
Abstract
The global epidemic of non-alcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH) and the high prevalence among individuals with type 2 diabetes has attracted the attention of clinicians specialising in liver disorders. Many drugs are in the pipeline for the treatment of NAFLD/NASH, and several glucose-lowering drugs are now being tested specifically for the treatment of liver disease. Among these are nuclear hormone receptor agonists (e.g. peroxisome proliferator-activated receptor agonists, farnesoid X receptor agonists and liver X receptor agonists), fibroblast growth factor-19 and -21, single, dual or triple incretins, sodium–glucose cotransporter inhibitors, drugs that modulate lipid or other metabolic pathways (e.g. inhibitors of fatty acid synthase, diacylglycerol acyltransferase-1, acetyl-CoA carboxylase and 11β-hydroxysteroid dehydrogenase type-1) or drugs that target the mitochondrial pyruvate carrier. We have reviewed the metabolic effects of these drugs in relation to improvement of diabetic hyperglycaemia and fatty liver disease, as well as peripheral metabolism and insulin resistance.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the treatment of type 2 diabetes the following organs are considered main targets: (1) the pancreas and the skeletal muscle, for the improvement of beta cell function and peripheral insulin sensitivity; (2) the adipose tissue, to reduce obesity and lipotoxicity; (3) the gut, since it secretes incretin hormones; and (4) the kidney, heart, endothelium, brain and eye, to prevent or reduce the micro- and macrovascular complications associated with type 2 diabetes. The liver is often not included among the target organs, although it is well established that hepatic insulin resistance is responsible for fasting hyperglycaemia and contributes to glucose intolerance. The global epidemic of non-alcoholic fatty liver disease (NAFLD) affects more than 25% of the general population [1] and more than 55% of individuals with type 2 diabetes [2], while the prevalence of non-alcoholic steatohepatitis (NASH) has been estimated to be 1–6% in the general population [1] and 37% in individuals with type 2 diabetes [2]. This has focused attention on the liver as a main target to combat these metabolic disorders as well as type 2 diabetes [3, 4]. It is now evident that it is not only simple steatosis but also hepatic inflammation that drives NASH and the progression of liver damage (i.e. fibrosis [3, 4]); however, the drivers of hepatic inflammation are still unknown. Tissues other than the liver may be important in the development and progression of NAFLD/NASH and should be targeted to treat this disease. The crosstalk between the liver, intestine and adipose tissue has shown that alterations in the release of intestinal hormones, such as incretins [5], or dysregulation of the gut microbiota [6] play an important role in the development and progression of NAFLD/NASH. Adipose tissue insulin resistance resulting in excess release of NEFA is associated with more severe forms of NAFLD/NASH [7] as well as with decompensated type 2 diabetes [8]. New drugs that are in the pipeline, and older drugs already approved for type 2 diabetes (since most individuals with NAFLD have type 2 diabetes or prediabetes), have shown promising effects on liver metabolism. The aim of this paper is to review the current literature on the metabolic effects of these drugs in relation to improvement of diabetic hyperglycaemia and/or fatty liver disease, as well as peripheral metabolism and insulin resistance.
Metformin and sulfonylureas
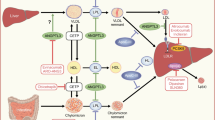
Metformin reduces hepatic glucose production by decreasing gluconeogenesis [9] and treatment with metformin is possibly protective against hepatocellular carcinoma, although its effect on adiponectin levels and hepatic fat oxidation is weak [10]. However, current guidelines consider the effect of metformin on NAFLD to be neutral [10]. On the other hand, sulfonylureas act on hepatic glucose metabolism through the stimulation of insulin secretion (Fig. 1) and treatment with sulfonylureas is associated with presence of significant fibrosis (OR 2.04, p = 0.022) but not NASH [11].

Pharmacological treatments that directly or indirectly target hepatic glucose and lipid metabolism, inflammation and fibrosis. The arrows indicate the different actions on insulin exerted by some glucose-lowering drugs on hepatic metabolism. GLP-1RA, dual GIP/GLP-1 agonists, DPP4 inhibitors and sulfonylureas increase insulin levels by stimulating insulin release, while during treatment with SGLT-2 inhibitors the insulin levels are reduced. GK, glucokinase; GKRP, glucokinase regulatory protein; MPC, mitochondrial pyruvate carrier. This figure is available as a downloadable slide
Nuclear hormone receptor agonists
Peroxisome proliferator-activated receptor agonists
Several peroxisome proliferator-activated receptor (PPAR) agonists target the liver (see Table 1 for information on specific drugs and references). PPAR-γ is expressed in many tissues, mainly in adipose tissue, but hepatic effects have been described (Fig. 1). PPAR-γ agonists approved for treatment of type 2 diabetes include the thiazolidinediones pioglitazone and rosiglitazone, which act by reducing endogenous glucose production (EGP) and gluconeogenesis [12]. Pioglitazone also improves hepatic steatosis, hepatic and peripheral inflammation, NASH and fibrosis, although its effect is more pronounced in individuals with type 2 diabetes than in those without the disease [13]. It is currently the only drug that has been suggested for treatment of diabetic NAFLD in the guidelines published by the European Association for the Study of Liver Disease, EASD and European Association for the Study of Obesity (EASL-EASD-EASO) [10]. Although the side effects of these drugs (weight gain, fluid retention, fractures, bladder cancer) must be considered, these are diminished at lower doses. Moreover, pioglitazone is a potent insulin sensitiser, retards onset of type 2 diabetes by protecting beta cell function, and reduces CVD, which is a frequent comorbidity in individuals with type 2 diabetes and/or NAFLD [14].
MSDC-0602 K is a novel thiazolidinedione designed to minimise binding to PPAR-γ, preferentially targeting the mitochondrial pyruvate carrier while still producing insulin-sensitising effects [15, 16]. Mitochondrial pyruvate metabolism is essential for the process of gluconeogenesis from pyruvate and for the development of NAFLD after a diet high in fat, fructose and cholesterol. In one study, after 6 months of treatment with MSDC-0602 K, individuals showed a significant reduction in glucose, HbA1c, insulin, liver enzymes and improved liver histology (NAS score [NAFLD activity score]) but no improvement in fibrosis, when compared with placebo [15].
A new drug class, selective PPAR modulators (SPPARM), is now under development (e.g. INT-131 besylate [CHS-131 [17]], MK-0533 [18], YR4-42 [19]). Preclinical data have shown that SPPARM, compared with thiazolidinedione PPAR-γ full agonists, exert similar effects of glucose and lipid lowering at smaller doses but without causing weight gain and fluid retention, thus reducing side effects and serious safety concerns [18, 19]. However, although promising, the safety data in humans are still scarce.
PPAR-α is expressed mainly in the liver. PPAR-α agonists (fibrates, namely fenofibrate [20,21,22], bezafibrate [23,24,25] and pemafibrate [26]) increase hepatic fat oxidation and are used to decrease triacylglycerol concentrations, although their effect on NAFLD and hyperglycaemia is limited [21, 22]. In individuals with biopsy-proven NAFLD, 48 weeks of treatment with 200 mg/day of fenofibrate reduced liver enzymes but the grade of steatosis, lobular inflammation, fibrosis or NAFLD activity score did not change significantly [22].
Dual PPAR-α/γ agonists are potent insulin sensitisers that also act on lipid metabolism. Several compounds have been tested for treatment of type 2 diabetes but none has yet received US Food and Drug Administration approval. Clinical trials with tesaglitazar, aleglitazar and muraglitazar have been terminated due to side effects such as oedema and possible renal complications. Saroglitazar has been shown to significantly decrease both glucose and lipids [27] and has been approved recently in India for the treatment of NASH after the Phase III EVIDENCES-II trial showed histological improvement of NASH using liver biopsy after 52 weeks of treatment [28, 29]; However, these data were only presented at conferences and there is only evidence of reduction in liver stiffness measured using FibroScan [29]. The Phase II EVIDENCES-IV trial is currently investigating the effect of saroglitazar in US individuals with NAFLD/NASH.
Great interest has been shown in a new class of PPAR agonists being developed for the treatment of NASH (comprising the dual PPAR-α/δ agonist elafibranor [30] and the triple PPAR-α/γ/δ agonist lanifibranor [31]). These drugs improve not only hepatic histology but also diabetic hyperglycaemia. They are also associated with weight loss, since PPAR-δ activates fat metabolism and energy expenditure. The results of the Phase IIb NATIVE trial (NAsh Trial to Validate IVA337 Efficacy; data to be published) showed that lanifibranor met the primary (decrease of ≥2 points of SAF [steatosis, activity, fibrosis] score, combining hepatocellular inflammation and ballooning) and key secondary endpoints (NASH resolution without worsening and with improvement of fibrosis, in both dose groups [800 mg/day and 1200 mg/day]) [32]. Furthermore, the effect of lanifibranor on diabetic hyperglycaemia and on body weight are encouraging. On the contrary, elafibranor did not meet the predefined primary endpoint of NASH resolution without worsening of fibrosis in the Phase III RESOLVE-IT trial [33]. Nevertheless, elafibranor’s results in primary biliary cholangitis showed great promise and were far more convincing than its results in NASH. The combination of elafibranor with either a glucagon-like peptide-1 (GLP-1) receptor agonist (GLP-1RA) or a sodium–glucose cotransporter 2 (SGLT2) inhibitor is under investigation for NASH.
In summary, single PPAR agonists have been employed for several years. In clinical practice their beneficial effects need to be weighed against their side effects, which are well known. The dual PPARs or pan-PPARs are indeed new but results from studies are encouraging, especially for those PPARs that decrease liver fat content and hyperglycaemia without increasing body weight. However, most of the respective clinical data have not been published yet and will need careful evaluation.
