
Abstract
Aims/hypothesis
AGEs are involved in diabetic complications and might be responsible for the phenomenon of ‘hyperglycaemic memory’. d-Carnosine-octylester (DCO) has been shown to attenuate AGE formation and vascular and renal injury induced by high-fat diet in Apoe-null mice. This study aimed to verify the protective effect of DCO in atherosclerosis and renal disease induced by experimental diabetes and to discover whether reduction of AGE formation by early vs late DCO treatment provides better macro and microvascular protection.
Methods
Apoe-null mice were rendered diabetic by streptozotocin and were left untreated or were treated with DCO for 20 weeks (DCO-Extended), from week 1 to 11 (DCO-Early) or from week 9 to 19 (DCO-Late). Non-diabetic Apoe-null mice served as controls. Aortic and renal lesions were evaluated by morphometry and protein and gene expression of disease markers were assessed by immunohistochemistry and real-time PCR.
Results
DCO-Extended treatment produced a more stable plaque phenotype by markedly attenuating diabetes-induced increases in lesion size, necrotic core area and plaque content of Nε-carboxymethyllysine, levels of apoptotic cells and markers of inflammation and oxidative stress and also reductions in collagen and smooth muscle cells. DCO treatment for 11 weeks afforded partial protection and this was significantly better in DCO-Early mice than in DCO-Late mice. Renal disease was attenuated in DCO-Extended mice and to a lesser extent in those treated for 11 weeks, with no significant difference between DCO-Early mice and DCO-Late mice.
Conclusions/interpretation
These data show that DCO protects mice from diabetes-induced vascular and renal disease and that protection against atherosclerosis is more effectively achieved by early treatment than by late treatment, thus suggesting that early inhibition of AGE formation attenuates progression of macroangiopathy and favours development of more stable lesions.
Similar content being viewed by others


Introduction
In clinical trials intensive glucose-lowering treatment has been found to have different effects on micro- and macrovascular disease. Strict glycaemic control prevents or retards microvascular complications in patients with both types of diabetes [1–3] and has a beneficial effect, especially on nephropathy, in individuals with long-standing type 2 diabetes [4–6]. Conversely, in patients with type 1 diabetes who took part in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC), reduction of macrovascular risk arising from earlier glycaemic control emerged only after several years, despite loss of within-trial differences in HbA1c levels—a phenomenon called ‘hyperglycaemic memory’ [7]. Similar results were obtained in patients with newly-diagnosed type 2 diabetes from the UK Prospective Diabetes Study (UKPDS) [8] whereas strict glycaemic control was ineffective in reducing cardiovascular risk in individuals with long-standing diabetes [4–6], suggesting that there is a ‘legacy effect’ from early treatment [9].
The injurious effect of hyperglycaemia has been attributed to multiple biochemical consequences of intracellular metabolism of excess glucose, including non-enzymatic glycation with formation of AGEs [10]. AGEs are heterogeneous compounds that accumulate in sera and tissues of individuals suffering from several disease conditions, including diabetes and its complications [11]. Increased AGE formation is due to generation of reactive carbonyl species (RCS), which react with amino acid residues on proteins to generate stable adducts or crosslinks collectively referred as to AGEs [11–13]. Since AGEs are primarily formed on long-lived proteins such as collagen, thus causing sustained alterations of vascular structure and function, they have been implicated in mediating the phenomenon of ‘hyperglycaemic memory’ arising from prior glycaemic control [14].
Therapeutic strategies aimed at reducing RCS- and AGE-induced tissue injury, including quenching of RCS by carbonyl scavengers, have been proposed and tested successfully in experimental animals [13]. l-Carnosine is a histidine-containing dipeptide that serves as a major endogenous quencher of RCS, via intramolecular Michael addition [15]. Unfortunately, it has a short half-life in humans due to its rapid inactivation by serum and tissue carnosinase [15]. We have recently shown that a bioavailable pro-drug of the carnosinase-resistant d-carnosine, d-carnosine-octylester (DCO) [16], is highly effective in attenuating experimental atherosclerosis and renal disease induced by high-fat diet in Apoe-null mice by reducing carbonyl stress and inflammation [17].
The aim of this study was to investigate whether DCO provides protection against vascular and renal complications of experimental diabetes in Apoe-null mice and whether reduction of AGE formation by early vs late DCO treatment results in better macro- and microvascular protection, thus providing a molecular basis for the role of AGEs in the legacy from hyperglycaemia.
