota at D0 allows a reasonable distinction of patients who develop or not VAP. Results of the CAP for OPS and ETA samples from the first three sampling points (D0, D3, DVAP-3) are presented. The analyses were based on the relative abundance of genera in the dataset normalized to 1000 reads per sample. Bars represent the percentage of correct allocations of samples to VAP and control groups obtained by cross-validation. n number of samples included in the analysis, m number of PCo axes used for the discriminant analysis, ETA endotracheal aspirate, OPS oropharyngeal secretions, VAP ventilator-associated pneumonia, CAP Canonical Analysis of Principal Coordinates, D0 intubation day, D3 3 days after intubation, DVAP-3 3 days before VAP
Metataxonomics vs. culture analyses
A comparison of culture and 16S rDNA data in OPS, ETA and BAL samples showed that 16S rRNA-based community profiling had the potential to detect bacteria identified by culture and to yield many more overlooked by routine culture analysis (Fig. S6 and Table S3).
Bacterial and human DNA load
The association between bacterial load in respiratory samples and the development of VAP was also investigated using a qPCR approach. In DNA extracts from OPS and ETA taken from D0 to DVAP, bacterial and human DNA loads were higher in VAP patients in most cases (Table S4). This was particularly obvious with human DNA in ETA (Fig. 4c) and bacterial DNA in OPS sampled on the day of VAP (Fig. 4d). Using a cutoff of 124.7 ng/µL, human DNA concentrations in the ETA sample extracts allowed to differentiate VAP vs. controls with a sensitivity of 94.1% and a specificity of 83.3% (Fig. 4d). Likewise, in the extracts of 6/7 (81%) ETA samples taken on the same day as BAL samples that turned culture-negative (controls), human DNA concentrations were also below this cutoff (data not shown). Bacterial DNA load in ETA and OPS at DVAP was higher in VAP patients, but these measurements were less discriminant compared to the human DNA load in ETA at the same time point (Fig. 4d and Table S4). BAL samples collected at DVAP from VAP patients also had a significantly higher bacterial load (p = 0.013) compared to BAL fluids taken at different time points in controls (n = 7) (data not shown).
Discussion
Theoretical advantages of molecular tests over culture are: (1) a shorter time to bacteriological results; (2) identification of a larger repertoire of bacteria (microbiota); (3) rapid identification of bacterial resistance markers [32]; and (4) the possibility to concomitantly measure host markers (host RNA and DNA) [33]. Using metataxonomic techniques, we found that at the time of intubation: (1) a lower abundance of Bacilli (normal Gram-positive oropharyngeal flora) in OPS was associated with the development of late-onset VAP with a sensitivity and specificity > 80% (Fig. 4b); (2) canonical analysis of principal coordinates (CAP) of microbiota in OPS allowed a reasonable allocation of patients who eventually developed VAP or not.
On the day of VAP, we also observed that the bacterial DNA load was significantly higher in OPS and ETA compared with controls. This is in accordance with Conway Morris et al. who showed that BAL fluids of VAP patients had a higher bacterial burden than those who did not have criteria for VAP [18]. A significantly higher human DNA load was also measured in ETA compared with controls (Fig. 4d). We hypothesize that this result reflects the presence of host inflammatory cells in ETA samples on the day of VAP [24]. The level of significance of differences in human and bacterial DNA load between VAP patients and controls increased when getting closer to the day of VAP. Interestingly, CAP did not show this trend. CAP of OPS microbiota better differentiated VAP from controls on the day of intubation than in time points closer to the day of VAP. This may be due to patient-specific changes in microbiota profiles during intubation.
Molecular analysis correctly identified bacteria obtained by standard culture, but metataxonomics found many more bacteria, typically difficult to culture, such as anaerobes (e.g., Fusobacteria) and Mycoplasma [17, 21]. Mycoplasma was frequently detected in the three types of respiratory specimens obtained during intubation, i.e., OPS, ETA and BAL fluid. Although we observed a trend of more Mycoplasma in patients with VAP, the difference did not reach statistical significance. An increased abundance of Mycoplasma in BAL fluids from patients with VAP compared with intubated controls has recently been reported [34]. However, these were mainly commensal species of Mycoplasma and not M. pneumoniae. Mycoplasma represents a typical illustration of a bacterial taxon not cultured with standard techniques but detected by metataxonomics that could play a role in the pathogenicity of VAP. Further studies are needed to investigate the potential implication of commensal Mycoplasma and other uncultured bacteria in the development of VAP.
Overall, bacterial alpha-diversity tended to decrease over time during intubation in respiratory samples, irrespective of the sample types. This decrease of diversity may be related not only to antibiotic therapy, but also to changes caused by mechanical ventilation [21] which may favor the emergence of VAP bacterial pathogens with an advantage of growth. The dynamics of changes in microbiota profile varied between individuals (Fig. 2c, d and S5), similar to previous studies on the respiratory microbiome in intubated patients [16, 17, 21]. These individualized responses of the microbiota to intubation are blurred by averaging the data across patients, resulting in non-significant differences in beta-diversity measure (Bray–Curtis similarity at the genus level) between VAP patients and controls. While the similarity measures based on the relative genera abundance did not provide the evidence of a more profound dysbiosis in VAP patients, univariate analyses showed that several higher-level taxa had significantly higher absolute abundance in VAP patients, notably on the day of VAP (Fig. S3).