Farnesoid X receptor agonists and fibroblast growth factor-19 analogues
The farnesoid X receptor (FXR), a bile acid receptor, is a nuclear receptor encoded by the NR1H4 gene in humans and regulates bile acid synthesis, secretion and transport, and lipid and glucose metabolism (Fig. 1). The FXR contributes to inter-organ communication, in particular the enterohepatic signalling pathway, through bile acids and fibroblast growth factor (FGF)-19, a gastrointestinal growth hormone that is stimulated by FXR. Several FXR agonists have been developed for the treatment of NAFLD (see Table 2 for specific drugs and further references). Obeticholic acid (OCA) is the first FXR agonist to reach Phase III trials after showing promising results in Phase II trials for the treatment of liver fibrosis in NASH and in primary biliary cholangitis (25 mg was more effective than 50 mg dose) [34]. The 18 month interim analysis of the Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment (REGENERATE) trial showed that fibrosis improvement of at least one stage (with no worsening of NASH) or NASH resolution (with no worsening of fibrosis) was obtained in 23% and 12%, respectively, of individuals treated with OCA 25 mg (vs 12% and 8% in placebo-treated individuals; p = 0.0002 and p = 0.13, respectively) [34,35,36]. OCA treatment was associated with an early transient increase in glucose and HbA1c in individuals with type 2 diabetes, with return to levels similar to those seen with placebo by month 6 [35]. Moreover, OCA induced transient increase in total cholesterol and LDL-cholesterol and decrease in HDL-cholesterol, all of which reversed rapidly on discontinuation [37]. At the end of June 2020, the US Food and Drug Administration determined that since the interim results of Phase III trials were based on surrogate histopathological endpoints the predicted benefit of OCA remains uncertain and does not sufficiently outweigh the potential risks to support its accelerated approval for the treatment of individuals with liver fibrosis due to NASH.
Other FXR agonists currently under development include the FGF-19 analogue NGM282 (aldafermin), which in a Phase II trial reduced hepatic fat and liver enzymes but increased LDL-cholesterol and total cholesterol; triacylglycerols and weight were slightly decreased at the higher dose studied (6 mg), while no changes were observed in HbA1c or insulin resistance [38]. After administration for 24 weeks, aldafermin resulted in improvement of fibrosis (≥1 stage) with no worsening of NASH in 38% of participants (vs 18% with placebo) and produced resolution of NASH with no worsening of fibrosis in 24% of participants (vs 9% with placebo) [39]. However, the increase in total and LDL-cholesterol and the reduction in HDL-cholesterol, which had been observed in healthy volunteers during administration of FXR agonists and FGF-19 analogues [37], raises some concern, although this dyslipidaemia can effectively be managed with statins [40].
New partial FXR agonists (non-bile acids) are currently under development. These include tropifexor (LNJ452) [41, 42], EDP-305 [43] and nidufexor (LMB763), of which nidufexor seems the most potent [44]. However, most of the available data on non-bile acid FXR agonists are limited to studies in animal models, and resilient efficacy and safety data in humans are awaited.
Liver X receptor agonists
Liver X receptors (LXRs) act as oxysterol sensors and are involved in the regulation of cholesterol and lipid metabolism [45]. There are two types of LXR: LXRα (NR1H3), expressed mostly in the liver and to a lesser extent in the kidney, small intestine, spleen and adrenal gland; and LXRβ (NR1H2), expressed ubiquitously [45]. LXRs stimulate lipogenesis while suppressing gluconeogenesis (Fig. 1). It has also been shown that the insulin stimulation of hepatic lipogenic genes is mediated through LXR activation [46]. LXRαβ-deficient ob/ob (LOKO) mice are protected from hepatic steatosis despite being obese and glucose intolerant [47]. Employment of euglycaemic−hyperinsulinaemic clamp showed that the LOKO mice are insulin sensitive at the level of both muscle and liver. However, these mice showed reduced glucose tolerance with low insulin values and the authors found that the low insulin secretion was due to reduced beta cell mass rather than beta cell dysfunction [47]. While LXR agonists may cause hepatic fat accumulation, LXR inverse agonists have the ability to suppress the expression of the lipogenic LXR target genes Fasn and Srepb1. Several compounds that bind to both LXRα and LXRβ (LXR agonists) have been developed [48,49,50,51,52,53,54,55] (Table 3) and studied for the treatment of NAFLD and atherosclerosis, as they have been shown to reduce lipogenesis, inflammation, insulin resistance and hyperlipidaemia, but some like T0901317 and GW3965 are associated to increased hepatic fat accumulation [53, 54]. Only some LXR agonists have made it to Phase I clinical trials [50, 52, 56,57,58], none have progressed to Phase II studies due to unforeseen adverse reactions or undisclosed reasons. Among these are LXR-623/WAY 252623; BMS-779788; BMS-852927 [48, 50,51,52]. At the moment there are no indications that these compounds might be successful for treating metabolic diseases. However, they are important in the study of LXRα and LXRβ, leading to better understanding of the receptors’ role in the deterioration of lipid metabolism.
Incretins
Incretins are gut hormones released in response to food ingestion that augment the secretion of insulin released from pancreatic beta cells. Incretins include GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which are rapidly degraded by dipeptidyl peptidase 4 [DPP-4]. GLP-1RAs and DPP-4 inhibitors both target the liver (Fig. 1). Table 4 shows information on specific drugs with references. GLP-1RAs have become second-line therapy for individuals with type 2 diabetes; they help to restore normoglycaemia as well as promote weight loss and ameliorate the risk of CVD. Compared with native GLP-1, which is rapidly degraded by DPP-4, GLP-1RAs are resistant to DPP-4, allowing a more prolonged duration of action. DPP-4 inhibitors decrease hyperglycaemia by reducing the degradation of endogenous GLP-1 but they only have a small effect on reducing hepatic fat content [59,60,61,62]. On the other hand, GLP-1RAs have a more potent effect on liver histology, not only on liver fat but also on hepatic inflammation and ballooning and in part on fibrosis [63].
In the LEAN (Liraglutide Efficacy and Action in NASH) trial, 52 participants with NASH were randomised to receive treatment with the GLP-1RA liraglutide or placebo for 48 weeks [64]. Resolution of NASH was observed in 39% of the liraglutide-treated participants vs 9% of the placebo-treated participants, indicating that liraglutide is safe and should be used to treat diabetic individuals with NAFLD, although complete resolution of NASH might not be achieved. In the Phase II trial ‘Investigation of Efficacy and Safety of Three Dose Levels of Subcutaneous Semaglutide Once Daily Versus Placebo in Subjects With Non-alcoholic Steatohepatitis’, 320 individuals with NASH with or without type 2 diabetes were enrolled and 302 completed the 72 weeks of treatment [65]. Changes in liver histology were assessed in 277 individuals. The primary aim, NASH resolution without worsening of fibrosis, was achieved in about 40% of participants treated with semaglutide 0.1 mg and 0.2 mg and in 59% of those treated with semaglutide 0.4 mg (vs 17% of those given placebo) (OR 6.87, p < 0.0001). However, the percentage of participants with an improvement in fibrosis stage was similar among groups.
Reduction of both liver fat content and hyperglycaemia in individuals with NAFLD has also been reported with exenatide [66,67,68,69,70,71], lixisenatide [72] and dulaglutide [73, 74]. GLP-1RAs have several effects on liver function: they decrease liver enzymes [64, 72, 75,76,77], EGP [78], lipotoxicity [64, 66, 75, 79, 80] and postprandial triacylglycerol concentrations [81, 82]. Some of these hepatic effects might be mediated by a decrease in body weight. However, weight loss with GLP-1RAs is 4–5 kg (higher with semaglutide), which seems insufficient to explain by itself the improvement in liver histology considering that a weight loss of 7–10% is necessary to bring about resolution of NASH [83].
New unimolecular polyagonists based on GLP-1 have been developed and have shown superior metabolic action compared with single GLP-1RAs. Among these, one of the most promising is the GLP-1/GIP receptor agonist tirzepatide, which has shown better reduction of HbA1c, body weight and liver fat content when compared with placebo or dulaglutide [84], and better improvement in markers of liver fibrosis [84,85,86].
The main effects of dual GLP-1/glucagon receptor agonists, engineered from the sequence of the gut hormone oxyntomodulin [87], are reduction of body weight and liver fat content, and improvement in glycaemic control, lipid profile and energy expenditure. In this class of drugs cotadutide showed promising results for weight loss and glycaemic control [88], while for the other compounds, such as ZP2929/BI 456906 [87, 89], MK-8521 [87], NN9277 [87] and efinopegdutide [87], data in humans are lacking.
The triple GLP-1/GIP/glucagon receptor agonist HM15211 is under development. Preclinical data have shown that HM15211 has antifibrotic and anti-inflammatory properties [90] while in the Phase Ib/IIa trial in non-diabetic obese individuals with NAFLD HM15211 significantly decreased liver fat content and body weight after 8 and 12 weeks of treatment [91].
Given the results of the recent trials (including the data on semaglutide), it seems that use of incretins should be among the first-line treatment for individuals with diabetes and NAFLD. Moreover, these individuals are at higher risk of cardiovascular and chronic kidney disease even in the absence of type 2 diabetes [92] and it should be considered that GLP-1RAs have also shown beneficial effects on cardiovascular and renal systems.