Methods
Design
The study protocol was approved by the locally appointed Ethics Committee. The mice were housed and cared for according to the Principles of Laboratory Animal Care protocols (NIH publication No. 85-23, revised 1985) and national laws and regulations and had free access to water and food. Six-week-old adult female Apoe-null mice (Charles River, Calco, Italy) were rendered diabetic by streptozotocin (Sigma, St Louis, MO, USA) administered by intraperitoneal injection at a dose of 50 mg/kg body weight for 5 consecutive days. Mice injected with citrate buffer served as non-diabetic controls (Cont). Diabetic mice were divided into four groups, each consisting of ten mice: untreated (Diab); mice treated with 60 mg/kg body weight DCO (Flamma, Chignolo d’Isola, Italy) dissolved in the drinking water for the entire duration of the study (20 weeks, DCO-Extended); mice treated with DCO for 11 weeks only, either starting immediately after diabetes induction and continuing up to week 11 (DCO-Early) or started at week 9 and continuing to week 19 (DCO-Late) followed by 1 week of wash-out to avoid potential interference of DCO plasma levels on the comparison with DCO-Early.
At the end of the 20 week period, the mice were placed into metabolism cages to collect urine. The next day, mice were anaesthetised by intraperitoneal injection of ketamine (Imalgene, 60 mg/kg, Merial, Milan, Italy) and xylazine (Rompum, 7.5 mg/kg, Bayer, Milan, Italy) and the heart (with attached aorta) and both kidneys were sampled and weighed. The aortic sinus, the brachiocephalic artery (BCA) and the right kidney were fixed in 4% (wt/vol.) formaldehyde. Then, the aortic sinus was embedded in Optimal Cutting Temperature embedding medium (Tissue-Tek, Torrance, CA, USA) for frozen section preparation and the BCA and the right kidney specimens were embedded in paraffin. The abdominal aorta was opened longitudinally, layered on ovalbumin-coated glass slides, allowed to dry for 1 h and then fixed in formalin for 24 h for en face preparation [18]. The left kidney and the thoracic part of the descending aorta were frozen in liquid nitrogen for subsequent mRNA and protein extraction. In selected mice, termination was preceded by measurement of BP by the tail-cuff method, using the CODA System (Kent Scientific, Torrington, CT, USA).
Blood and urine measurements
Blood glucose was measured using an automated colorimetric instrument (Glucocard G meter; Menarini, Florence, Italy), and serum cholesterol and triacylglycerols by enzymatic colorimetric methods (Roche Diagnostics, Milan, Italy). Serum levels of isoprostane 8-epi-prostaglandin (PG) F2α were assessed using an ELISA kit (Cayman, Ann Arbor, MI, USA) and those of AGEs were measured by a competitive ELISA technique [17, 19, 20]. Serum and urine creatinine levels were measured by HPLC and proteinuria and albuminuria were assessed using the Bradford dye-binding protein assay kit (Pierce, Rockford, IL, USA) and the Mouse Albumin ELISA kit (Bethyl, Montgomery, TX, USA), respectively, and results were normalised to urine creatinine concentration [17, 20].
Morphology/morphometry
For the assessment of atherosclerotic lesions [17, 19], the extent of lipid accumulation was evaluated in en face preparations after staining with Oil Red O. Lesion area was quantified at the aortic sinus and BCA level after staining with Oil Red O and the Weigert–van Gieson method for elastic and collagen fibres [21], respectively. Five cryosections of aortic sinus (10 μm thick, every other section) and four sections (5 μm thick) at 200, 400, 600 and 800 μm from the branching point of the BCA were evaluated. The necrotic core area and collagen content were measured in five sections of the aortic sinus (50 μm apart) and in four sections of the BCA (200 μm apart) stained with the Weigert–van Gieson method. For the assessment of renal lesions [21], 4 μm sections were stained with periodic acid–Schiff (PAS) to allow glomerular and tubule-interstitial damage to be examined by light microscopy. Moreover, the areas of at least 60 glomerular tuft profiles per sample were measured and the harmonic mean of the profile area (mean glomerular area, mGA) was obtained. Then, PAS-positive material was quantified and expressed as a percentage of the glomerular tuft area (fractional mesangial area, fMA), and the mean mesangial area (mMA) was calculated by the formula: (fMA × mGA)/100. All measurements were performed with the aid of the Optimas 6.5 image analysis system (Bioscan, Washington, DC, USA).
