
Abstract
Aims/hypothesis
For circulating insulin to act on the brain it must cross the blood–brain barrier (BBB). Remarkably little is known about how circulating insulin crosses the BBB’s highly restrictive brain endothelial cells (BECs). Therefore, we examined potential mechanisms regulating BEC insulin uptake, signalling and degradation during BEC transcytosis, and how transport is affected by a high-fat diet (HFD) and by astrocyte activity.
Methods
125I–TyrA14-insulin uptake and transcytosis, and the effects of insulin receptor (IR) blockade, inhibition of insulin signalling, astrocyte stimulation and an HFD were tested using purified isolated BECs (iBECs) in monoculture and co-cultured with astrocytes.
Results
At physiological insulin concentrations, the IR, not the IGF-1 receptor, facilitated BEC insulin uptake, which required lipid raft-mediated endocytosis, but did not require insulin action on phosphoinositide-3-kinase (PI3K) or mitogen-activated protein kinase kinase (MEK). Feeding rats an HFD for 4 weeks decreased iBEC insulin uptake and increased NF-κB binding activity without affecting insulin PI3K signalling, IR expression or content, or insulin degrading enzyme expression. Using an in vitro BBB (co-culture of iBECs and astrocytes), we found insulin was not degraded during transcytosis, and that stimulating astrocytes with l-glutamate increased transcytosis, while inhibiting nitric oxide synthase decreased insulin transcytosis.
Conclusions/interpretation
Insulin crosses the BBB intact via an IR-specific, vesicle-mediated transport process in the BECs. HFD feeding, nitric oxide inhibition and astrocyte stimulation can regulate BEC insulin uptake and transcytosis.
Similar content being viewed by others


Introduction
Insulin’s actions on the central nervous system (CNS) have been well documented. Intracerebroventricular insulin administration robustly decreased food intake and body weight in rats [1] and baboons [2]. This effect was decreased in insulin-resistant Zucker rats or rats fed a moderate- or high-fat diet (HFD) [3,4,5], but was restored by weight loss [6]. In humans, insulin resistance is associated with increased rates of dementia and Alzheimer’s disease [7]. Despite important CNS actions, how (or whether) circulating insulin accesses the brain interstitial fluid (bISF) or neuronal tissues remains uncertain. Using autoradiographic methods, van Houten et al. [8,9,10,11] and others [12, 13] demonstrated saturable binding of labelled insulin to brain blood vessels, consistent with a receptor-dependent process. Isolated human and bovine brain capillaries also bound radiolabelled insulin and released it in a time-dependent manner [14, 15]. However, these studies did not resolve whether intact insulin was bound to the vessel exterior, internalised by brain endothelial cells (BECs), transported across vessels, or degraded by insulin degrading enzyme (IDE) or other proteases either on the BECs [16] or in brain tissue [17].
Recent imaging studies have delineated the ‘glymphatic’ circulation of cerebrospinal fluid (CSF) [18,19,20], whereby ventricular CSF flows to the subarachnoid space before entering the Virchow–Robin space (VRS) that parallels penetrating pial arterioles into brain tissue. Compounds crossing the BECs, the first layer of the blood–brain barrier (BBB), mix with CSF in the VRS and must then cross astrocyte endfeet that form the VRS’s lateral boundary to reach the bISF. This admixture within the bISF may flow into the VRS along venules and function to clear waste from the brain to the lymphatics along dural veins or to the venous circulation via arachnoid granulations (reviewed [20]).
Numerous studies have reported low insulin concentrations in CSF, as well as a slow transport of circulating insulin into CSF [21,22,23]. Mathematical modelling of circulating insulin entry into CSF suggested a three-compartment model in which bISF was predicted to be an unsampled ‘intermediate’ compartment with plasma insulin crossing the BBB, then passing through bISF and eventually entering CSF [24]. The frequent use of CSF in multiple studies as a bISF ‘surrogate’ assumes some validity to this model. However, no study has validated this model of intact insulin entry into CSF by transport across the BBB. This is understandable as neither bISF nor the VRS compartment is accessible to sampling without tissue damage.
We recently compared the rate of 125I-TyrA14-insulin transport into brain tissue with its movement into CSF and found its appearance in brain tissue preceded its appearance in CSF and that this was mediated by the insulin receptor (IR) and blunted by HFD feeding [25]. However, these studies were unable to differentiate insulin appearance in brain tissue from its binding and potential uptake into the BECs. To date, no work has elucidated the path by which BECs internalise insulin or clarified whether 125I-TyrA14-insulin crosses the brain endothelium without degradation.
Given the importance of brain insulin action and paucity of information on BBB insulin transport, we undertook a detailed study of the mechanisms that regulate insulin uptake by, action on, and transcytosis across BECs using human and rat BECs and an in vitro BBB model. The rat provides a historical model for examining brain insulin transport and sufficient tissue to compare dietary effects between individual animals. We hypothesised that, similar to the peripheral endothelium [26], insulin transport would be mediated by its receptor and downstream signalling through phosphoinositide-3-kinase (PI3K) and NO generation, and that transport would be decreased in rats fed an HFD. Additionally, we hypothesised that astrocyte stimulation would increase BEC insulin transcytosis.
