
Abstract
Articular cartilage damage caused by sports injury or osteoarthritis (OA) has gained increased attention as a worldwide health burden. Pharmaceutical treatments are considered cost-effective means of promoting cartilage regeneration, but are limited by their inability to generate sufficient functional chondrocytes and modify disease progression. Small molecular chemical compounds are an abundant source of new pharmaceutical therapeutics for cartilage regeneration, as they have advantages in design, fabrication, and application, and, when used in combination, act as powerful tools for manipulating cellular fate. In this review, we present current achievements in the development of small molecular drugs for cartilage regeneration, particularly in the fields of chondrocyte generation and reversion of chondrocyte degenerative phenotypes. Several clinically or preclinically available small molecules, which have been shown to facilitate chondrogenesis, chondrocyte dedifferentiation, and cellular reprogramming, and subsequently ameliorate cartilage degeneration by targeting inflammation, matrix degradation, metabolism, and epigenetics, are summarized. Notably, this review introduces essential parameters for high-throughput screening strategies, including models of different chondrogenic cell sources, phenotype readout methodologies, and transferable advanced systems from other fields. Overall, this review provides new insights into future pharmaceutical therapies for cartilage regeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Articular cartilage possesses a unique ability for weight-bearing and joint surface lubrication during an individual’s lifetime. However, cartilage injury may lead to permanent joint disability if not properly treated [1]. The incidence of knee cartilage surgery is 56 per 100,000 person years [2] and osteoarthritis, a worse condition caused by cartilage defects, currently affects approximately 250 million people worldwide [3]. Unlike other tissues, adult articular cartilage is neither innervated nor vascularized, and has extremely poor self-renewal capacity [4]. Articular cartilage consists of abundant extracellular matrix (ECM) but limited chondrocytes, which occupy only 1–3% of the total tissue volume [5]. Thus, defective tissue is rarely replaced by functional cartilage and additional therapeutic interventions are often required.
In recent years, the fields of cell-based therapy and tissue engineering have made remarkable improvements in the treatment of cartilage damage [6]. However, the clinical outcomes are unsatisfactory and challenges still exist. First, functional chondrocytes for articular cartilage recovery are lacking, hindering the application of chondrocyte implantation and necessitating the development of alternative cell sources. Second, chondrocytes with pathological phenotypes, representing either degradative or fibrotic cartilage ECM, cause functional impairment in cartilage diseases. This demands effective manipulation of pathological cellular phenotypes or processes.
Efforts have been made to address these problems. Among them, chemical compounds with molecular weights in the range of 0.1–1 kDa have gained increased attention as powerful tools for tissue regeneration and cell-fate control. Because of their small sizes, they easily cross the outer plasma membrane to target extracellular components such as cell surface receptors, protein domains attached to cell membranes, such as glycoproteins, and intracellular proteins such as kinases [7]. As a result, small molecular weight chemical cocktails have been demonstrated to drive direct reprogramming of fibroblasts into induced pluripotent stem cells (iPSCs), neural progenitor cells, cardiomyocytes, and other somatic cells [8,9,10]. Small molecular drugs have also been adopted for the treatment of various diseases, including cancers [11], fibrotic diseases [12], and asthma [13]. From a clinical perspective, small molecular drugs have considerable advantages: they are classified as low-immunogenic molecules (compared with biomacromolecules), and are easily synthesized, stored, transported, and standardized [14]. They offer rapid and temporal control over protein function, allowing their effects to be fine-tuned based on different concentrations and combinations [15]. In terms of drug discovery and design, small molecules typically mimic the interactions formed by peptides and place groups into hotspot pockets with a smaller volume and tighter interaction in space [16]. Therefore, this review will highlight current achievements in the use of small molecules for cartilage regeneration, as well as rational high-throughput screening strategies used for the discovery of small molecules that facilitate chondrocyte generation or manipulation of chondrocyte degenerative phenotypes.
Generating chondrocytes using small molecules
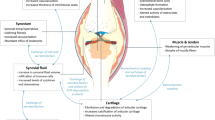
Great advances have been made in the development of cartilage cell-based therapies and tissue engineering since the 1990s, providing new perspectives for achieving cartilage function recovery using alternative chondrocyte sources. Technically, chondrocytes can be expanded in vitro, differentiated from stem/progenitor cells, and reprogrammed from somatic cells. In other words, cell sources for cartilage regeneration include chondrocytes (autologous or allogeneic), stem cells (multipotent cells or pluripotent cells), and other trans-differentiated somatic cells [6]. Here, we discuss the routines to obtain functional chondrocytes, which can be effectively facilitated using small molecular compounds (Fig. 1).

Major applicable routines of small molecules for generating functional chondrocytes. BMSCs bone marrow mesenchymal stem cells, iPSCs induced pluripotent stem cells, KGN kartogenin, VPA valproic acid
Small molecules facilitating chondrogenesis of stem/progenitor cells
Stem cells with chondrogenic differentiation ability are of great interest in the field of cartilage regeneration. For example, mesenchymal stem cells (MSCs) have gained increasing popularity as an alternative cell source for the treatment of cartilage injuries in clinical trials [17]. However, many studies have reported inferior outcomes. There are still gaps in the precise regulation of MSC chondrogenesis because MSCs can differentiate into fibrochondrocytes, resulting in a mixture of fibrous cartilaginous tissue [4].