Immunohistochemistry
Immunohistochemical analysis [17, 19, 20] was performed to assess the aortic and kidney content and distribution of markers of murine macrophage activation (F4/80), oxidative stress (nitrotyrosine) and carbonyl stress (N ε-carboxymethyllysine, CML). Nitrotyrosine was chosen as a marker of free radical damage since its production does not depend on RCS levels and, hence, is not directly influenced by DCO treatment, at variance with CML and the adducts of malonylaldehyde or 4-hydroxy-2-nonenal. The expression of active caspase-3, a marker of apoptotic cells, α-smooth muscle actin (α-SMA), a marker of vascular smooth muscle cells (VSMCs) and receptor for AGEs (RAGE) in the aorta, and of collagen IV in the kidney, were also assessed (see electronic supplementary material [ESM] Table 1 for primary antibodies). Sections were analysed using the Optimas 6.5 image analysis system.
Quantitative real-time PCR
Total RNA was extracted from aorta and kidneys by the guanidine thiocyanate–phenol–chloroform method using TRIzol Reagent (Invitrogen Italia, San Giuliano Milanese, Italy). Quantitative real-time PCR (qRT-PCR) was used for assessing the gene expression level of multiple disease markers [17]. The transcripts for the following proteins were measured in the aorta: the inflammatory markers F4/80, monocyte chemoattractant protein-1 (MCP-1) and IL-1β, the antioxidant enzymes CuZn-superoxide dismutase (CuZn-SOD), catalase and glutathione peroxidase 1 (GPX1) and hypoxia inducible factor-1α (HIF-1α), the transcriptional activity of which is increased by AGEs [22] and inhibited by carnosine [23]. The transcripts for the following were measured in the kidney: fibronectin, collagen IV, TGF-β, MCP-1, CuZn-SOD, catalase, GPX1 and HIF-1α.
Statistical analysis
Results are expressed as means ± SD. Statistical significance was evaluated by one-way ANOVA followed by the Student–Newman–Keuls test for multiple comparisons. A p value <0.05 was considered significant. All statistical tests were performed on raw data.
Results
Metabolic and haemodynamic variables
Body weight decreased and blood glucose, triacylglycerol and cholesterol levels increased in Diab mice and were unaffected by DCO treatment (Table 1).
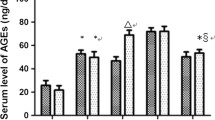
Serum AGE and isoprostane 8-epi-PGF2α levels increased markedly (∼ two- to threefold) in Diab mice compared with Cont mice. These levels were almost normalised in DCO-Extended mice, although isoprostane levels remained significantly higher than in Cont mice, possibly as a consequence of residual oxidative stress directly attributable to hyperglycaemia. Values in DCO-Early mice were similar to those found in Diab mice, whereas they were partially reduced in DCO-Late mice (Table 1). BP did not differ among groups (not shown).
Atherosclerosis
Oil Red O staining of en face aorta preparations (ESM Fig. 1) showed a significant increase (5.5×) in the extent of lipid accumulation in Diab vs Cont mice. Treatment with DCO for 20 weeks reduced the increase in lipid content by 62%. A more modest (yet significant) reduction was observed in mice treated with DCO for 11 weeks, with a more marked (though not significantly different) improvement seen in DCO-Early than in DCO-Late mice (33 vs 20% reduction). Likewise, lesion areas at the aortic sinus and the BCA (Fig. 1) were significantly higher in Diab than in Cont mice (2.6× and 3.4×, respectively). The increased lesion areas in diabetic mice were reduced by treatment with DCO for 20 weeks (by 55% and 66%, respectively) and, less markedly, 11 weeks. The extent of reduction was significantly higher with the DCO-Early than with the DCO-Late regimen (by 33% and 44% vs 16% and 21%, respectively).