Methods
Cell culture
Three preparations of BECs were used: commercially available rat brain microvascular endothelial cells (RBMVECs; Cell Applications, San Diego, CA, USA), freshly isolated rat BECs (iBECs) and human BECs (hBECs). Isolated astrocytes were employed in a transwell co-culture system.
RBMVECs, passages 5–10, were cultured as per the manufacturer’s recommendations. iBECs were isolated from Sprague Dawley rats using a previously established method [27] (see ESM Methods ‘Harvest of iBECs’). iBECs were 98% CD31+ by flow cytometry, expressed claudin-5, and had increased phosphorylated Akt in response to insulin stimulation (see ESM Methods ‘Flow cytometry’ and ‘Immunofluorescence’, ESM Fig. 1). hBECs were a gift from J. Catravas and were 99% positive for DiI-Ac-LDL (acetylated DiI-labelled LDL) uptake and endothelial nitric oxide synthase (eNOS) expression [28]. hBECs were cultured in DMEM/Ham’s F12 medium (F12) with HEPES and l-glutamine, 10% FBS, endothelial cell growth supplement (Corning, Corning, NY, USA), heparin (Sigma-Aldrich, St Louis, MO, USA) and Anti-Anti [29]. Astrocytes were isolated from Sprague Dawley rat pups at postnatal day 3–10 as per established protocols [30]. Astrocytes were positive for GFAP (glial fibrillary acidic protein) by immunofluorescence (see ESM Methods ‘Immunofluorescence’, ESM Fig. 2) and western blot.
Animals
To test whether HFD feeding affected iBEC insulin uptake and signalling, male Sprague Dawley rats (Charles River, Wilmington, MA, USA) aged 8 weeks were fed a normal chow diet (ND) or HFD (60% lipid, Research Diets, New Brunswick, NJ, USA) for 4 weeks before BEC isolation (see ESM Methods ‘Animal studies’). Prior to tissue collection, rats were weighed and trunk blood collected for non-fasting body weights and serum insulin concentrations, respectively. In total, 44 animals were used (21 ND/23 HFD).
125I-TyrA14-insulin uptake assay
When 90–95% confluent, cells were serum starved for 4, 2 or 1 h (RBMVECs, iBECs, hBECs, respectively) then incubated with 200 pmol/l 125I-TyrA14-insulin (Perkin Elmer, Waltham, MA, USA) for 15 min in HEPES binding buffer (HBB) and uptake measured as previously described [31] (see ESM Methods ‘125I-TyrA14-insulin uptake assay’). For pharmacological studies, cells were pretreated with drug 30 min prior to insulin addition.
Serum insulin concentrations
Blood was collected from ND and HFD rats immediately prior to the rats being killed. Insulin was measured with a rat-specific insulin ELISA (Mercodia, Uppsala, Sweden) or an ultrasensitive ELISA (Alpco, Salem, NH, USA).
Real-time RT-PCR
RNA was extracted from iBECs and real-time quantitative RT-PCR was used to determine expression levels of IR-β (Insr β) and IDE (Ide). See ESM Methods ‘RNA/cDNA preparation and RT-PCR’.
Immunoblotting
IR expression and downstream insulin signalling was assessed by immunoblot (see ESM Methods ‘Immunoblotting’). For pharmacological experiments, drugs were added 30 min prior to incubation with 10 nmol/l insulin.
NF-κB binding activity
Nuclei were isolated from cultured ND and HFD iBECs using a nuclear extraction kit (Active Motif, Carlsbad, CA, USA) and were positive for Lamin A/C (Cell Signaling 2032, 1:500 dilution) nuclear marker upon western blot. We measured nuclear NF-κB p65 binding activity using a TransAM Transcription Factor ELISA (Active Motif), normalised to protein and compared with a standard curve of recombinant NF-κB p65 protein (Active Motif) as per the manufacturer’s instructions.
In vitro BBB co-culture and transcytosis
For transcytosis assays, co-cultures of isolated astrocytes and iBECs were used as an in vitro BBB [32, 33] (see ESM Methods ‘In vitro blood–brain barrier co-cultures’). Barrier integrity was assessed by measuring transendothelial electrical resistance (TEER) (EndOhm, WPI, Sarasota, FL, USA) and expressed as Ω × cm2. Peak TEER (reported as mean ± SD) was achieved on day 5 when transcytosis experiments were performed. Prior to the experiment, the iBEC monolayer and astrocytes were washed once in HBB. Then, the basolateral chamber received equivolume mixture of DMEM/F12 with IGF-1 and astrocyte-conditioned media and the apical chamber received HBB.
In transcytosis experiments, either native or heat-denatured (HD; 70°C for 90 min) 125I-TyrA14-insulin (200 pmol/l) was added to the apical or basolateral chamber. HD-125I-TyrA14-insulin is a control for paracellular leak [31]. Following insulin incubation, the total fluid volume from the apical and basolateral chambers was collected, quantified on a gamma counter, and adjusted for efficiency. To estimate insulin transcytosis for all conditions, HD-125I-TyrA14-insulin was subtracted from 125I-TyrA14-insulin.