SGLT2 inhibitors
SGLT2 inhibitors are among the most widely used drugs for the treatment of type 2 diabetes, as second-line agents along with GLP-1RAs. Not only do they reduce hyperglycaemia but they also promote cardiorenal protection and weight loss [93]. Although SGLT2 inhibitors decrease fasting and postprandial glucose, they do not suppress EGP, which has been found to be increased in several studies (Fig. 1). A number of studies reported significant benefits in individuals with type 2 diabetes, with respect to reversal of liver steatosis and reduction in plasma aminotransferase levels, following treatment with the SGLT2 inhibitors empagliflozin [94, 95], dapagliflozin [96], canagliflozin [97, 98], luseogliflozin [99, 100], ipragliflozin [101, 102] and ertugliflozin [103] (see Table 5 for further information and references). The effect of SGLT2 inhibitors on hepatocyte necrosis, inflammation and/or fibrosis is less studied but there are indications that treatment with some SGLT2 inhibitors, such as dapagliflozin [96], canagliflozin [98] and ipragliflozin [102] (but not empagliflozin [104] or ertugliflozin [103]), improves variables of liver fibrosis. Typically, the reduction in plasma alanine aminotransferase concentration and liver fat content is proportional to the magnitude of weight loss and is greater with higher baseline plasma aminotransferases [70].
Combinations of SGLT2 inhibitors with GLP-1RAs are now under investigation and have been shown to potentiate the actions of each of the drugs on glucose metabolism [105], as well as each drug’s effect of improving liver function and indices of steatosis and fibrosis [70].
The effects of SGLT2 inhibitors on NAFLD seem to be related mainly to weight loss [70]. However, their effects of rapidly lowering blood glucose levels and reducing CVD risk should make this class of drugs one of the first choices, together with GLP-1RAs, for the treatment of type 2 diabetes with NAFLD/NASH, until new drugs specifically for the treatment of NASH become available.
Insulin
The effect of insulin treatment on liver fat content and liver enzymes in individuals with uncontrolled type 2 diabetes and NAFLD has not been extensively studied and no prospective trial has examined its effect on liver histology (Table 6). In one study, the prevalence of NAFLD was low in individuals with type 1 diabetes (8.8%) and in those with type 2 diabetes NAFLD prevalence was lower in insulin-treated (61.7%) than in insulin-naive (75.6%) individuals [106]. Basal exogenous insulin decreases liver steatosis [71, 80, 107, 108], likely by improving both lipotoxicity and glucotoxicity. However, not all studies have reported a decrease in hepatic fat content [68, 79]. One study showed that acute normalisation of fasting glucose concentrations in individuals with type 2 diabetes with variable insulin infusion for 67 h decreased EGP without changing peripheral insulin resistance but increased hepatic triacylglycerol content [109]. Furthermore, in a cross-sectional study involving 346 individuals with type 2 diabetes and biopsy-proven NAFLD, multivariate models analysis showed that treatment with insulin (but not metformin) was significantly associated with a higher prevalence of NASH (OR 2.24, p = 0.025) but not fibrosis [11]. However, the individuals treated with insulin had HbA1c 8.2% vs 6.9% (66 vs 52 mmol/mol) and it cannot be excluded that worse metabolic control and longer duration of diabetes may contribute to these findings. Insulin is known to decrease FFA concentrations by stimulating triacylglycerol re-esterification, not only in the adipose tissue, but also in other organs such as the liver or the muscle [109]. However, insulin also stimulates de novo lipogenesis (DNL). Thus, it is likely that in conditions of excess substrates (e.g. in more obese individuals with adipose tissue insulin resistance) high insulin concentrations favour hepatic triacylglycerol accumulations.
Drugs targeting hepatokines
Hepatokines are proteins secreted by hepatocytes involved in the regulation of metabolic processes through autocrine, paracrine and endocrine pathways [110] and might become a target for the treatment of hepatic metabolic diseases (see Table 7 for more information on specific drugs targeting hepatokines). Among the hepatokines we can find fetuin-A, FGF-21 and angiopoietin-like protein 3 (ANGPTL3).
Fetuin-A is involved in the pathophysiology of type 2 diabetes and CVD [110]. Among the drugs approved for the treatment of diabetes, liraglutide [111] and pioglitazone [112], but not metformin [112], reduce circulating levels of fetuin-A.
Individuals with metabolic disease (i.e. diabetes, NAFLD and obesity) display increased circulating levels of FGF-21; this has been attributed to a resistance to this hormone, and the administration of pharmacological doses of exogenous FGF-21 would overcome the resistance. FGF-21 is considered to have acute insulin-sensitising effects via activation of the FGF receptor-1/transmembrane protein β-klotho complex in adipose tissue. In contrast, the long-term metabolic benefits of FGF-21 treatment (in particular weight loss) are thought to be primarily caused by binding of FGF-21 to the FGF receptor-1/transmembrane protein β-klotho complex in the brain [113].
Several FGF-21 analogues are in the pipeline of pharma companies [114,115,116,117,118,119] and preclinical studies indicate that they reduce body weight, hepatic fat, circulating lipids, insulin and glucose in a dose-dependent manner by reducing hepatic gluconeogenesis and lipogenesis and improving hepatic and peripheral insulin resistance [120]. A recent trial that employed pegbelfermin, a pegylated FGF-21, administered subcutaneously for 16 weeks [115], showed that there was a significant decrease in hepatic fat content and an increase in adiponectin levels compared with placebo.
NGM313 (now MK3655) is a humanised monoclonal antibody activator of β-klotho/FGF receptor-1c that, by once-monthly administration, boosts the effect of FGF-21 [121]. Results of a Phase 1 trial employing NGM313 vs pioglitazone for 36 days showed a reduction in absolute and relative liver fat content, HbA1c and ALT for both treatment arms, compared with baseline, but effects were more robust in individuals treated with NGM313, although the data are published only as an abstract [121].
Angiopoietin-like protein 3 (ANGPTL3) is secreted mainly by the liver and, in this sense, it might be considered a hepatokine. ANGPTL3 acts as dual inhibitor of lipoprotein lipase and endothelial lipase, thereby increasing plasma NEFA, triacylglycerols, LDL-cholesterol and HDL-cholesterol, and its plasma concentration is associated with clinical/histological markers of NAFLD/NASH and with hepatic ANGPTL3 expression [122]. Evinacumab is an investigational monoclonal antibody that blocks ANGPTL3. Results of a Phase III trial showed that evinacumab reduced LDL-cholesterol, apolipoprotein B, non-HDL-cholesterol and total cholesterol, compared with placebo [123, 124]. Moreover, evinacumab decreased odds of atherosclerotic CVD [125] and, although data on fatty liver are lacking, it is likely that inhibition of ANGPTL3 might improve NAFLD.
Drugs that modulate lipid metabolic pathways
Several drugs that target hepatic lipid metabolism have recently been developed (see Table 7 for more information on specific drugs and references).
Stearoyl CoA desaturase-1 (SCD-1) is a key enzyme in the formation of monounsaturated fatty acids, specifically oleate and palmitoleate from stearoyl CoA and palmitoyl CoA. Inhibitors of SCD-1 are being tested in the treatment of NAFLD. In one trial, the SCD-1 inhibitor Aramchol (developed by Galmed, Israel) decreased liver fat content in individuals with NAFLD at a dose of 300 mg but not 100 mg, compared with placebo, but no significant change was observed in ALT, adiponectin or HOMA-IR [126]. However, no such effect on liver fat content was seen in individuals with HIV-associated NAFLD and lipodystrophy [127].
Ketohexokinase (KHK), also known as hepatic fructokinase, catalyses the first step in the metabolism of dietary fructose, comprising the conversion of fructose to fructose-1-phosphate, with the potential to decrease DNL. The KHK inhibitor PF-06835919 reduced hepatic fat and improved insulin resistance in individuals with NAFLD [128, 129].
Fatty acid synthase (FAS) is involved in DNL, since it catalyses the synthesis of palmitate (C16:0, a long-chain saturated fatty acid), from acetyl-CoA and malonyl-CoA. The FAS inhibitor TVB-2640 is reported to reduce DNL and hepatic fat when administered for 10 days [130].
Acyl-CoA:diacylglycerol acyltransferase-1 and -2 (DGAT-1 and DGAT-2) catalyse the formation of triacylglycerols from diacylglycerol and Acyl-CoA. DGAT inhibitors are under study for the treatment of diabetes, obesity and NAFLD and exert effects on both endogenous and meal-induced triacylglycerol turnover [131,132,133].
Epeleuton is a synthetic n-3 fatty acid derivative of eicosapentaenoic acid that decreased triacylglycerols, improved glycaemic control and decreased markers of inflammation in a Phase II exploratory study (16 weeks) in individuals with obesity and NAFLD [134]. Epeleuton at the highest dose significantly decreased hepatic fat from baseline, although not significantly, vs placebo but it did not meet the primary endpoints of decreased ALT concentrations or liver stiffness.
Acetyl-CoA carboxylase (ACC) is a key enzyme in fatty acid synthesis since it catalyses the irreversible carboxylation of acetyl-CoA to produce malonyl-CoA. The ACC inhibitor GS-0976 is reported to reduce hepatic fat and markers of fibrosis but increases the concentration of triacylglycerols [135, 136].
Liver-directed selective thyroid hormone receptor-β agonists are in the pipeline for the treatment of NAFLD. Resmetirom [137, 138] and VK2809 [138, 139] have been shown to improve hepatic lipid metabolism and ameliorate NAFLD in Phase II studies although in vitro their effect seems less potent than the native thyroid hormone receptor ligand, triiodothyronine (T3) [137, 138].
Several drugs target important enzymes like 11β-hydroxysteroid dehydrogenase type-1 (11β-HSD1) [140,141,142,143,144,145,146,147,148] and protein tyrosine phosphatase-1B (PTP1B) [149]. 11β-HSD1 reduces cortisone to the active hormone cortisol, which activates glucocorticoid receptors. 11β-HSD1 inhibitors, not only reduce HbA1c and fasting plasma glucose but also, if present, improve hyperlipidaemia and hypertriacylglycerolaemia and reduce hepatic steatosis [143]. PTP1B is a soluble non-transmembrane and cytosolic tyrosine-specific phosphatase; it is a negative regulator of insulin signalling. Liver-specific deletion of PTP1B in mice brought about improvement in both glucose and lipid metabolism, with suppression of gluconic and lipogenic genes (Fig. 1) [150]. PTP1B inhibitors, such as IONIS-PTP-1BRx, have demonstrated sustained effects on HbA1c and glucose variables and increased adiponectin levels in humans [149].