The strengths of our study are: (1) a large cohort of intubated patients studied prospectively with respiratory specimens sampled daily and (2) culture and metataxonomic analyses performed on the same samples. Metataxonomic analyses were performed in serial samples from the day of intubation until 3 days after VAP on the subset of late-onset “definite VAP” patients and in matched controls, corresponding to 473 analyzed samples.
Our study has some limitations: (1) despite the complex matching algorithm, the matching accuracy was not perfect and one cannot exclude that controls were not completely appropriate; (2) although we used a classical definition of late-onset VAP, most VAP occurred quite early after intubation (median day 7; IQR 6–9), which may explain a lower frequency of multidrug-resistant organisms compared with other VAP studies; (3) samples were not strictly obtained every day for all patients due to practical issues related to a “real life” study; (4) the metataxonomic technique is difficult to standardize, which currently makes it difficult to compare studies from different groups; in particular, pre-analytical aspects, contaminant removal and bioinformatics pipelines significantly influence results [35]; (5) virtually all patients received at least one dose of antibiotics between intubation (D0) and day VAP-1. This is in line with ICU prevalence studies showing that antibiotics are used in 71% of patients on a particular day [36]. Therefore, the effect of antibiotics on the evolution of respiratory microbiota could unfortunately not be easily studied.
Conclusions
Molecular analyses of respiratory samples identified early markers associated with the subsequent development of VAP, as well as important differences in the taxa abundance between VAP and controls. Further prospective trials are needed to test the predictive value of these markers, as well as the relevance of uncultured bacteria in the pathogenesis of VAP.
Metataxonomic analyses remain complicated, resource- and time-consuming; quantitative PCR targeting of bacterial markers identified by metataxonomics (or metagenomics) combined with measurement of the host response, such as human DNA load or expression level of specific genes [33], appears as more promising tools for rapid molecular diagnostics.
These approaches may allow to diagnose VAP more accurately and rapidly in the future and help to tailor effective preventive and treatment strategies [37, 38].
References
Nguile-Makao M, Zahar JR, Francais A et al (2010) Attributable mortality of ventilator-associated pneumonia: respective impact of main characteristics at ICU admission and VAP onset using conditional logistic regression and multi-state models. Intensive Care Med 36:781–789
Timsit JF, Zahar JR, Chevret S (2011) Attributable mortality of ventilator-associated pneumonia. Curr Opin Crit Care 17:464–471
Torres A, Niederman MS, Chastre J et al (2017) International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: guidelines for the management of hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) of the European respiratory society (ERS), European society of intensive care medicine (ESICM), European society of clinical microbiology and infectious diseases (ESCMID) and asociacion latinoamericana del torax (ALAT). Europ Respir J. https://doi.org/10.1183/13993003.00582-2017
Kalil AC, Metersky ML, Klompas M et al (2016) Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of America and the American thoracic society. Clin Infect Dis 63:e61–e111
Berton DC, Kalil AC, Teixeira PJ (2014) Quantitative versus qualitative cultures of respiratory secretions for clinical outcomes in patients with ventilator-associated pneumonia. Cochrane Database Syst Rev 10:CD006482
O’Horo JC, Thompson D, Safdar N (2012) Is the gram stain useful in the microbiologic diagnosis of VAP? A meta-analysis. Clin Infect Dis 55:551–561
Luna CM, Sarquis S, Niederman MS et al (2013) Is a strategy based on routine endotracheal cultures the best way to prescribe antibiotics in ventilator-associated pneumonia? Chest 144:63–71
Pirracchio R, Mateo J, Raskine L et al (2009) Can bacteriological upper airway samples obtained at intensive care unit admission guide empiric antibiotherapy for ventilator-associated pneumonia? Crit Care Med 37:2559–2563
Brusselaers N, Labeau S, Vogelaers D, Blot S (2013) Value of lower respiratory tract surveillance cultures to predict bacterial pathogens in ventilator-associated pneumonia: systematic review and diagnostic test accuracy meta-analysis. Intensive Care Med 39:365–375
Depuydt P, Benoit D, Vogelaers D et al (2008) Systematic surveillance cultures as a tool to predict involvement of multidrug antibiotic resistant bacteria in ventilator-associated pneumonia. Intensive Care Med 34:675–682
Dennis BM, Betzold RD, Patton D et al (2018) Bacterial burden in critically injured ventilated patients does not correlate with progression to pneumonia. Surg Infect 19:369–375