To investigate iBEC regulation of transcytosis, apical chambers were pretreated for 15 min (S-961) or 30 min (all other drugs) before addition of native or HD-125I-TyrA14-insulin (200 pmol/l) for 30 min. To investigate astrocyte-mediated regulation of insulin transcytosis, glutamate was added to the basolateral chamber 30 min before addition of insulin. Thapsigargin was added 30 min before glutamate.
Pharmacological reagents
Drug manufacturers are listed in ESM Table 1.
Insulin degradation
125I-TyrA14-insulin degradation was determined by elution profile from a Sephadex G-50 (medium, Sigma-Aldrich) column as previously established [34]. Briefly, samples were loaded onto a 50 cm column and eluted with 0.15 M ammonium bicarbonate buffer, 0.02% BSA and 5% sodium azide, pH 8.0. Column elution fractions were quantified on a gamma counter, adjusted for efficiency, and expressed as disintegrations per min (DPM). To verify that the column elution profile discriminates intact from degraded insulin, 125I-TyrA14-insulin (200 pmol/l) was incubated with or without HepG2 cells overnight at 37°C. 125I-TyrA14-insulin samples collected after transcytosis assays were compared with native and HepG2-degraded insulin elution peaks.
Ethics statement
Animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, NRC 2011 and approved by the University of Virginia Animal Care & Use Committee.
Statistical analysis
Experimental replicates and individual rats are shown as dot plots in each figure. Experimenters were not blinded. 125I-TyrA14-insulin uptake data were log10-transformed and analysed with a one-way ANOVA or hierarchical linear mixed model if there were missing values. Data were excluded in cases of experimental error. Non-normally distributed groups were compared with the Mann–Whitney U test. Normally distributed groups were compared using an unpaired t test, paired t test or one-way ANOVA. Post hoc testing is specified where employed. Significance was declared at p < 0.05 and a trend towards significance at p ≤ 0.10. TEER values are presented as mean ± SD and all other data are presented as means ± SEM.
Results
RBMVECs and hBECs take up insulin and signal through Akt
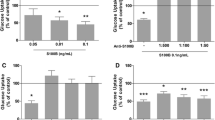
RBMVECs exposed to physiological concentrations of 125I-TyrA14-insulin (200 pmol/l) internalised the tracer within 5 min and uptake continued for 60 min (ESM Fig. 3). Uptake was rapid and approximately linear between 0 and 15 min; therefore 15 min incubations were used for subsequent experiments. We previously reported that the IR-specific antagonist S-961 blocked 125I-TyrA14-insulin uptake by RBMVECs, while IGF-I receptor blockade did not [25]. Figure 1a shows similar findings in hBECs. Excess unlabelled insulin (2 μmol/l) and S-961 pretreatment (10 nmol/l or 20 nmol/l) blunted 125I-TyrA14-insulin uptake. Pretreatment with an IGF-1 receptor antibody, Ab-3 (1 μg/ml), did not decrease hBEC insulin uptake (Fig. 1a).

IR blockade blunts insulin uptake and signalling in human and rat BECs. IR blockade by S-961 blunts 125I-TyrA14-insulin (125I-insulin) uptake (200 pmol/l, 15 min) (a) and AktSer473 phosphorylation in hBECs (vehicle vs 20 nmol/l S-961, p = 0.08) (b) and RBMVECs (c), while the IGF-1 receptor antibody Ab-3 did not significantly decrease uptake. Data presented are means ± SEM. One-way ANOVA with Dunnett’s (a) or Sidak (b, c) post hoc test. *p < 0.05, **p < 0.01 vs vehicle
Insulin (10 nmol/l) treatment for 15 min increased AktSer473 phosphorylation in hBECs and RBMVECs, which was blocked by pretreatment with 10 nmol/l or 20 nmol/l S-961 in both human and rat cells (Fig. 1b, c). Importantly, IGF-1 receptor blockade did not decrease insulin-stimulated Akt phosphorylation. These results confirm our earlier rodent studies and identify a role for the IR in hBEC insulin transport.
Insulin uptake is not affected by PI3K or mitogen-activated protein kinase kinase (MEK) signalling but requires intact lipid rafts
In bovine aortic endothelial cells, blocking insulin signalling with wortmannin, genistein or PD98059 decreased FITC-insulin endocytosis [26]. Treating RBMVECs with these agents did not affect 125I-TyrA14-insulin uptake (ESM Fig. 4). The brain endothelium is highly restrictive because of its tight junctions and multiple efflux pumps, with the latter preventing BEC accumulation of numerous compounds and lipophilic pharmacological agents [35]. Therefore, we confirmed that wortmannin (100 nmol/l) and genistein (50 μmol/l) inhibited insulin-induced AktSer473 phosphorylation in RBMVECs (ESM Fig. 5a, b). Insulin did not consistently increase extracellular signal-regulated kinase (ERK) phosphorylation and PD98059 (25 μmol/l) inhibited basal ERKThr202/Tyr204 phosphorylation (ESM Fig. 5c). In aggregate, these findings suggest insulin uptake by RBMVECs does not require insulin-stimulated signalling.