Summary and conclusions
The high prevalence of NAFLD, NASH and type 2 diabetes has made the liver a central target for drug development. It is now evident that not only reducing glucotoxicity and lipotoxicity but also improving insulin resistance and inflammation is beneficial for the liver in both type 2 diabetes and NAFLD/NASH. Many drugs are in the pipeline for the treatment of NAFLD/NASH, also having effects on hyperglycaemia and insulin resistance. Similarly, several (but not all) drugs already approved to treat type 2 diabetes are effective in improving hepatic lipid metabolism and are now being tested specifically for treatment of NAFLD/NASH. The effect of these drugs on hepatic inflammation is less clear, mainly because of lack of standard methods, besides liver biopsy, to specifically evaluate tissue inflammation.
Abbreviations
- 11β-HSD:
-
11β-Hydroxysteroid dehydrogenase type-1
- ACC:
-
Acetyl-CoA carboxylase
- ANGPTL3:
-
Angiopoietin-like protein 3
- DGAT-1:
-
Diacylglycerol acyltransferase-1
- DNL:
-
De novo lipogenesis
- DPP-4:
-
Dipeptidyl peptidase-4
- EGP:
-
Endogenous glucose production
- FAS:
-
Fatty acid synthase
- FGF:
-
Fibroblast growth factor
- FXR:
-
Farnesoid X receptor
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1RA:
-
GLP-1 receptor agonist
- KHK:
-
Ketohexokinase
- LOKO:
-
LXRαβ-deficient ob/ob
- LXR:
-
Liver X receptor
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- OCA:
-
Obeticholic acid
- PPAR:
-
Peroxisome proliferator-activated receptor
- PTP1B:
-
Protein tyrosine phosphatase-1B
- SCD-1:
-
Stearoyl CoA desaturase-1
- SGLT2:
-
Sodium–glucose cotransporter 2
- SPPARM:
-
Selective PPAR modulators
References
Younossi Z, Tacke F, Arrese M et al (2019) Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 69(6):2672–2682. https://doi.org/10.1002/hep.30251
Younossi ZM, Golabi P, de Avila L et al (2019) The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol 71(4):793–801. https://doi.org/10.1016/j.jhep.2019.06.021
Gastaldelli A, Cusi K (2019) From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep 1(4):312–328. https://doi.org/10.1016/j.jhepr.2019.07.002
Stefan N, Haring HU, Cusi K (2019) Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol 7(4):313–324. https://doi.org/10.1016/S2213-8587(18)30154-2
Junker AE, Gluud LL, van Hall G, Holst JJ, Knop FK, Vilsboll T (2016) Effects of glucagon-like peptide-1 on glucagon secretion in patients with non-alcoholic fatty liver disease. J Hepatol 64(4):908–915. https://doi.org/10.1016/j.jhep.2015.11.014
Marra F, Svegliati-Baroni G (2018) Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 68(2):280–295. https://doi.org/10.1016/j.jhep.2017.11.014
Rosso C, Kazankov K, Younes R et al (2019) Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J Hepatol 71(5):1012–1021. https://doi.org/10.1016/j.jhep.2019.06.031
Gastaldelli A, Gaggini M, DeFronzo RA (2017) Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes 66(4):815–822. https://doi.org/10.2337/db16-1167
Madiraju AK, Erion DM, Rahimi Y et al (2014) Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510(7506):542–546. https://doi.org/10.1038/nature13270
European Association for the Study of the Liver, European Association for the Study of Diabetes, European Association for the Study of Obesity (2016) EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 59(6):1121–1140. https://doi.org/10.1007/s00125-016-3902-y
Nascimbeni F, Aron-Wisnewsky J, Pais R et al (2016) Statins, antidiabetic medications and liver histology in patients with diabetes with non-alcoholic fatty liver disease. BMJ Open Gastroenterol 3(1):e000075. https://doi.org/10.1136/bmjgast-2015-000075
Gastaldelli A, Miyazaki Y, Mahankali A et al (2006) The effect of pioglitazone on the liver: role of adiponectin. Diabetes Care 29(10):2275–2281. https://doi.org/10.2337/dc05-2445
Bril F, Kalavalapalli S, Clark VC et al (2018) Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin Gastroenterol Hepatol 16(4):558–566 e552. https://doi.org/10.1016/j.cgh.2017.12.001
DeFronzo RA, Inzucchi S, Abdul-Ghani M, Nissen SE (2019) Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab Vasc Dis Res 16(2):133–143. https://doi.org/10.1177/1479164118825376
Harrison SA, Alkhouri N, Davison BA et al (2020) Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol 72(4):613–626. https://doi.org/10.1016/j.jhep.2019.10.023
Chen Z, Vigueira PA, Chambers KT et al (2012) Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione. J Biol Chem 287(28):23537–23548. https://doi.org/10.1074/jbc.M112.363960
DePaoli AM, Higgins LS, Henry RR, Mantzoros C, Dunn FL, INT131-007 Study Group (2014) Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 37(7):1918–1923. https://doi.org/10.2337/dc13-2480
Acton JJ 3rd, Akiyama TE, Chang CH et al (2009) Discovery of (2R)-2-(3-{3-[(4-Methoxyphenyl)carbonyl]-2-methyl-6-(trifluoromethoxy)-1H-indol-1 -yl}phenoxy)butanoic acid (MK-0533): a novel selective peroxisome proliferator-activated receptor gamma modulator for the treatment of type 2 diabetes mellitus with a reduced potential to increase plasma and extracellular fluid volume. J Med Chem 52(13):3846–3854. https://doi.org/10.1021/jm900097m
Huan Y, Pan X, Peng J et al (2019) A novel specific peroxisome proliferator-activated receptor gamma (PPARgamma) modulator YR4-42 ameliorates hyperglycaemia and dyslipidaemia and hepatic steatosis in diet-induced obese mice. Diabetes Obes Metab 21(11):2553–2563. https://doi.org/10.1111/dom.13843
Belfort R, Berria R, Cornell J, Cusi K (2010) Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab 95(2):829–836. https://doi.org/10.1210/jc.2009-1487
Yaghoubi M, Jafari S, Sajedi B et al (2017) Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 29(12):1385–1388. https://doi.org/10.1097/MEG.0000000000000981
Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA (2008) A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis 40(3):200–205. https://doi.org/10.1016/j.dld.2007.10.002
Corpechot C, Chazouilleres O, Rousseau A et al (2018) A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med 378(23):2171–2181. https://doi.org/10.1056/NEJMoa1714519
Karhapaa P, Uusitupa M, Voutilainen E, Laakso M (1992) Effects of bezafibrate on insulin sensitivity and glucose tolerance in subjects with combined hyperlipidemia. Clin Pharmacol Ther 52(6):620–626. https://doi.org/10.1038/clpt.1992.200
Day AP, Feher MD, Chopra R, Mayne PD (1993) The effect of bezafibrate treatment on serum alkaline phosphatase isoenzyme activities. Metabolism 42(7):839–842. https://doi.org/10.1016/0026-0495(93)90056-t
Ida S, Kaneko R, Murata K (2019) Efficacy and safety of pemafibrate administration in patients with dyslipidemia: a systematic review and meta-analysis. Cardiovasc Diabetol 18(1):38. https://doi.org/10.1186/s12933-019-0845-x
Krishnappa M, Patil K, Parmar K et al (2020) Effect of saroglitazar 2 mg and 4 mg on glycemic control, lipid profile and cardiovascular disease risk in patients with type 2 diabetes mellitus: a 56-week, randomized, double blind, phase 3 study (PRESS XII study). Cardiovasc Diabetol 19(1):93. https://doi.org/10.1186/s12933-020-01073-w
Zydus Cadila (2020) World’s first drug for the treatment of Non-Cirrhotic NASH. Press release March 5, 2020. Available from https://zyduscadila.com/public/pdf/pressrelease/Zydus_announces_world’s_first_drug_for_the_treatment_of_Non_Cirrhotic_NASH.pdf. Accessed 23 June 2020
Kaul U, Parmar D, Manjunath K et al (2019) New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: integrated analysis of the real world evidence. Cardiovasc Diabetol 18(1):80. https://doi.org/10.1186/s12933-019-0884-3
Ratziu V, Harrison SA, Francque S et al (2016) Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 150(5):1147–1159 e1145. https://doi.org/10.1053/j.gastro.2016.01.038
Boubia B, Poupardin O, Barth M et al (2018) Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) alpha/gamma/delta Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J Med Chem 61(6):2246–2265. https://doi.org/10.1021/acs.jmedchem.7b01285
Inventiva (2020) Lanifibranor meets the primary and key secondary endpoints in the Phase IIb NATIVE clinical trial in non-alcoholic steatohepatitis (NASH). Press release June 15, 2020. Available from https://www.globenewswire.com/news-release/2020/06/15/2048284/0/en/Inventiva-s-lanifibranor-meets-the-primary-and-key-secondary-endpoints-in-the-Phase-IIb-NATIVE-clinical-trial-in-non-alcoholic-steatohepatitis-NASH.html. Accessed 23 June 2020
Genfit (2020) Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis. Press release May 11 2020. Available from https://ml-eu.globenewswire.com/Resource/Download/38e085e1-66f5-4251-8abe-648d0e7b9ed1. Accessed 23 June 2020
Mudaliar S, Henry RR, Sanyal AJ et al (2013) Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145(3):574–582 e571. https://doi.org/10.1053/j.gastro.2013.05.042
Younossi ZM, Ratziu V, Loomba R et al (2019) Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394(10215):2184–2196. https://doi.org/10.1016/S0140-6736(19)33041-7