Craven DE, Hudcova J, Lei Y, Craven KA, Waqas A (2016) Pre-emptive antibiotic therapy to reduce ventilator-associated pneumonia: “thinking outside the box”. Crit Care 20:300
Landelle C, Nocquet Boyer V, Abbas M et al (2018) Impact of a multifaceted prevention program on ventilator-associated pneumonia including selective oropharyngeal decontamination. Intensive Care Med 44:1777–1786
Bouza E, Granda MJ, Hortal J, Barrio JM, Cercenado E, Munoz P (2013) Pre-emptive broad-spectrum treatment for ventilator-associated pneumonia in high-risk patients. Intensive Care Med 39:1547–1555
Lazarevic V, Gaia N, Emonet S et al (2014) Challenges in the culture-independent analysis of oral and respiratory samples from intubated patients. Front Cell Infect Microbiol 4:65
Bousbia S, Papazian L, Saux P et al (2012) Repertoire of intensive care unit pneumonia microbiota. PLoS One 7:e32486
Kelly BJ, Imai I, Bittinger K et al (2016) Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome 4:7
Conway Morris A, Gadsby N, McKenna JP et al (2017) 16S pan-bacterial PCR can accurately identify patients with ventilator-associated pneumonia. Thorax 72:1046–1048
Lung M, Codina G (2012) Molecular diagnosis in HAP/VAP. Curr Opin Crit Care 18:487–494
Pendleton KM, Erb-Downward JR, Bao Y et al (2017) Rapid pathogen identification in bacterial pneumonia using real-time metagenomics. Am J Respir Crit Care Med 196:1610–1612
Zakharkina T, Martin-Loeches I, Matamoros S et al (2017) The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax 72:803–810
Huttner A, Emonet S, Harbarth S, Renzi G, Kaiser L, Schrenzel J (2014) Polymerase-chain reaction/electrospray ionization-mass spectrometry for the detection of bacteria and fungi in bronchoalveolar lavage fluids: a prospective observational study. Clin Microbiol Infect 20:O1059–1066
Stralin K, Ehn F, Giske CG et al (2016) The IRIDICA PCR/electrospray ionization-mass spectrometry assay on bronchoalveolar lavage for bacterial etiology in mechanically ventilated patients with suspected pneumonia. PLoS One 11:e0159694
Pugin J (2002) Clinical signs and scores for the diagnosis of ventilator-associated pneumonia. Minerva Anestesiol 68:261–265
McDonald D, Price MN, Goodrich J et al (2012) An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Edgar RC (2010) Search and clustering orders of magnitude faster than Blast. Bioinformatics 26:2460–2461
Schloss PD, Westcott SL, Ryabin T et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Cole JR, Wang Q, Fish JA et al (2014) Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–642
Lazarevic V, Gaia N, Girard M, Schrenzel J (2016) Decontamination of 16S rRNA gene amplicon sequence datasets based on bacterial load assessment by qPCR. BMC Microbiol 16:73
DeLong ER, DeLong DM, Clarke-Pearson DL (1988) Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44:837–845
Anderson MJ, Willis TJ (2003) Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84:511–525
Yan Q, Cui S, Chen C et al (2016) Metagenomic analysis of sputum microbiome as a tool toward culture-independent pathogen detection of patients with ventilator-associated pneumonia. Am J Respir Crit Care Med 194:636–639
Langelier C, Kalantar KL, Moazed F et al (2018) Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci USA 115:E12353–E12362
Nolan TJ, Gadsby NJ, Hellyer TP et al (2016) Low-pathogenicity Mycoplasma spp. alter human monocyte and macrophage function and are highly prevalent among patients with ventilator-acquired pneumonia. Thorax 71:594–600
Emonet S, Lazarevic V, Pugin J, Schrenzel J, Ruppe E (2017) Clinical metagenomics for the diagnosis of hospital-acquired infections: promises and hurdles. Am J Respir Crit Care Med 196:1617–1618
Vincent JL, Rello J, Marshall J et al (2009) International study of the prevalence and outcomes of infection in intensive care units. JAMA 302:2323–2329
Klompas M (2017) What is new in the prevention of nosocomial pneumonia in the ICU? Curr Opin Crit Care 23:378–384
Bos LD, Stips C, Schouten LR et al (2017) Selective decontamination of the digestive tract halves the prevalence of ventilator-associated pneumonia compared to selective oral decontamination. Intensive Care Med 43:1535–1537
Acknowledgements
The authors are deeply indebted to the patients and their next-of-kin, as well as the medical and nursing staff of the ICU and the staff of the bacteriology laboratory of Geneva University Hospitals for their participation in this study. The authors thank Rosemary Sudan for outstanding editorial support. This study was investigator-initiated and sponsored by Geneva University Hospitals. An unrestricted grant from bioMérieux (Marcy l’Étoile, France) was used to financially support this study, which was independently designed, executed, analyzed and reported by the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Emonet, S., Lazarevic, V., Leemann Refondini, C. et al. Identification of respiratory microbiota markers in ventilator-associated pneumonia. Intensive Care Med 45, 1082–1092 (2019). https://doi.org/10.1007/s00134-019-05660-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-019-05660-8