Next we tested whether inhibiting lipid rafts or vesicle endocytosis affected insulin uptake. Pretreating RBMVECs with filipin (5 μg/ml) or methyl-β-cyclodextrin (MβCD, 10 mmol/l) prior to 125I-TyrA14-insulin exposure decreased tracer uptake (Fig. 2). MβCD had a greater effect than filipin (p = 0.004, Tukey’s post hoc). MβCD depletes and filipin binds cholesterol in lipid rafts, suggesting that intact raft formation/function is necessary for insulin endocytosis. Neither pretreatment with dynasore (40 μmol/l), which inhibits dynamin-mediated scission of internalised endocytic vesicles from the plasma membrane, nor with PP1 or PP2 (10 μmol/l), inhibitors of Src-kinase, which is implicated in facilitating caveolae- and clathrin-mediated endocytosis, decreased 125I-TyrA14-insulin uptake.

Insulin uptake requires lipid rafts. 125I-TyrA14-insulin (125I-insulin) uptake decreased in RBMVECs pretreated with MβCD and filipin. Dynasore, PP1 and PP2 did not decrease uptake. Data presented are means ± SEM, compared using Dunnett’s test. *p < 0.05, ***p < 0.001 vs vehicle
Since NO regulates insulin uptake in aortic endothelial cells [36, 37], we tested whether eNOS inhibition would affect insulin uptake in RBMVECs. Pretreatment with N ω-nitro-l-arginine methyl ester (l-NAME, 200 μmol/l) did not significantly decrease 125I-TyrA14-insulin uptake. Pretreatment of RBMVECs with NO donors, (S)-nitroso-N-acetylpenicillamine (SNAP) or sodium nitroprusside (SNP), at several concentrations did not affect 125I-TyrA14-insulin uptake (ESM Fig. 6). These results suggest that the brain endothelium regulates insulin uptake differently from peripheral vasculature.
HFD decreases iBEC insulin uptake and increases NF-κB binding
In vivo, 4 weeks of HFD feeding decreased brain insulin clearance [25], so we purified iBECs from male Sprague Dawley rats fed a ND or an HFD (60% lipid) ad lib for 4 weeks. iBECs were cultured for 5–7 days before experiments. HFD rats weighed more than ND rats (479 ± 10 vs 430 ± 9 g, p < 0.01, n = 17 per diet). Non-fasting insulin concentrations (HFD = 1.53 ± 0.31 ng/ml, ND = 0.94 ± 0.23 ng/ml, n = 17 per diet) did not differ (Mann–Whitney test, p = 0.14).
iBECs from HFD rats had 30% lower 125I-TyrA14-insulin (200 pmol/l) uptake compared with ND rats (p < 0.01, Fig. 3a). In aortic tissues, HFD feeding can increase NF-κB p65 activity [38]. Nuclei from HFD iBECs had increased NF-κB p65 binding activity compared with ND iBEC nuclei (p < 0.05, Fig. 3b). Insulin (10 nmol/l, 15 min) increased AktSer473 (Fig. 3c, d) and eNOSSer1177 (ESM Fig. 7a) phosphorylation comparably in iBECs from ND and HFD rats (n = 4 per diet). Insulin did not affect ERKThr202/Tyr204 phosphorylation in either group (ESM Fig. 7b). Expression of endocytotic proteins caveolin-1 and clathrin heavy chain did not differ between diet groups and insulin did not increase caveolin-1Tyr14 phosphorylation in either group (data not shown). We also compared IR and IDE expression to assess whether this might be driving differences in insulin uptake. We did not detect differences in IR-β mRNA, IDE mRNA or IR-β protein between iBECs from ND and HFD rats (Fig. 3e–g).

HFD feeding decreases insulin uptake and increases NF-κB nuclear binding activity in iBECs despite intact IR expression and signalling. HFD feeding decreased 125I-TyrA14-insulin (125I-insulin) uptake (a) and increased NF-κB nuclear binding activity (b) in iBECs. Representative blot (line denotes crop) (c) and quantification (d) of insulin-stimulated (10 nmol/l, 15 min) Akt phosphorylation (p-Akt, normalised to total Akt) did not differ between ND and HFD rats. IR-β (e, f) and Ide expression (g) did not differ between diet groups. White circles, rats fed an HFD for 4 weeks; black circles, rats fed an ND for 4 weeks. Mann–Whitney U test (a, b), Welch’s t test for non-equal variance (d) and t tests (e, f, g) were used to analyse data. Data presented are means ± SEM. *p < 0.05, **p < 0.01
iBECs transcytose intact insulin
Using our in vitro BBB, we first measured the time course for transcytosis of 200 pmol/l 125I-TyrA14-insulin or HD-125I-TyrA14-insulin added to the apical chamber (TEER 362 ± 38 Ω × cm2). Transcytosis of 125I-TyrA14-insulin (corrected for paracellular leak) increased linearly, with 0.4% of the added 125I-TyrA14-insulin (132 ± 33 fmol) transcytosed at 15 min, rising to 2.4% (728 ± 241 fmol) at 60 min (Fig. 4a). Using linear regression (R 2 = 0.99), we estimated this transport rate at 13.1 fmol/min. To assess the symmetry of this transport, we measured insulin transcytosis 30 min after adding 125I-TyrA14-insulin to the apical or basolateral chamber (TEER 439 ± 55 Ω × cm2). We found insulin’s movement from apical to basolateral or from basolateral to apical was comparable (Fig. 4b), indicating that transport is bidirectional.