Oil Red O staining of aortic roots (a) and Weigert–van Gieson’s staining of BCAs (e; black, elastic fibres; red, collagen; *necrotic core) from representative mice and quantification of lesion area (b and f, respectively), necrotic core (c and g, respectively) and collagen content (d and h, respectively) in non-diabetic Apoe-null control mice (Cont, white bars) and diabetic Apoe-null mice either untreated (Diab, black bars) or treated with DCO-Extended (hatched bars), -Early (light grey bars) or -Late (dark grey bars) protocol (mean ± SD; n = 10 per group). Scale bar, 500 μm in (a) and 200 μm in (e). Post hoc multiple comparison: ***p < 0.001 vs Cont; † p < 0.05 and ††† p < 0.001 vs Diab; ‡‡ p < 0.01 and ‡‡‡ p < 0.001 for DCO-Late vs DCO-Early
Diabetes increased the complexity of lesions, as defined by the presence of cholesterol clefts, necrosis and/or fibrous cap formation, whereas Cont mice showed predominantly fatty streaks. DCO treatment increased features of plaque stability, as shown by the smaller necrotic cores and the more extensive fibrosis as compared with untreated mice. Again, lesions showed a more stable phenotype in mice treated for 20 weeks than in those treated for 11 weeks and in those on the DCO-Early vs DCO-Late protocol (Fig. 1).
Immunohistochemical detection of active caspase-3 in specimens from Diab mice revealed the presence of apoptotic cells, especially around the necrotic core (Fig. 2). Staining for F4/80 was almost uniform, from the lumen to the media, suggesting that plaques in these mice were composed mainly of foam cells. Conversely, staining for α-SMA was faint or absent. Positivity for RAGE, CML and nitrotyrosine was similar to that for F4/80 (Fig. 3). DCO-Extended treatment was very effective in reducing the number of active caspase-3 positive cells and the content of monocyte/macrophages while increasing the fraction of the plaque occupied by VSMCs, especially at the luminal side and all around the lesions (Fig. 2). Likewise, this intervention significantly reduced the lesion content of RAGE, CML and nitrotyrosine (Fig. 3). The 11-week DCO treatment also affected these variables positively, though to a different extent according to the type of protocol. In fact, with DCO-Late regimen the reductions in active caspase-3, F4/80, RAGE, CML and nitrotyrosine were relatively modest and the increase in α-SMA was not significant (Figs 2, 3). Conversely, the DCO-Early protocol provided a significant protection from diabetes-induced changes in these variables, also producing a unique plaque phenotype. In particular, staining for apoptotic cells, F4/80, RAGE, CML and nitrotyrosine was confined to the luminal side of the plaque, whereas the distribution of α-SMA positivity resembled that in plaques from DCO-Extended mice, with VSMCs forming a fibrous cap surrounding the lesion (Figs 2, 3).

Immunohistochemical detection of active caspase-3 (a, arrows indicate active caspase-3 positive cells), F4/80 (c) and α-SMA (e) in aortic specimens from representative mice and quantification of staining (b, d and f, respectively) in diabetic Apoe-null mice either untreated (Diab, black bars) or treated with DCO-Extended (hatched bars), -Early (light grey bars) or -Late (dark grey bars) protocol (mean ± SD; n = 10 per group). L, lumen; TM, tunica media. Scale bar, 80 μm in (a) and (e) and 50 μm in (c). Post hoc multiple comparison using the Student–Newman–Keuls test: ***p < 0.001 vs Diab; ††† p < 0.001 for DCO-Late vs DCO-Early

Immunohistochemical detection of RAGE (a), CML (c) and nitrotyrosine (e) in aortic specimens from representative mice and quantification of staining (b, d and f, respectively) in diabetic Apoe-null mice either untreated (Diab, black bars) or treated with DCO-Extended (hatched bars), -Early (light grey bars) or -Late (dark grey bars) protocol (mean ± SD; n = 10 per group). Scale bar, 80 μm in (a) and 50 μm in (c) and (e). L, lumen; TM, tunica media. Post hoc multiple comparison: ***p < 0.001 vs Diab; ††† p < 0.001 for DCO-Late vs DCO-Early
qRT-PCR analysis of mRNA levels showed that F4/80 (also known as Emr1), Mcp-1 (also known as Ccl2), Il-1β, CuZn-Sod (also known as Sod1), catalase (also known as Cat), Gpx1 and Hif-1α were higher in Diab mice than in Cont mice and were significantly reduced by DCO-Extended treatment (ESM Fig. 2). The gene expression of inflammatory markers tended to decrease in DCO-Early mice (though the extent of reduction was significant only for Il-1β) and to increase in DCO-Late mice (to a significant extent only for F4/80). Moreover, the DCO-Early protocol reduced the mRNA levels of genes encoding antioxidant enzymes (except Gpx1) and Hif-1α to the same extent as observed with DCO-Extended therapy, whereas the DCO-Late regimen decreased significantly only the expression of genes encoding catalase and, particularly, Hif-1α. Finally, significant differences between DCO-Late and DCO-Early protocols were observed for F4/80, Il-1β, Sod and catalase.