Neuschwander-Tetri BA, Loomba R, Sanyal AJ et al (2015) Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385(9972):956–965. https://doi.org/10.1016/S0140-6736(14)61933-4
Pencek R, Marmon T, Roth JD, Liberman A, Hooshmand-Rad R, Young MA (2016) Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes Metab 18(9):936–940. https://doi.org/10.1111/dom.12681
Harrison SA, Rossi SJ, Paredes AH et al (2020) NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients With Nonalcoholic Steatohepatitis. Hepatology 71(4):1198–1212. https://doi.org/10.1002/hep.30590
Harrison SA, Neff G, Guy CD et al (2021) Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 160(1):219–231 e211. https://doi.org/10.1053/j.gastro.2020.08.004
Rinella ME, Trotter JF, Abdelmalek MF et al (2019) Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol 70(4):735–744. https://doi.org/10.1016/j.jhep.2018.11.032
Tully DC, Rucker PV, Chianelli D et al (2017) Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J Med Chem 60(24):9960–9973. https://doi.org/10.1021/acs.jmedchem.7b00907
Lucas KJ, Lopez P, Lawitz E et al (2020) Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig Liver Dis 52:e38. https://doi.org/10.1016/j.dld.2019.12.129
An P, Wei G, Huang P et al (2020) A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int 40(7):1655–1669. https://doi.org/10.1111/liv.14490
Chianelli D, Rucker PV, Roland J et al (2020) Nidufexor (LMB763), a Novel FXR Modulator for the Treatment of Nonalcoholic Steatohepatitis. J Med Chem 63(8):3868–3880. https://doi.org/10.1021/acs.jmedchem.9b01621
Burris TP, Solt LA, Wang Y et al (2013) Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev 65(2):710–778. https://doi.org/10.1124/pr.112.006833
Chen G, Liang G, Ou J, Goldstein JL, Brown MS (2004) Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A 101(31):11245–11250. https://doi.org/10.1073/pnas.0404297101
Beaven SW, Matveyenko A, Wroblewski K et al (2013) Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab 18(1):106–117. https://doi.org/10.1016/j.cmet.2013.04.021
Dou F, Chen J, Cao H et al (2019) Anti-atherosclerotic effects of LXRalpha agonist through induced conversion of M1 macrophage to M2. Am J Transl Res 11(6):3825–3840
Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, Burris TP (2015) The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol Metab 4(4):353–357. https://doi.org/10.1016/j.molmet.2015.01.009
Kirchgessner TG, Sleph P, Ostrowski J et al (2016) Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils. Cell Metab 24(2):223–233. https://doi.org/10.1016/j.cmet.2016.07.016
Nambi P, Basso MD, Chen L et al (2007) Abstract 1453: LXR-623, a novel Liver X Receptor modulator, displays neutral lipid effects in Cholesteryl Ester Transfer Protein-expressing species and inhibits atherosclerotic lesion progression in Low Density Lipoprotein receptor knockout mice. Circulation 116(suppl_16):II_299–II_299. https://doi.org/10.1161/circ.116.suppl_16.II_299-a
Katz A, Udata C, Ott E et al (2009) Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J Clin Pharmacol 49(6):643–649. https://doi.org/10.1177/0091270009335768
Grefhorst A, van Dijk TH, Hammer A et al (2005) Differential effects of pharmacological liver X receptor activation on hepatic and peripheral insulin sensitivity in lean and ob/ob mice. Am J Physiol Endocrinol Metab 289(5):E829–E838. https://doi.org/10.1152/ajpendo.00165.2005
Chisholm JW, Hong J, Mills SA, Lawn RM (2003) The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44(11):2039–2048. https://doi.org/10.1194/jlr.M300135-JLR200
Griffett K, Burris TP (2016) Promiscuous activity of the LXR antagonist GSK2033 in a mouse model of fatty liver disease. Biochem Biophys Res Commun 479(3):424–428. https://doi.org/10.1016/j.bbrc.2016.09.036
Liu Y, Yan C, Wang Y et al (2006) Liver X receptor agonist T0901317 inhibition of glucocorticoid receptor expression in hepatocytes may contribute to the amelioration of diabetic syndrome in db/db mice. Endocrinology 147(11):5061–5068. https://doi.org/10.1210/en.2006-0243
Dong Y, Gao G, Fan H, Li S, Li X, Liu W (2015) Activation of the Liver X Receptor by Agonist TO901317 Improves Hepatic Insulin Resistance via Suppressing Reactive Oxygen Species and JNK Pathway. PLoS One 10(4):e0124778. https://doi.org/10.1371/journal.pone.0124778
Schultz JR, Tu H, Luk A et al (2000) Role of LXRs in control of lipogenesis. Genes Dev 14(22):2831–2838. https://doi.org/10.1101/gad.850400
Cui J, Philo L, Nguyen P et al (2016) Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J Hepatol 65(2):369–376. https://doi.org/10.1016/j.jhep.2016.04.021
Watanabe T, Tamura Y, Kakehi S et al (2015) Effects of sitagliptin on ectopic fat contents and glucose metabolism in type 2 diabetic patients with fatty liver: A pilot study. J Diabetes Investig 6(2):164–172. https://doi.org/10.1111/jdi.12262
Joy TR, McKenzie CA, Tirona RG et al (2017) Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J Gastroenterol 23(1):141–150. https://doi.org/10.3748/wjg.v23.i1.141
Seko Y, Sumida Y, Tanaka S et al (2017) Effect of sodium glucose cotransporter 2 inhibitor on liver function tests in Japanese patients with non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatol Res 47(10):1072–1078. https://doi.org/10.1111/hepr.12834
Dong Y, Lv Q, Li S et al (2017) Efficacy and safety of glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 41(3):284–295. https://doi.org/10.1016/j.clinre.2016.11.009
Armstrong MJ, Gaunt P, Aithal GP et al (2016) Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387(10019):679–690. https://doi.org/10.1016/S0140-6736(15)00803-X
Newsome PN, Buchholtz K, Cusi K et al (2020) A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N Engl J Med. https://doi.org/10.1056/NEJMoa2028395
Bi Y, Zhang B, Xu W et al (2014) Effects of exenatide, insulin, and pioglitazone on liver fat content and body fat distributions in drug-naive subjects with type 2 diabetes. Acta Diabetol 51(5):865–873. https://doi.org/10.1007/s00592-014-0638-3
Gastaldelli A, Gaggini M, Daniele G et al (2016) Exenatide improves both hepatic and adipose tissue insulin resistance: A dynamic positron emission tomography study. Hepatology 64(6):2028–2037. https://doi.org/10.1002/hep.28827
Shao N, Kuang HY, Hao M, Gao XY, Lin WJ, Zou W (2014) Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes Metab Res Rev 30(6):521–529. https://doi.org/10.1002/dmrr.2561
Klonoff DC, Buse JB, Nielsen LL et al (2008) Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin 24(1):275–286. https://doi.org/10.1185/030079908x253870
Gastaldelli A, Repetto E, Guja C et al (2020) Exenatide and dapagliflozin combination improves markers of liver steatosis and fibrosis in patients with type 2 diabetes. Diabetes Obes Metab 22(3):393–403. https://doi.org/10.1111/dom.13907
Liu L, Yan H, Xia M et al (2020) Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab Res Rev 36(5):e3292. https://doi.org/10.1002/dmrr.3292
Gluud LL, Knop FK, Vilsboll T (2014) Effects of lixisenatide on elevated liver transaminases: systematic review with individual patient data meta-analysis of randomised controlled trials on patients with type 2 diabetes. BMJ Open 4(12):e005325. https://doi.org/10.1136/bmjopen-2014-005325
Cusi K, Sattar N, Garcia-Perez LE et al (2018) Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: a post hoc analysis of the AWARD programme. Diabet Med 35(10):1434–1439. https://doi.org/10.1111/dme.13697
Seko Y, Sumida Y, Tanaka S et al (2017) Effect of 12-week dulaglutide therapy in Japanese patients with biopsy-proven non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatol Res 47(11):1206–1211. https://doi.org/10.1111/hepr.12837
Kuchay MS, Krishan S, Mishra SK et al (2020) Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: randomised controlled trial (D-LIFT trial). Diabetologia 63(11):2434–2445. https://doi.org/10.1007/s00125-020-05265-7
Armstrong MJ, Hull D, Guo K et al (2016) Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol 64(2):399–408. https://doi.org/10.1016/j.jhep.2015.08.038
Newsome P, Francque S, Harrison S et al (2019) Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment Pharmacol Ther 50(2):193–203. https://doi.org/10.1111/apt.15316
Seghieri M, Rebelos E, Gastaldelli A et al (2013) Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia 56(1):156–161. https://doi.org/10.1007/s00125-012-2738-3
Yan J, Yao B, Kuang H et al (2019) Liraglutide, Sitagliptin, and Insulin Glargine Added to Metformin: The Effect on Body Weight and Intrahepatic Lipid in Patients With Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Hepatology 69(6):2414–2426. https://doi.org/10.1002/hep.30320
Tang A, Rabasa-Lhoret R, Castel H et al (2015) Effects of Insulin Glargine and Liraglutide Therapy on Liver Fat as Measured by Magnetic Resonance in Patients With Type 2 Diabetes: A Randomized Trial. Diabetes Care 38(7):1339–1346. https://doi.org/10.2337/dc14-2548