125I–TyrA14-insulin (125I–insulin) is transported across iBECs and remains intact. 125I–TyrA14-insulin transcytosis over 60 min (a). Dotted lines are individual experiments (n = 3) and solid line is mean ± SEM. 125I-TyrA14-insulin transcytosis after 30 min is bidirectional and directions did not differ significantly (b). Data are presented as means ± SEM, paired t test. Basolateral media radioactivity (disintegrations per min, DPM) from transwell experiments sampled 30 min after apical 125I-TyrA14-insulin addition eluted from a Sephadex G-50 column in the same fractions in presence (solid line) or absence (dashed line) of iBECs (c). Thin dotted lines represent individual experiments without iBECs and thin solid lines represent individual experiments with iBECs (n = 3). These peaks corresponded to intact insulin (insert, solid line) and did not appear in the same fraction as HepG2-degraded insulin (insert, dotted line)
The brain expresses IDE [17, 39], some of which is localised to the BEC [16], and previous reports hypothesise the BEC lysosomes degrade insulin [40]. We confirmed that 125I-TyrA14-insulin incubated with HepG2 cells was rapidly degraded [34], giving an elution peak distinctly different from intact 125I-TyrA14-insulin (Fig. 4c, insert). By contrast, media collected from basolateral chambers 30 min after adding 125I-TyrA14-insulin to the apical transwell chamber eluted similar to 125I-TyrA14-insulin that was not exposed to BECs (Fig. 4c). Similar results were observed in the apical chamber (ESM Fig. 8). Thus, we conclude that insulin is transported across iBECs without being degraded.
IR and endocytosis machinery regulate transcytosis
To test IR’s role in transcytosis, iBECs were pretreated with 10 nmol/l S-961 for 30 min before adding either 200 pmol/l 125I–TyrA14-insulin or HD-125I-TyrA14-insulin to the apical side of the transwell insert (TEER 284 ± 72 Ω × cm2). S-961 specifically decreased insulin transcytosis (p < 0.05, Fig. 5a), affirming IR’s role in insulin uptake and transcytosis. Dynasore decreased transcytosis by 40% (p < 0.05), while filipin did not decrease transcytosis significantly (Fig. 5b, TEER 440 ± 84 Ω × cm2). MβCD greatly increased paracellular leak (>200%) and therefore was not investigated further.

125I-TyrA14-insulin (125I-insulin) is transported across iBECs by IR and dynamin-mediated vesicles. IR antagonist S-961 blunted 125I-TyrA14-insulin transcytosis across iBECs (a). 125I-TyrA14-insulin transcytosis was decreased by pretreatment with dynasore, but the effect of filipin was not significant (b). Data presented are means ± SEM, paired t test vs vehicle control. *p < 0.05
Neurovascular coupling can affect insulin transcytosis
Knowing neurovascular coupling regulates IGF-1 transcytosis [41], we investigated whether astrocyte stimulation affected insulin transcytosis. Using our BBB model, we treated the astrocytes in the basolateral (‘brain’) chamber with l-glutamate (100 μM) before adding 200 pmol/l 125I-TyrA14-insulin to iBECs in the apical (‘blood’) chamber. Glutamate increased insulin transcytosis across the iBEC monolayer by 30% (p < 0.05, TEER 473 ± 182 Ω × cm2, Fig. 6a), without affecting HD-125I-TyrA14-insulin transport. Apical pretreatment with l-NAME for 30 min decreased 125I-TyrA14-insulin transcytosis (p < 0.05, TEER 429 ± 115 Ω × cm2, Fig. 6b) and adding glutamate (basolateral) with l-NAME (apical) restored insulin transcytosis to control rates (TEER 534 ± 42 Ω × cm2, Fig. 6c). l-NAME and glutamate treatment did not affect HD-125I-TyrA14-insulin transcytosis. Glutamate can increase calcium signalling in astrocytes [42]. We pretreated astrocytes with thapsigargin (1 μmol/l) to increase cytosolic calcium 30 min prior to glutamate or vehicle treatment for an additional 30 min (TEER 476 ± 45 Ω × cm2). Thapsigargin alone trended towards increasing 125I-TyrA14-insulin transcytosis (p = 0.07, Fig. 6d). Thapsigargin and glutamate co-treatment increased transcytosis vs vehicle (p < 0.05, Fig. 6d) but the effect was not additive with glutamate, consistent with glutamate acting by increasing astrocyte cytosolic calcium. Thapsigargin increased HD-125I-TyrA14-insulin transcytosis, but increased transcytosis persisted after correcting for this.