Renal disease
Kidney weights, both absolute and relative to body weight, were significantly increased in Diab mice and reduced by treatment with DCO for 20 weeks and, to a lesser extent, for 11 weeks, irrespective of protocol (DCO-Early or -Late). Serum creatinine levels did not differ among groups, whereas proteinuria and albuminuria increased significantly in Diab mice (by 68% and 104%, respectively) compared with Cont mice. The increases were significantly attenuated (by 54% and 68%, respectively) by DCO-Extended treatment. Slight, non-significant, reductions were observed with both DCO-Early and DCO-Late protocols (Table 1). Glomerulopathy was detected in Diab mice, with significantly higher mGA, fMA and mMA (+41, 71 and 142%, respectively). All these increases were significantly reduced by 20 weeks of DCO treatment (by 69%, 79% and 79%, respectively), and to a lesser and comparable extent by both 11 week protocols (by ∼30%, 40% and 40%, respectively) (Fig. 4). Also, the increased renal content of collagen IV, F4/80, CML and nitrotyrosine caused by diabetes was significantly reduced by DCO treatment, again more markedly in mice treated for 20 weeks than in those treated for 11 weeks (Fig. 5). Likewise, diabetes-induced increases in mRNA levels of fibronectin (also known as Fn1), collagen IV (also known as Col4a1), Tgf-β (also known as Tgfb1), Mcp-1 and Hif-1α were significantly reduced by the 20 week DCO treatment and also by the 11 week protocols, with the exception of Mcp-1 and particularly of Tgf-β, which was even increased in DCO-Late mice (ESM Fig. 3). CuZn-Sod, catalase and Gpx1 gene expression was reduced, increased and unaffected by diabetes, respectively, and was significantly reduced by all DCO protocols.

PAS staining of kidneys from representative mice (a) and quantification of mGA (b), fMA (c) and mMA (d) in non-diabetic Apoe-null control mice (Cont, white bars) and diabetic Apoe-null mice either untreated (Diab, black bars) or treated with DCO-Extended (hatched bars), -Early (light grey bars) or -Late (dark grey bars) protocol (mean ± SD; n = 10 per group). Scale bar, 80 μm. Post hoc multiple comparison: ***p < 0.001 vs Cont; †† p < 0.01 and ††† p < 0.001 vs Diab; ‡‡‡ p < 0.001 vs DCO-Extended

Immunohistochemical detection of collagen IV (a), F4/80 (b, arrows indicate F4/80 positive cells), CML (c) and nitrotyrosine (d) in kidney specimens from representative mice and quantification of staining (e, f, g and h, respectively) in non-diabetic Apoe-null control mice (Cont, white bars) and diabetic Apoe-null mice either untreated (Diab, black bars) or treated with DCO-Extended (hatched bars), -Early (light grey bars) or -Late (dark grey bars) protocol (mean ± SD; n = 10 per group). Scale bar, 50 μm in (a), (c) and (d) and 80 μm in (b). Post hoc multiple comparison: **p < 0.01 vs Cont; ††† p < 0.001 vs Diab; ‡‡‡ p < 0.001 vs DCO-Extended. Glom, glomerular
Discussion
In diabetic mice, treatment with DCO for 20 weeks significantly reduced the size and extent of atherosclerotic lesions, which presented with a more stable phenotype, and ameliorated abnormalities in renal function and structure. These effects were associated with reduced circulating and tissue AGE levels, which, in the absence of changes in metabolic variables, support the concept that the efficacy of DCO is related to its RCS-quenching activity which results in decreased AGE formation, as previously shown in a diet-induced, non-diabetic model of atherosclerosis [17]. In turn, reduced AGE accumulation prompted attenuated inflammation and blunted oxidative stress, a well-recognised pathogenic mechanism of diabetes-induced atherosclerosis and renal disease in Apoe-null mice [24]. The concept that attenuation of oxidative stress by DCO treatment is related to its RCS-quenching activity is supported by several lines of evidence. First, an indirect antioxidant effect of DCO via reduction of AGE formation has been postulated on the basis of the known effect of these byproducts in promoting generation of reactive oxygen species via receptor-mediated mechanisms [25, 26]. Second, previous data from our group [17] and other investigators [27, 28] argue against a direct antioxidant action for carnosine. Finally, DCO did not appear to reduce the burden from oxidative stress by upregulating antioxidant enzymes. In fact, in the aorta, treatment with DCO decreased the mRNA levels of genes encoding antioxidant enzymes, which were increased in Diab mice probably as part of an adaptive response to enhanced oxidative stress. Conversely, administration of DCO did not normalise the reduced gene expression of renal CuZn-Sod, the downregulation of which has been previously reported to occur in experimental diabetic nephropathy [29]. Nor did it significantly modified the mRNA levels of catalase or Gpx1. In addition, the increased mRNA expression of Hif-1α was prevented by DCO treatment, probably via reduction of AGEs, which are known to enhance Hif-1α gene transcription and transcriptional activity [22].