Matikainen N, Soderlund S, Bjornson E et al (2019) Liraglutide treatment improves postprandial lipid metabolism and cardiometabolic risk factors in humans with adequately controlled type 2 diabetes: A single-centre randomized controlled study. Diabetes Obes Metab 21(1):84–94. https://doi.org/10.1111/dom.13487
Whyte MB, Shojaee-Moradie F, Sharaf SE et al (2019) Lixisenatide Reduces Chylomicron Triacylglycerol by Increased Clearance. J Clin Endocrinol Metab 104(2):359–368. https://doi.org/10.1210/jc.2018-01176
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L et al (2015) Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 149(2):367–378 e365; quiz e314-365. https://doi.org/10.1053/j.gastro.2015.04.005
Frias JP, Nauck MA, Van J et al (2018) Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392(10160):2180–2193. https://doi.org/10.1016/S0140-6736(18)32260-8
Hartman ML, Sanyal AJ, Loomba R et al (2020) Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 43(6):1352–1355. https://doi.org/10.2337/dc19-1892
Frias JP, Nauck MA, Van J et al (2020) Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens. Diabetes Obes Metab 22(6):938–946. https://doi.org/10.1111/dom.13979
Brandt SJ, Gotz A, Tschop MH, Muller TD (2018) Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides 100:190–201. https://doi.org/10.1016/j.peptides.2017.12.021
Ambery P, Parker VE, Stumvoll M et al (2018) MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet 391(10140):2607–2618. https://doi.org/10.1016/S0140-6736(18)30726-8
Skarbaliene J, Madsen AN, Mouritzen U, Bak HH, Just R (2017) Exploring the therapeutic potential of Glucagon/GLP-1 dual agonist ZP2929 in a mouse model of diet induced and biopsy-confirmed non-alcoholic steatohepatitis. J Hepatol 66(1):S102. https://doi.org/10.1016/s0168-8278(17)30466-x
Choi J, Kim JK, Lee SM et al (2020) 1830-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-Induced NASH and Fibrosis Mice. Diabetes 69(Supplement 1):1830. https://doi.org/10.2337/db20-1830-P
Abdelmalek M, Choi J, Kim Y, Seo K, Hompesch M, Baek S (2020) HM15211, a novel GLP-1/GIP/Glucagon triple-receptor co-agonist significantly reduces liver fat and body weight in obese subjects with non-alcoholic fatty liver disease: A Phase 1b/2a, multi-center, randomized, placebo-controlled trial. J Hepatol 73:S124. https://doi.org/10.1016/S0168-8278(20)30765-0
Mantovani A, Scorletti E, Mosca A, Alisi A, Byrne CD, Targher G (2020) Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 111S:154170. https://doi.org/10.1016/j.metabol.2020.154170
Verma S, McMurray JJV (2018) SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia 61(10):2108–2117. https://doi.org/10.1007/s00125-018-4670-7
Kahl S, Gancheva S, Strassburger K et al (2020) Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 43(2):298–305. https://doi.org/10.2337/dc19-0641
Kuchay MS, Krishan S, Mishra SK et al (2018) Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 41(8):1801–1808. https://doi.org/10.2337/dc18-0165
Eriksson JW, Lundkvist P, Jansson PA et al (2018) Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia 61(9):1923–1934. https://doi.org/10.1007/s00125-018-4675-2
Cusi K, Bril F, Barb D et al (2019) Effect of canagliflozin treatment on hepatic triglyceride content and glucose metabolism in patients with type 2 diabetes. Diabetes Obes Metab 21(4):812–821. https://doi.org/10.1111/dom.13584
Akuta N, Watanabe C, Kawamura Y et al (2017) Effects of a sodium-glucose cotransporter 2 inhibitor in nonalcoholic fatty liver disease complicated by diabetes mellitus: Preliminary prospective study based on serial liver biopsies. Hepatol Commun 1(1):46–52. https://doi.org/10.1002/hep4.1019
Sumida Y, Murotani K, Saito M et al (2019) Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective, single-arm trial (LEAD trial). Hepatol Res 49(1):64–71. https://doi.org/10.1111/hepr.13236
Shibuya T, Fushimi N, Kawai M et al (2018) Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective randomized controlled pilot study. Diabetes Obes Metab 20(2):438–442. https://doi.org/10.1111/dom.13061
Miyake T, Yoshida S, Furukawa S et al (2018) Ipragliflozin Ameliorates Liver Damage in Non-alcoholic Fatty Liver Disease. Open Med (Wars) 13:402–409. https://doi.org/10.1515/med-2018-0059
Komiya C, Tsuchiya K, Shiba K et al (2016) Ipragliflozin Improves Hepatic Steatosis in Obese Mice and Liver Dysfunction in Type 2 Diabetic Patients Irrespective of Body Weight Reduction. PLoS One 11(3):e0151511. https://doi.org/10.1371/journal.pone.0151511
Gallo S, Calle RA, Terra SG, Pong A, Tarasenko L, Raji A (2020) Effects of Ertugliflozin on Liver Enzymes in Patients with Type 2 Diabetes: A Post-Hoc Pooled Analysis of Phase 3 Trials. Diabetes Ther 11(8):1849–1860. https://doi.org/10.1007/s13300-020-00867-1
Taheri H, Malek M, Ismail-Beigi F et al (2020) Effect of Empagliflozin on Liver Steatosis and Fibrosis in Patients With Non-Alcoholic Fatty Liver Disease Without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv Ther 37(11):4697–4708. https://doi.org/10.1007/s12325-020-01498-5
Castellana M, Cignarelli A, Brescia F et al (2019) Efficacy and safety of GLP-1 receptor agonists as add-on to SGLT2 inhibitors in type 2 diabetes mellitus: A meta-analysis. Sci Rep 9(1):19351. https://doi.org/10.1038/s41598-019-55524-w
Cusi K, Sanyal AJ, Zhang S et al (2017) Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab 19(11):1630–1634. https://doi.org/10.1111/dom.12973
Juurinen L, Tiikkainen M, Hakkinen AM, Hakkarainen A, Yki-Jarvinen H (2007) Effects of insulin therapy on liver fat content and hepatic insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab 292(3):E829–E835. https://doi.org/10.1152/ajpendo.00133.2006
Cusi K, Sanyal AJ, Zhang S et al (2016) Different effects of basal insulin peglispro and insulin glargine on liver enzymes and liver fat content in patients with type 1 and type 2 diabetes. Diabetes Obes Metab 18(Suppl 2):50–58. https://doi.org/10.1111/dom.12751
Anderwald C, Bernroider E, Krssak M et al (2002) Effects of insulin treatment in type 2 diabetic patients on intracellular lipid content in liver and skeletal muscle. Diabetes 51(10):3025–3032. https://doi.org/10.2337/diabetes.51.10.3025
Stefan N, Haring HU (2013) The role of hepatokines in metabolism. Nat Rev Endocrinol 9(3):144–152. https://doi.org/10.1038/nrendo.2012.258
Zhang LY, Qu XN, Sun ZY, Zhang Y (2020) Effect of liraglutide therapy on serum fetuin A in patients with type 2 diabetes and non-alcoholic fatty liver disease. Clin Res Hepatol Gastroenterol 44(5):674–680. https://doi.org/10.1016/j.clinre.2020.01.007
Esteghamati A, Afarideh M, Feyzi S, Noshad S, Nakhjavani M (2015) Comparative effects of metformin and pioglitazone on fetuin-A and osteoprotegerin concentrations in patients with newly diagnosed diabetes: A randomized clinical trial. Diabetes Metab Syndr 9(4):258–265. https://doi.org/10.1016/j.dsx.2014.09.009
Lan T, Morgan DA, Rahmouni K et al (2017) FGF19, FGF21, and an FGFR1/beta-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab 26(5):709–718 e703. https://doi.org/10.1016/j.cmet.2017.09.005
Charles ED, Neuschwander-Tetri BA, Pablo Frias J et al (2019) Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity (Silver Spring) 27(1):41–49. https://doi.org/10.1002/oby.22344
Sanyal A, Charles ED, Neuschwander-Tetri BA et al (2019) Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392(10165):2705–2717. https://doi.org/10.1016/S0140-6736(18)31785-9
Rosenstock M, Ayalon M, Mansbach H, Liu Y, Margalit M (2019) LBP29 BIO89–100, a novel PEG-FGF21 analogue, is efficacious following weekly and every 2-week subcutaneous dosing in spontaneous diabetic cynomolgus monkeys. J Hepatol 70(LBP):e141–e382
Talukdar S, Zhou Y, Li D et al (2016) A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab 23(3):427–440. https://doi.org/10.1016/j.cmet.2016.02.001
Yin J, Bao L, Chen R, Gao W, Gao X, Yao W (2018) Enhanced expression and distinctive characterization of a long-acting FGF21 and its potential to alleviate nonalcoholic steatohepatitis. Biochimie 151:166–175. https://doi.org/10.1016/j.biochi.2018.05.020
Bao L, Yin J, Gao W, Wang Q, Yao W, Gao X (2018) A long-acting FGF21 alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis partly through an FGF21-adiponectin-IL17A pathway. Br J Pharmacol 175(16):3379–3393. https://doi.org/10.1111/bph.14383
Xu J, Lloyd DJ, Hale C et al (2009) Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58(1):250–259. https://doi.org/10.2337/db08-0392
Depaoli A, Phung VAN, Bashir MR et al (2019) 140-LB: NGM313, a Novel Activator of b-Klotho/FGFR1c, Improves Insulin Resistance and Reduces Hepatic Fat in Obese, Nondiabetic Subjects. Diabetes 68(Supplement 1):140-LB. https://doi.org/10.2337/db19-140-LB
Barchetta I, Cimini FA, Chiappetta C et al (2020) Relationship between hepatic and systemic angiopoietin-like 3, hepatic vitamin D receptor expression and NAFLD in obesity. Liver Int 40(9):2139–2147. https://doi.org/10.1111/liv.14554