125I-TyrA14-insulin (125I-insulin) transcytosis is increased by glutamate stimulation and cytosolic calcium at the astrocyte and inhibited by NO blockade. 125I-TyrA14-insulin transcytosis was stimulated by basolateral glutamate pretreatment (a) and decreased by apical l-NAME pretreatment (b). Combined basolateral glutamate and apical l-NAME restored l-NAME-induced decreases in transcytosis (c). Basolateral thapsigargin pretreatment tended to increase transcytosis (p = 0.07), and combination of basolateral thapsigargin and subsequent glutamate increased transcytosis (d). Thapsigargin treatment did not differ significantly from thapsigargin and glutamate treatments combined. Data presented are means ± SEM, paired t test vs vehicle (a–c), one-way ANOVA with Tukey’s post hoc test (d). † p = 0.07, *p < 0.05
Discussion
The results presented here demonstrate that BEC insulin transport is IR-dependent, but does not require intact insulin signalling through PI3K. Second, insulin transport requires lipid raft functionality, consistent with a vesicular endocytotic process. Third, despite the presence of proteases, such as IDE, insulin remains intact during BEC transcytosis. Fourth, insulin transport can be regulated by astrocyte stimulation, as evidenced by glutamate-induced increases in transport. Finally, in vivo, HFD feeding has sustained effects to diminish insulin uptake and increase basal NF-κB binding activity in cultured iBECs.
Multiple studies have demonstrated saturable insulin binding to brain microvessels or retention by brain tissue, consistent with IR-mediated transport. However, these studies did not eliminate a role for IGF-1R or IGF-1R/IR hybrid receptors present on endothelial cells [43]. Our use of the specific IR-blocker S-961 and physiological insulin concentrations strongly support IR-facilitated insulin transport at physiological insulin concentrations. Inhibition of insulin-stimulated PI3K signalling did not decrease insulin uptake, suggesting BECs regulate insulin transport differently from aortic endothelial cells [26]. Insulin stimulates ERK phosphorylation within 15 min in bovine aortic endothelial cells. We did not detect ERK phosphorylation and cannot implicate any role for this in BEC insulin transport. However, vesicular transport of insulin appears to be conserved between peripheral and brain ECs. Our studies emphasise the importance of lipid raft formation and dynamin-mediated vesicle budding for insulin uptake and transcytosis. Filipin did not decrease insulin transcytosis despite inhibiting uptake. This may be due to residual serum in the conditioned media in the basolateral chamber, which may have reversed filipin action [44].
We are unaware of prior studies of insulin transport across an intact in vitro BBB. Miller et al. preloaded bovine BECs for 30 min with 125I-TyrA14-insulin before measuring an additional 90 min of tracer efflux. After 30 min efflux, they found <5% of insulin was degraded as assessed by trichloroacetic acid precipitation [40]. Unfortunately, neither TEER nor permeability markers were measured, making it difficult to resolve whether the luminal or abluminal membrane mediated efflux. In our studies, TEER and HD-125I-TyrA14-insulin measurements indicated tight iBEC monolayers and insulin degradation was not detected in the apical or basolateral media of iBECs.
Glutamate’s action to increase insulin BEC transcytosis suggests astrocytes can regulate BBB insulin transport. Neuronal activation and subsequent downstream release of astrocyte-derived factors facilitates IGF-1 BBB transcytosis [41]. In response to glutamate, astrocytes increase cytosolic calcium and secrete factors such as prostaglandin E2 or epoxyeicosatrienoic acids, which may influence BEC transcytosis [41, 45]. While NOS blockade reduces astrocyte glutamate uptake [46], we observed that glutamate stimulation of astrocytes rescued l-NAME-induced decreases in insulin transcytosis. It is possible that these are acting through unrelated mechanisms and that the glutamate action on astrocytes induces the secretion of factors that counteract the eNOS inhibition in iBECs. l-NAME and NO donors did not affect insulin uptake, which may be due to methodological differences or may suggest a role for NO in regulating either intracellular vesicle trafficking or exocytosis (i.e. steps beyond uptake). Thapsigargin treatment of astrocytes tended to increase transcytosis, yet did not differ significantly from combination of thapsigargin and glutamate. This suggests elevated cytosolic calcium may mediate glutamate-stimulated increases in insulin transcytosis across the BEC layer. Elucidating the regulation of BEC transcytosis merits further investigation.
The decrease in 125I-TyrA14-insulin uptake by iBECs from rats fed an HFD for 4 weeks is particularly intriguing. Mechanistically, we did not find evidence for altered canonical insulin signalling to Akt or eNOS. However, increased oxidative stress, acting via increased NF-κB binding activity, may contribute. While we [25] and others [3, 4, 6, 47] found a delay of insulin entry into or action in the brains of HFD-fed animals, this is the first study to our knowledge to report impaired insulin transport at the level of the BEC. Our co-culture findings, as well as recent work regarding insulin signalling in astrocytes [48, 49], may suggest a role for astrocytes in the effect of an HFD on BBB insulin transport. Given the clinical importance of brain insulin resistance, the latter possibly warrants further investigation.