Both of the 11 week protocols provided a modest, yet significant, protection. However, while DCO-Late was not as effective as DCO-Early treatment in attenuating atherosclerosis, the two protocols ameliorated renal disease to a similar extent. These discordant effects on target organ damage are consistent with the discordant effects of the two treatment regimens on aortic and kidney staining for CML, F4/80 and nitrotyrosine and mRNA levels for inflammatory markers and antioxidant enzymes. The finding that, in the aorta, the two protocols reduced Hif-1α mRNA levels to a similar extent, despite lower attenuation of AGE accumulation in DCO-Late than in DCO-Early mice, suggests that the known direct effect of carnosine on HIF-1α [23] played an important role in DCO-Late mice. Moreover, in these mice, data on protein and gene expression levels of F4/80 were contrasting, since F4/80 staining was reduced, whereas its mRNA expression was further increased when compared with Diab mice. This might suggest that the composition of the monocyte/macrophage population differs between lesions in DCO-Late and Diab mice, with the former containing fewer but more mature cells, which are known to express more F4/80 [30]. Furthermore, cessation of the DCO-Late treatment 1 week before mice were killed might have resulted in a prompt reactivation of inflammation, with a sort of rebound effect, as suggested by the upward trend observed for MCP-1 and IL-1β levels. This interpretation is consistent with the finding that the mRNA expression of inflammatory markers in DCO-Early mice was significantly reduced compared with DCO-Late mice, but not Diab mice, except for Il-1β, suggesting that, after 9 weeks of uncontrolled diabetes and unrestrained AGE formation following cessation of DCO-Early treatment, pro-inflammatory mechanisms are almost fully reactivated at the molecular level. The histological correlate of this finding is the immunohistochemical pattern noted for F4/80, but also for RAGE, CML and nitrotyrosine staining, which was limited to the luminal edge of plaques, suggesting new monocyte recruitment and AGE deposition after the end of treatment. Similar data were obtained in kidneys from both DCO-Early and DCO-Late mice, in which reduction in the mRNA levels for fibronectin and collagen IV were not accompanied by decreases in the gene expression for Tgf-β and Mcp-1, thus suggesting that both pro-fibrotic and pro-inflammatory pathways were fully reactivated after cessation of DCO treatment, though the extent of fibrosis and cellular inflammation, as of AGE accumulation and oxidative stress, was still lower than in Diab mice.
Although a conclusive explanation for the different outcomes of early vs late intervention in the aorta and kidney cannot be provided, it is tempting to speculate that they may reflect profound differences in the histological nature of atherosclerotic and renal lesions. On the one hand, AGEs and macrophages that accumulate during initial atheroma formation participate in a vicious cycle of inflammation and damage, frustrating attempts at tissue repair and promoting further AGE formation and monocyte/macrophage infiltration in the vessel wall [31]. Although DCO-Late intervention might have blocked deposition of new AGEs, preformed AGEs that accumulated prior to treatment could have sustained plaque inflammation, growth and progression during subsequent administration of DCO, since this agent has no effect on preformed AGEs [32]. On the other hand, the inhibition of AGE deposition (and monocyte recruitment) by DCO-Early treatment during initial atheroma formation might have attenuated subsequent plaque progression through a reduced inflammatory burden, which would also contribute to decreased AGE formation during the treatment-free period. Conversely, in diabetic glomerulopathy, both deposition of AGEs and infiltration by monocyte/macrophages are less marked than in atherosclerotic lesions and are not topographically segregated into a lipid-rich, pro-oxidant environment as occurs in atheroma. Thus, the accumulation of AGEs and inflammatory cells before initiation of DCO-Late treatment might have not been sufficient, per se, to sustain further progression of glomerulopathy during DCO administration. This interpretation is in agreement with the observation that glomerular lesions, including Kimmelstiel–Wilson nodules, are reversible in individuals with type 1 diabetes undergoing isolated pancreas transplantation [33], whereas fewer and more elusive data exist on the reversibility of the atherosclerotic process [34].