Ahmad Z, Banerjee P, Hamon S et al (2019) Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation 140(6):470–486. https://doi.org/10.1161/CIRCULATIONAHA.118.039107
Raal FJ, Rosenson RS, Reeskamp LF et al (2020) Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med 383(8):711–720. https://doi.org/10.1056/NEJMoa2004215
Dewey FE, Gusarova V, Dunbar RL et al (2017) Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med 377(3):211–221. https://doi.org/10.1056/NEJMoa1612790
Safadi R, Konikoff FM, Mahamid M et al (2014) The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 12(12):2085–2091 e2081. https://doi.org/10.1016/j.cgh.2014.04.038
Ajmera VH, Cachay E, Ramers C et al (2019) MRI Assessment of Treatment Response in HIV-associated NAFLD: A Randomized Trial of a Stearoyl-Coenzyme-A-Desaturase-1 Inhibitor (ARRIVE Trial). Hepatology 70(5):1531–1545. https://doi.org/10.1002/hep.30674
Calle R, Bergman A, Somayaji V, Chidsey K, Kazierad D (2019) PS-110-Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J Hepatol 70(1, Supplement):e69–e70. https://doi.org/10.1016/S0618-8278(19)30122-7
Futatsugi K, Smith AC, Tu M et al (2020) Discovery of PF-06835919: A Potent Inhibitor of Ketohexokinase (KHK) for the Treatment of Metabolic Disorders Driven by the Overconsumption of Fructose. J Med Chem 63(22):13546–13560. https://doi.org/10.1021/acs.jmedchem.0c00944
Syed-Abdul MM, Parks EJ, Gaballah AH et al (2020) Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males With Metabolic Abnormalities. Hepatology 72(1):103–118. https://doi.org/10.1002/hep.31000
Loomba R, Morgan E, Watts L et al (2020) Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol 5(9):829–838. https://doi.org/10.1016/s2468-1253(20)30186-2
Okour M, Brigandi RA, Tenero D (2019) A population analysis of the DGAT1 inhibitor GSK3008356 and its effect on endogenous and meal-induced triglyceride turnover in healthy subjects. Fundam Clin Pharmacol 33(5):567–580. https://doi.org/10.1111/fcp.12455
Seo H, Choi SH, Kwak EY et al (2014) Determination of a novel diacylglycerol acyltransferase 1 inhibitor, 2-[4-(4-{5-[2-phenyl-5-(trifluoromethyl) oxazole-4-carboxamido]-1H-benzo[d]imidazol-2-yl} phenyl) cyclohexyl] acetic acid (KR-69232) in rat plasma using liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. Biomed Chromatogr 28(3):324–326. https://doi.org/10.1002/bmc.3053
Climax J, Newsome PN, Hamza M et al (2020) Effects of Epeleuton, a Novel Synthetic Second-Generation n-3 Fatty Acid, on Non-Alcoholic Fatty Liver Disease, Triglycerides, Glycemic Control, and Cardiometabolic and Inflammatory Markers. J Am Heart Assoc 9(16):e016334. https://doi.org/10.1161/JAHA.119.016334
Loomba R, Kayali Z, Noureddin M et al (2018) GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 155(5):1463–1473 e1466. https://doi.org/10.1053/j.gastro.2018.07.027
Lawitz EJ, Coste A, Poordad F et al (2018) Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol 16(12):1983–1991 e1983. https://doi.org/10.1016/j.cgh.2018.04.042
Harrison SA, Bashir MR, Guy CD et al (2019) Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 394(10213):2012–2024. https://doi.org/10.1016/S0140-6736(19)32517-6
Luong XG, Stevens SK, Jekle A et al (2020) Regulation of gene transcription by thyroid hormone receptor beta agonists in clinical development for the treatment of non-alcoholic steatohepatitis (NASH). PLoS One 15(12):e0240338. https://doi.org/10.1371/journal.pone.0240338
Loomba R, Neutel J, Mohseni R et al (2019) LBP-20-VK2809, a Novel Liver-Directed Thyroid Receptor Beta Agonist, Significantly Reduces Liver Fat with Both Low and High Doses in Patients with Non-Alcoholic Fatty Liver Disease: A Phase 2 Randomized, Placebo-Controlled Trial. Hepatol 70(1):e150–e151. https://doi.org/10.1016/s0618-8278(19)30266-x
Feig PU, Shah S, Hermanowski-Vosatka A et al (2011) Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab 13(6):498–504. https://doi.org/10.1111/j.1463-1326.2011.01375.x
Rosenstock J, Banarer S, Fonseca VA et al (2010) The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33(7):1516–1522. https://doi.org/10.2337/dc09-2315
Hollis G, Huber R (2011) 11beta-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes Obes Metab 13(1):1–6. https://doi.org/10.1111/j.1463-1326.2010.01305.x
Stefan N, Ramsauer M, Jordan P et al (2014) Inhibition of 11beta-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2(5):406–416. https://doi.org/10.1016/S2213-8587(13)70170-0
Heise T, Morrow L, Hompesch M et al (2014) Safety, efficacy and weight effect of two 11beta-HSD1 inhibitors in metformin-treated patients with type 2 diabetes. Diabetes Obes Metab 16(11):1070–1077. https://doi.org/10.1111/dom.12317
Bellaire S, Walzer M, Wang T, Krauwinkel W, Yuan N, Marek GJ (2019) Safety, Pharmacokinetics, and Pharmacodynamics of ASP3662, a Novel 11beta-Hydroxysteroid Dehydrogenase Type 1 Inhibitor, in Healthy Young and Elderly Subjects. Clin Transl Sci 12(3):291–301. https://doi.org/10.1111/cts.12618
Markey KA, Ottridge R, Mitchell JL et al (2017) Assessing the Efficacy and Safety of an 11beta-Hydroxysteroid Dehydrogenase Type 1 Inhibitor (AZD4017) in the Idiopathic Intracranial Hypertension Drug Trial, IIH:DT: Clinical Methods and Design for a Phase II Randomized Controlled Trial. JMIR Res Protoc 6(9):e181. https://doi.org/10.2196/resprot.7806
Scott JS, Bowker SS, Deschoolmeester J et al (2012) Discovery of a potent, selective, and orally bioavailable acidic 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitor: discovery of 2-[(3S)-1-[5-(cyclohexylcarbamoyl)-6-propylsulfanylpyridin-2-yl]-3-piperidyl]acet ic acid (AZD4017). J Med Chem 55(12):5951–5964. https://doi.org/10.1021/jm300592r
Harno E, Cottrell EC, Yu A et al (2013) 11beta-Hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors still improve metabolic phenotype in male 11beta-HSD1 knockout mice suggesting off-target mechanisms. Endocrinology 154(12):4580–4593. https://doi.org/10.1210/en.2013-1613
Digenio A, Pham NC, Watts LM et al (2018) Antisense Inhibition of Protein Tyrosine Phosphatase 1B With IONIS-PTP-1BRx Improves Insulin Sensitivity and Reduces Weight in Overweight Patients With Type 2 Diabetes. Diabetes Care 41(4):807–814. https://doi.org/10.2337/dc17-2132
Delibegovic M, Zimmer D, Kauffman C et al (2009) Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 58(3):590–599. https://doi.org/10.2337/db08-0913
Belfort R, Harrison SA, Brown K et al (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355(22):2297–2307. https://doi.org/10.1056/NEJMoa060326
Sanyal AJ, Chalasani N, Kowdley KV et al (2010) Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 362(18):1675–1685. https://doi.org/10.1056/NEJMoa0907929
Cusi K, Orsak B, Bril F et al (2016) Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med 165(5):305–315. https://doi.org/10.7326/M15-1774
Ratziu V, Charlotte F, Bernhardt C et al (2010) Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 51(2):445–453. https://doi.org/10.1002/hep.23270
Juurinen L, Kotronen A, Graner M, Yki-Jarvinen H (2008) Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab 93(1):118–124. https://doi.org/10.1210/jc.2007-1825
Lee YH, Kim JH, Kim SR et al (2017) Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J Korean Med Sci 32(1):60–69. https://doi.org/10.3346/jkms.2017.32.1.60
Colca JR, McDonald WG, Adams WJ (2018) MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin Investig Drugs 27(7):631–636. https://doi.org/10.1080/13543784.2018.1494153
Roy S, Ghosh A (2019) Significant Reduction of Elevated Triglycerides and Liver Fibrosis in Diabetic Dyslipidemia with Saroglitazar: A Case Report. Cureus 11(12):e6361. https://doi.org/10.7759/cureus.6361
Stirban AO, Andjelkovic M, Heise T et al (2016) Aleglitazar, a dual peroxisome proliferator-activated receptor-alpha/gamma agonist, improves insulin sensitivity, glucose control and lipid levels in people with type 2 diabetes: findings from a randomized, double-blind trial. Diabetes Obes Metab 18(7):711–715. https://doi.org/10.1111/dom.12620
Fagerberg B, Edwards S, Halmos T et al (2005) Tesaglitazar, a novel dual peroxisome proliferator-activated receptor alpha/gamma agonist, dose-dependently improves the metabolic abnormalities associated with insulin resistance in a non-diabetic population. Diabetologia 48(9):1716–1725. https://doi.org/10.1007/s00125-005-1846-8
Coletta DK, Fernandez M, Cersosimo E, Gastaldelli A, Musi N, DeFronzo RA (2015) The effect of muraglitazar on adiponectin signalling, mitochondrial function and fat oxidation genes in human skeletal muscle in vivo. Diabet Med 32(5):657–664. https://doi.org/10.1111/dme.12664
Fernandez M, Gastaldelli A, Triplitt C et al (2011) Metabolic effects of muraglitazar in type 2 diabetic subjects. Diabetes Obes Metab 13(10):893–902. https://doi.org/10.1111/j.1463-1326.2011.01429.x