We previously reported that brain insulin uptake in vivo is ∼40-fold greater than its transfer to CSF [25]. While production of insulin within brain tissue has been reported [50], our studies strongly indicate that insulin detected in the brain can originate from plasma. Insulin’s transport across the BBB occurs first at the BEC, involves IR (not IGF-1R or IGF-1R/IR hybrids) and lipid rafts, and there is no detectable degradation of insulin during transcytosis despite the presence of IDE and other proteases. Finally, the ability of HFD feeding and astrocyte activation to affect insulin transport highlights the complex and multifaceted regulation of insulin’s entry into brain. These results materially advance our understanding of the pathways involved in brain insulin transport, highlight the previously unrecognised role of the astrocyte in BBB insulin transport, and offer potential pathways for ameliorating central insulin resistance.
Abbreviations
- BBB:
-
Blood–brain barrier
- BEC:
-
Brain endothelial cell
- bISF:
-
Brain interstitial fluid
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- eNOS:
-
Endothelial nitric oxide synthase
- ERK:
-
Extracellular signal-regulated kinase
- F12:
-
Ham’s F12 medium
- hBEC:
-
Human brain endothelial cell
- HD:
-
Heat-denatured
- HFD:
-
High-fat diet
- iBEC:
-
Isolated brain endothelial cell
- IDE:
-
Insulin degrading enzyme
- IR:
-
Insulin receptor
- IR-β:
-
Insulin receptor, β subunit
- l-NAME:
-
N ω-nitro-l-arginine methyl ester
- MEK:
-
Mitogen-activated protein kinase kinase
- MβCD:
-
Methyl-β-cyclodextrin
- ND:
-
Normal chow diet
- NOS:
-
Nitric oxide synthase
- PI3K:
-
Phosphoinositide-3-kinase
- RBMVEC:
-
Rat brain microvascular endothelial cell
- TEER:
-
Transendothelial electrical resistance
- VRS:
-
Virchow–Robin space
References
Chavez M, Kaiyala K, Madden LJ, Schwartz MW, Woods SC (1995) Intraventricular insulin and the level of maintained body weight in rats. Behav Neurosci 109:528–531
Woods SC, Lotter EC, McKay LD, Porte D Jr (1979) Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282:503–505
Clegg DJ, Benoit SC, Reed JA, Woods SC, Dunn-Meynell A, Levin BE (2005) Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Phys Regul Integr Comp Phys 288:R981–R986
Clegg DJ, Gotoh K, Kemp C et al (2011) Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol Behav 103:10–16
Ikeda H, West DB, Pustek JJ et al (1986) Intraventricular insulin reduces food intake and body weight of lean but not obese Zucker rats. Appetite 7:381–386
Begg DP, Mul JD, Liu M et al (2013) Reversal of diet-induced obesity increases insulin transport into cerebrospinal fluid and restores sensitivity to the anorexic action of central insulin in male rats. Endocrinology 154:1047–1054
Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM (2010) Insulin metabolism and the risk of Alzheimer disease: the Rotterdam study. Neurology 75:1982–1987
van Houten M, Posner BI (1979) Insulin binds to brain blood vessels in vivo. Nature 282:623–625
van Houten M, Posner BI, Kopriwa BM, Brawer JR (1980) Insulin binding sites localized to nerve terminals in rat median eminence and arcuate nucleus. Science 207:1081–1083
van Houten M, Posner B, Kopriwa B, Brawer J (1979) Insulin-binding sites in the rat brain: in vivo localization to the circumventricular organs by quantitative radioautography. Endocrinology 105:666–673
van Houten M, Posner B (1981) Specific binding and internalization of blood-borne [125I]-iodoinsulin by neurons of the rat area postrema. Endocrinology 109:853–859
Banks WA, Kastin AJ (1998) Differential permeability of the blood–brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19:883–889
Duffy KR, Pardridge WM (1987) Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res 420:32–38
Frank HJ, Pardridge WM, Morris WL, Rosenfeld RG, Choi TB (1986) Binding and internalization of insulin and insulin-like growth factors by isolated brain microvessels. Diabetes 35:654–661
Pardridge WM, Eisenberg J, Yang J (1985) Human blood-brain barrier insulin receptor. J Neurochem 44:1771–1778
Lynch JA, George AM, Eisenhauer PB et al (2006) Insulin degrading enzyme is localized predominantly at the cell surface of polarized and unpolarized human cerebrovascular endothelial cell cultures. J Neurosci Res 83:1262–1270
Miners JS, Kehoe PG, Love S (2008) Immunocapture-based fluorometric assay for the measurement of insulin-degrading enzyme activity in brain tissue homogenates. J Neurosci Methods 169:177–181
Iliff JJ, Wang M, Liao Y et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111
Yang L, Kress BT, Weber HJ et al (2013) Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med 11:107
Jessen N, Munk A, Lundgaard I, Nedergaard M (2015) The glymphatic system: a beginner’s guide. Neurochem Res 40:2583–2599
Schwartz MW, Sipols A, Kahn SE et al (1990) Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am J Phys 259:E378–E383
Porte D (2006) Central regulation of energy homeostasis. Diabetes 55:S155–S160
Heni M, Schopfer P, Peter A et al (2014) Evidence for altered transport of insulin across the blood-brain barrier in insulin-resistant humans. Acta Diabetol 51:679–681
Baura GD, Foster DM, Porte D Jr et al (1993) Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest 92:1824–1830