Altogether, our data support the hypothesis of a main role for AGEs in the ‘hyperglycaemic memory’ or ‘legacy effect’, as postulated some years ago [35], although this hypothesis is not yet proven. In fact, the lower efficacy of late vs early intervention with the inhibitor of AGE formation DCO implies that early vs late deposition of AGEs, which results in tissue accumulation of these byproducts for longer vs shorter periods, respectively, is responsible for the different therapeutic effects of the two regimens. Deposition of AGEs during the initial phase of atherogenesis favours plaque instability and further lesion progression even during subsequent intervention, whereas early inhibition of AGE formation produces smaller and more stable plaques despite cessation of treatment. This interpretation is consistent with the evidence that AGE levels are associated with a rupture-prone plaque phenotype [36]. In this view, our findings suggest that inhibition of AGE formation, achieved with early institution of strict glycaemic control, might be important for preventing cardiovascular events, as observed in the post-trial long-term follow-up of participants formerly assigned to the intensive vs conventional group of the DCCT/EDIC and UKPDS [7, 8]. Conversely, unrestrained generation of AGEs during previous poor glycaemic control might explain the failure of intensive treatment to provide cardiovascular benefit in individuals with long-standing diabetes [4–6].
In conclusion, these data show that DCO protects against diabetes-induced vascular and renal disease and that early treatment is more effective than late treatment against atherosclerosis. In particular, the data indicate that early inhibition of AGE formation attenuates progression of macroangiopathy and favours development of more stable lesions, consistent with the evidence that early aggressive therapy provides protection from cardiovascular events.
Abbreviations
- BCA:
-
Brachiocephalic artery
- CML:
-
N ε-Carboxymethyllysine
- DCCT/EDIC:
-
Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications
- DCO:
-
d-Carnosine-octylester
- fMA:
-
Fractional mesangial area
- GPX1:
-
Glutathione peroxidase 1
- HIF-1α:
-
Hypoxia inducible factor-1α
- MCP-1:
-
Monocyte chemoattractant protein-1
- mGA:
-
Mean glomerular area
- mMA:
-
Mean mesangial area
- PAS:
-
Periodic acid–Schiff
- PG:
-
Prostaglandin
- qRT-PCR:
-
Quantitative real-time PCR
- RAGE:
-
Receptor for AGEs
- RCS:
-
Reactive carbonyl species
- α-SMA:
-
α-Smooth muscle actin
- SOD:
-
Superoxide dismutase
- UKPDS:
-
UK Prospective Diabetes Study
- VSMC:
-
Vascular smooth muscle cell
References
The Diabetes Control and Complications Trial (DCCT) Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study Group (1998) Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Shichiri M, Kishikawa H, Ohkubo Y, Wake N (2000) Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 23(suppl 2):B21–B29
The ADVANCE Collaborative Group (2008) Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 358:2560–2572
The Action to Control Cardiovascular Risk in Diabetes Study Group (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358:2545–2559
Duckworth W, Abraira C, Moritz T et al (2009) Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 360:129–139
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 22:2643–2653
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
del Prato S (2009) Megatrials in type 2 diabetes. From excitement to frustration? Diabetologia 52:1219–1226
Haneda M, Koya D, Isono M, Kikkawa R (2003) Overview of glucose signaling in mesangial cells in diabetic nephropathy. J Am Soc Nephrol 14:1374–1382
Ahmed N (2005) Advanced glycation endproducts—role in pathology of diabetic complications. Diabetes Res Clin Pract 67:3–21
Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R (2008) Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol 153:6–20
Ellis EM (2007) Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 115:13–24
Goh SY, Cooper ME (2008) The role of advance glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab 93:1143–1152
Aldini G, Maffei Facino R, Beretta G, Carini M (2005) Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. Biofactors 24:77–87
Vistoli G, Orioli M, Pedretti A et al (2009) Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. ChemMedChem 4:967–975