Ding SY, Tigno XT, Braileanu GT, Ito K, Hansen BC (2007) A novel peroxisome proliferator--activated receptor alpha/gamma dual agonist ameliorates dyslipidemia and insulin resistance in prediabetic rhesus monkeys. Metabolism 56(10):1334–1339. https://doi.org/10.1016/j.metabol.2007.05.019
Doebber TW, Kelly LJ, Zhou G et al (2004) MK-0767, a novel dual PPARalpha/gamma agonist, displays robust antihyperglycemic and hypolipidemic activities. Biochem Biophys Res Commun 318(2):323–328. https://doi.org/10.1016/j.bbrc.2004.04.032
Trauner M, Gulamhusein A, Hameed B et al (2019) The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 70(3):788–801. https://doi.org/10.1002/hep.30509
Patel K, Harrison SA, Elkhashab M et al (2020) Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 72(1):58–71. https://doi.org/10.1002/hep.31205
Aspinall R, Shennak M, Stocia G et al (2020) Nidufexor, a non-bile acid FXR agonist, decreases ALT and hepatic fat fraction in patients with NASH after 12 weeks dosing. J Hepatol 73:S4-S5
Erstad DJ, Farrar CT, Ghoshal S et al (2018) Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP-305, a novel farnesoid X receptor agonist. Hepatol Commun 2(7):821–835. https://doi.org/10.1002/hep4.1193
Harrison SA, Rinella ME, Abdelmalek MF et al (2018) NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391(10126):1174–1185. https://doi.org/10.1016/S0140-6736(18)30474-4
Hirschfield GM, Chazouilleres O, Drenth JP et al (2019) Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol 70(3):483–493. https://doi.org/10.1016/j.jhep.2018.10.035
DePaoli AM, Zhou M, Kaplan DD et al (2019) FGF19 Analog as a Surgical Factor Mimetic That Contributes to Metabolic Effects Beyond Glucose Homeostasis. Diabetes 68(6):1315–1328. https://doi.org/10.2337/db18-1305
Frias JP, Bastyr EJ 3rd, Vignati L et al (2017) The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090-2746, in Patients with Type 2 Diabetes. Cell Metab 26(2):343–352 e342. https://doi.org/10.1016/j.cmet.2017.07.011
Nahra R, Gadde K, Hirshberg B et al (2020) AS076 - Effects of cotadutide on biomarkers of non-alcoholic steatohepatitis in overweight or obese subjects with type 2 diabetes mellitus: a 54-week analysis of a randomized phase 2b study. J Hepatol 73:S54–S55. https://doi.org/10.1016/S0168-8278(20)30655-3
Kim JK, Kim J, Lee SM, Lee J, Lee SH, Choi IY (2019) 991-P: Potential of a Novel Long-Acting Glucagon Analog, HM15136, for the Treatment of Obesity. Diabetes 68(Supplement 1):991. https://doi.org/10.2337/db19-991-P
Lai LL, Vethakkan SR, Nik Mustapha NR, Mahadeva S, Chan WK (2020) Empagliflozin for the Treatment of Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes Mellitus. Dig Dis Sci 65(2):623–631. https://doi.org/10.1007/s10620-019-5477-1
Inoue M, Hayashi A, Taguchi T et al (2019) Effects of canagliflozin on body composition and hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease. J Diabetes Investig 10(4):1004–1011. https://doi.org/10.1111/jdi.12980
Itani T, Ishihara T (2018) Efficacy of canagliflozin against nonalcoholic fatty liver disease: A prospective cohort study. Obes Sci Pract 4(5):477–482. https://doi.org/10.1002/osp4.294
Leiter LA, Forst T, Polidori D, Balis DA, Xie J, Sha S (2016) Effect of canagliflozin on liver function tests in patients with type 2 diabetes. Diabetes Metab 42(1):25–32. https://doi.org/10.1016/j.diabet.2015.10.003
Li B, Wang Y, Ye Z et al (2018) Effects of Canagliflozin on Fatty Liver Indexes in Patients with Type 2 Diabetes: A Meta-analysis of Randomized Controlled Trials. J Pharm Pharm Sci 21(1):222–235. https://doi.org/10.18433/jpps29831
Seko Y, Nishikawa T, Umemura A et al (2018) Efficacy and safety of canagliflozin in type 2 diabetes mellitus patients with biopsy-proven nonalcoholic steatohepatitis classified as stage 1-3 fibrosis. Diabetes Metab Syndr Obes 11:835–843. https://doi.org/10.2147/DMSO.S184767
Lee PCH, Gu Y, Yeung MY et al (2018) Dapagliflozin and Empagliflozin Ameliorate Hepatic Dysfunction Among Chinese Subjects with Diabetes in Part Through Glycemic Improvement: A Single-Center, Retrospective, Observational Study. Diabetes Ther 9(1):285–295. https://doi.org/10.1007/s13300-017-0355-3
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF (2010) Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet 375(9733):2223–2233. https://doi.org/10.1016/S0140-6736(10)60407-2
Tobita H, Sato S, Miyake T, Ishihara S, Kinoshita Y (2017) Effects of Dapagliflozin on Body Composition and Liver Tests in Patients with Nonalcoholic Steatohepatitis Associated with Type 2 Diabetes Mellitus: A Prospective, Open-label, Uncontrolled Study. Curr Ther Res Clin Exp 87:13–19. https://doi.org/10.1016/j.curtheres.2017.07.002
Liu J, Tarasenko L, Terra SG et al (2019) Efficacy of ertugliflozin in monotherapy or combination therapy in patients with type 2 diabetes: A pooled analysis of placebo-controlled studies. Diab Vasc Dis Res 16(5):415–423. https://doi.org/10.1177/1479164119842513
Han E, Lee YH, Lee BW, Kang ES, Cha BS (2020) Ipragliflozin Additively Ameliorates Non-Alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Controlled with Metformin and Pioglitazone: A 24-Week Randomized Controlled Trial. J Clin Med 9(1). https://doi.org/10.3390/jcm9010259
Ohki T, Isogawa A, Toda N, Tagawa K (2016) Effectiveness of Ipragliflozin, a Sodium-Glucose Co-transporter 2 Inhibitor, as a Second-line Treatment for Non-Alcoholic Fatty Liver Disease Patients with Type 2 Diabetes Mellitus Who Do Not Respond to Incretin-Based Therapies Including Glucagon-like Peptide-1 Analogs and Dipeptidyl Peptidase-4 Inhibitors. Clin Drug Investig 36(4):313–319. https://doi.org/10.1007/s40261-016-0383-1
Ito D, Shimizu S, Inoue K et al (2017) Comparison of Ipragliflozin and Pioglitazone Effects on Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Randomized, 24-Week, Open-Label, Active-Controlled Trial. Diabetes Care 40(10):1364–1372. https://doi.org/10.2337/dc17-0518
Takase T, Nakamura A, Miyoshi H, Yamamoto C, Atsumi T (2017) Amelioration of fatty liver index in patients with type 2 diabetes on ipragliflozin: an association with glucose-lowering effects. Endocr J 64(3):363–367. https://doi.org/10.1507/endocrj.EJ16-0295
Ohta A, Kato H, Ishii S et al (2017) Ipragliflozin, a sodium glucose co-transporter 2 inhibitor, reduces intrahepatic lipid content and abdominal visceral fat volume in patients with type 2 diabetes. Expert Opin Pharmacother 18(14):1433–1438. https://doi.org/10.1080/14656566.2017.1363888
Bando Y, Ogawa A, Ishikura K et al (2017) The effects of ipragliflozin on the liver-to-spleen attenuation ratio as assessed by computed tomography and on alanine transaminase levels in Japanese patients with type 2 diabetes mellitus. Diabetol Int 8(2):218–227. https://doi.org/10.1007/s13340-016-0302-y
Ozaki A, Yoneda M, Kessoku T et al (2020) Effect of tofogliflozin and pioglitazone on hepatic steatosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: A randomized, open-label pilot study (ToPiND study). Contemp Clin Trials Commun 17:100516. https://doi.org/10.1016/j.conctc.2019.100516
He YL, Haynes W, Meyers CD et al (2019) The effects of licogliflozin, a dual SGLT1/2 inhibitor, on body weight in obese patients with or without diabetes. Diabetes Obes Metab 21(6):1311–1321. https://doi.org/10.1111/dom.13654
Yokote K, Sano M, Tsumiyama I, Keefe D (2020) Dose-dependent reduction in body weight with LIK066 (licogliflozin) treatment in Japanese patients with obesity. Diabetes Obes Metab 22(7):1102–1110. https://doi.org/10.1111/dom.14006
Iruarrizaga-Lejarreta M, Varela-Rey M, Fernandez-Ramos D et al (2017) Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun 1(9):911–927. https://doi.org/10.1002/hep4.1107
Nio Y, Hasegawa H, Okamura H, Miyayama Y, Akahori Y, Hijikata M (2016) Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis C treatment. Antivir Res 132:262–267. https://doi.org/10.1016/j.antiviral.2016.07.003
Funding
Open Access funding enabled and organized by Projekt DEAL.
Authors’ relationships and activities
AG reports acting as a consultant for Boehringer Ingelheim, Eli-Lilly, Genentech, Gilead, Inventiva, Novo Nordisk and Pfizer. NS acts as a consultant and is a member of the speaker bureaus for Allergan, AstraZeneca, Boehringer Ingelheim, Gilead, Genkyotex, Intercept Pharma, MSD, Novartis, Novo Nordisk, Pfizer and Sanofi. NS has conducted clinical trials with support from AstraZeneca, Boehringer Ingelheim, Sanofi, DSM Nutritional Products and Roche Diagnostics. The authors declare that none of the above interactions affected the writing of this article.
Author information
Authors and Affiliations
Contributions
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Figure slide
(PPTX 252 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gastaldelli, A., Stefan, N. & Häring, HU. Liver-targeting drugs and their effect on blood glucose and hepatic lipids. Diabetologia 64, 1461–1479 (2021). https://doi.org/10.1007/s00125-021-05442-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05442-2