Meijer RI, Gray SM, Aylor KW, Barrett EJ (2016) Pathways for insulin access to the brain: the role of the microvascular endothelial cell. Am J Physiol Heart Circ Physiol 311:H1132–H1138
Wang H, Wang AX, Liu Z, Barrett EJ (2008) Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes 57:540–547
Burek M, Salvador E, Forster CY (2012) Generation of an immortalized murine brain microvascular endothelial cell line as an in vitro blood brain barrier model. J Vis Exp 66:e4022
Catravas JD, Snead C, Dimitropoulou C et al (2010) Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vasc Pharmacol 52:175–181
Bernas MJ, Cardoso FL, Daley SK et al (2010) Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protoc 5:1265–1272
Schildge S, Bohrer C, Beck K, Schachtrup C (2013) Isolation and culture of mouse cortical astrocytes. J Vis Exp 71:e50079
Genders AJ, Frison V, Abramson SR, Barrett EJ (2013) Endothelial cells actively concentrate insulin during its transendothelial transport. Microcirculation 20:434–439
Deli M (2007) Blood–brain barrier models. In: Lajtha A, Reith MEA (eds) Handbook of neurochemistry and molecular neurobiology: neural membranes and transport. Springer, New York, pp 29–55
Wuest DM, Wing AM, Lee KH (2013) Membrane configuration optimization for a murine in vitro blood–brain barrier model. J Neurosci Methods 212:211–221
Keller S, Schmid C, Zapf J, Froesch ER (1989) Inhibition of insulin degradation by insulin-like growth factors I and II in human hepatoma (HepG2) cells. Acta Endocrinol 121:279–285
Löscher W, Potschka H (2005) Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 6:591–602
Wang H, Wang AX, Aylor K, Barrett EJ (2013) Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes 62:4030–4042
Barrett EJ, Wang H, Upchurch CT, Liu Z (2011) Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab 301:E252–E263
Zhao L, Fu Z, Wu J et al (2015) Inflammation-induced microvascular insulin resistance is an early event in diet-induced obesity. Clin Sci (Lond) 129:1025–1036
Kuo WL, Montag AG, Rosner MR (1993) Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 132:604–611
Miller DW, Keller BT, Borchardt RT (1994) Identification and distribution of insulin receptors on cultured bovine brain microvessel endothelial cells: possible function in insulin processing in the blood–brain barrier. J Cell Physiol 161:333–341
Nishijima T, Piriz J, Duflot S et al (2010) Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 67:834–846
Koehler RC, Gebremedhin D, Harder DR (2006) Role of astrocytes in cerebrovascular regulation. J Appl Physiol 100:307–317
Li G, Barrett EJ, Wang H, Chai W, Liu Z (2005) Insulin at physiological concentrations selectively activates insulin but not insulin-like growth factor I (IGF-I) or insulin/IGF-I hybrid receptors in endothelial cells. Endocrinology 146:4690–4696
Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R (1997) A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol 138:877–889
Iadecola C, Nedergaard M (2007) Glial regulation of the cerebral microvasculature. Nat Neurosci 10:1369–1376
Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC (2000) Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7:153–159
Chavez M, Riedy CA, van Dijk G, Woods SC (1996) Central insulin and macronutrient intake in the rat. Am J Phys 271:R727–R731
García-Cáceres C, Quarta C, Varela L et al (2016) Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166:867–880
Fernandez AM, Hernandez-Garzón E, Perez-Domper P et al (2016) Insulin regulates astrocytic glucose handling through cooperation with insulin-like growth factor I. Diabetes 66:64–74
Molnar G, Farago N, Kocsis AK et al (2014) GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci 34:1133–1137
Acknowledgements
The authors thank J. Catravas (Old Dominion University, Norfolk, VA, USA) for the gift of human cerebral endothelial cells, Novo Nordisk for the gift of S-961, M. Thorner (University of Virginia) for thoughtful critique of the manuscript, J. Patrie (University of Virginia) for statistical assistance, and P. Xu (University of Virginia), J. Gildea (University of Virginia) and L. Zhao (Sun Yat-sen University, Guangdong, China) for technical assistance. We thank the staff of the UVA Keck Center for Cellular Imaging for imaging support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Funding
This work was supported by NIH (DK073059) and ADA (11-BS6) to EJB, and NIH (T32 HL007284, F31 DK104521) and AHA (PRE20100048) to SMG. Funding for the UVA Keck Center for Cellular Imaging for the usage of the Leica SP5 and Zeiss LSM 780 was provided by NIH (RR025616 and OD016446).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
SMG and EJB conceptualised and designed the experiments. SMG and KWA performed the experiments and acquired the data. SMG, KWA and EJB analysed and interpreted the data, wrote the manuscript and approved the final version for publication. EJB is the guarantor of this work.
Electronic supplementary material
ESM
(PDF 542 kb)
Rights and permissions
About this article

Cite this article
Gray, S.M., Aylor, K.W. & Barrett, E.J. Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 60, 1512–1521 (2017). https://doi.org/10.1007/s00125-017-4285-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4285-4