Menini S, Iacobini C, Ricci C et al (2012) D-Carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br J Pharmacol 166:1344–1356
Branen L, Pettersson L, Lindholm M, Zaina S (2001) A procedure for obtaining whole mount mouse aortas that allows atherosclerotic lesions to be quantified. Histochem J 33:227–229
Iacobini C, Menini S, Ricci C et al (2009) Accelerated lipid-induced atherogenesis in galectin-3 deficient mice: role of lipoxidation via receptor-mediated mechanisms. Arterioscler Thromb Vasc Biol 29:831–836
Iacobini C, Menini S, Oddi G et al (2004) Galectin-3/AGE-receptor 3 knockout mice show accelerated AGE-induced glomerular injury. Evidence for a protective role of galectin-3 as an AGE-receptor. FASEB J 18:1773–1775
Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK (2000) Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 106:523–531
Bondeva T, Heinzig J, Ruhe C, Wolf G (2013) Advanced glycated end-products affect HIF-transcriptional activity in renal cells. Mol Endocrinol 27:1918–1933
Bharadwaj LA, Davies GF, Xavier IJ, Ovsenek N (2002) l-carnosine and verapamil inhibit hypoxia-induced expression of hypoxia inducible factor (HIF-1 alpha) in H9c2 cardiomyoblasts. Pharmacol Res 45:175–181
Tan SM, Sharma A, Stefanovic N et al (2014) A derivative of Bardoxolone methyl, dh404, in an inverse dose-dependent manner, lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 63:3091–3103
Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM (1997) Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 272:17810–17814
Menini S, Iacobini C, Ricci C et al (2007) Ablation of the gene encoding p66Shc protects mice against AGE-induced glomerulopathy by preventing oxidant-dependent tissue injury and further AGE accumulation. Diabetologia 50:1997–2007
Decker EA, Livisay SA, Zhou S (2000) A re-evaluation of the antioxidant activity of purified carnosine. Biochemistry (Mosc) 65:766–770
Velez S, Nair NG, Reddy VP (2008) Transition metal ion binding studies of carnosine and histidine: biologically relevant antioxidants. Colloids Surf Biointerfaces 66:291–294
Fujita H, Fujishima H, Chida S et al (2009) Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol 20:1303–1313
Gordon S, Lawson L, Rabinowitz S, Crocker PR, Morris L, Perry VH (1992) Antigen markers of macrophage differentiation in murine tissues. Curr Top Microbiol Immunol 181:1–37
Basta G, Schmidt AM, de Caterina R (2004) Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 63:582–592
Aldini G, Orioli M, Rossoni G et al (2011) The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med 15:1339–1354
Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M (1998) Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 339:69–75
Nissen SE, Nicholls SJ, Sipahi I et al (2006) Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 295:1556–1565
Chalmers J, Cooper ME (2008) UKPDS and the legacy effect. N Engl J Med 359:1618–1620
Hanssen NM, Wouters K, Huijberts MS et al (2014) Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur Heart J 35:1137–1146
Funding
This work was supported by the EFSD and Sanofi-Aventis European Research Programme in Micro- and Macrovascular Complications of Diabetes 2011 and by a research grant from the Research Foundation of the Italian Society of Diabetology (Fo.Ri.SID).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
SM and CI contributed to conception and design, acquisition of data, analysis and interpretation of data and critical revision of the article for important intellectual content. CR and CBF contributed to acquisition of data and critical revision of the article for important intellectual content. GP contributed to conception and design, analysis and interpretation of data and drafting the article. All authors gave their final approval to the version to be published. GP is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Additional information
Stefano Menini and Carla Iacobini contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 171 kb)
ESM Fig. 2
(PDF 305 kb)
ESM Fig. 3
(PDF 353 kb)
ESM Table 1
(PDF 62 kb)
Rights and permissions
About this article

Cite this article
Menini, S., Iacobini, C., Ricci, C. et al. Protection from diabetes-induced atherosclerosis and renal disease by d-carnosine-octylester: effects of early vs late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia 58, 845–853 (2015). https://doi.org/10.1007/s00125-014-3467-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3467-6