A 2012 study highlighted the exploration of a small molecule, kartogenin (KGN), which was dubbed a “game-changer in regenerative medicine” [18] for its ability to induce human MSC chondrogenesis in vitro by regulating CBFβ-RUNX1. It was demonstrated to be an efficacious disease-modifying OA drug (DMOAD) in two OA mouse models as it did not enhance cartilage hypertrophy or calcification [19]. KGN is extensively used as a chondroprotective agent against cartilage degeneration and injury. To establish a controllable drug delivery system, Wang et al. combined KGN with PLGA microspheres for the superficial repair of articular cartilage in a rabbit model [20]. KGN can also be conjugated to Pluronic F127 thermoresponsive nanospheres with the anti-inflammatory drug diclofenac (DCF) [21], or to hyaluronic acid (HA)/PEG to suppress OA progression in rats [22]. Recently, other groups have conjugated KGN to polyurethane nanoparticles; their intra-articular injection increased type II collagen expression levels and decreased type I collagen expression, alleviating OA in a rat model [23]. In 2013, the compound TD-198946, discovered by Yano et al., showed remarkable promotion of chondrogenic differentiation of mouse stem cells and the cartilage progenitor cell line ATDC5 by activating Runx1 to upregulate Col2a1 and Acan gene expression without promoting chondrocyte hypertrophy [24, 25]. They also showed that, in combination with TGF-β3, TD-198946 improved chondrogenesis of human synovium-derived stem cells [26].
In addition, through high-throughput screening, Hwang et al. discovered a protein kinase A inhibitor, H-89, which enhanced the chondrogenic differentiation ability of rat MSCs [27]. Using the structure of H-89, they synthesized a series of analogs and showed that, among them, compound 6 had the strongest ability to induce chondrogenesis of adipose-derived stem cells (ADSCs). Articular cartilage thickness was maintained with increased aggrecan levels by the injection of compound 6 or compound 6-treated-ADSCs in collagenase II-induced osteoarthritis rat models [28]. Heck et al. reported that GW0742, a PPAR-δ agonist, enhanced human MSC chondrogenesis and alleviated cartilage fibrosis/inflammation in an in vitro model by stimulating type II collagen/glycosaminoglycan secretion [29]. As Sirtuin-1 (SIRT1) is required for stem cell chondrogenesis, the SIRT1 activator resveratrol partially inhibits the inflammatory effect of IL-1β on human MSC differentiation in vitro [30] and promotes human ADSC proliferation in a three-dimensional chondrogenic culture [31]. The small molecule BIO (6-bromoindirubin-3-oxim) inhibits glycogen synthase kinase-3 (GSK-3) in the Wnt pathway and can upregulate cartilage-specific genes in mouse MSC in in vitro chondrogenesis [32]. Using a Col2GFP-ATDC5 system, Hojo et al. identified oxytetracycline, which acts as a chondrogenic inducer in a BMP-dependent manner and suppresses chondrocyte hypertrophy [33]. Similarly, the small molecule tetradecylthioacetic acid (TTA) was shown to improve ECM production in a fluorescently labeled collagen-binding probe system in ATDC5 cultures [34].
With the rapid development in stem cell-based regenerative medicine, small molecules are promising tools for regulating stem cell behavior. However, challenges still exist in the precise regulation of chondrogenesis, particularly the functional phenotype of stem cell-derived chondrocytes. There is a need to uncover relevant critical targets for optimizing chondrogenesis efficiency, to harvest hyaline chondrocytes, rather than fibrotic or hypertrophic chondrocytes.
Small molecules facilitating chondrocyte expansion
To obtain sufficient cell numbers for chondrocyte implantation, in vitro expansion is necessary. However, chondrocyte dedifferentiation, representing a loss-of-function phenotype of expanded chondrocytes, results in fibrocartilage formation and has severely hindered the clinical application of chondrocyte implantation [35]. This limitation motivated Kobayashi et al. to select A-674563 from 5822 compounds based on mouse chondrocyte dedifferentiated phenotypes. Based on the RNA sequencing data, A-674563 could inhibit the degradation of SOX9 protein by activating the gene that induces protein deubiquitination [36]. Simvastatin, a lipid-lowering agent used to treat hypercholesterolemia [37], has the ability to maintain the phenotype of passaged human chondrocytes by stimulating the expression of SOX9 [38]. Clodronate, a bisphosphonate, [39] exerts an anabolic effect on articular chondrocytes [40]. It can upregulate the expression of SOX9 in an in vitro pathological human MSC model, reducing osteoarticular pain and improving mental and physical performance in human patients 6 months after treatment [41].
Although the regulation of SOX9 is critical to the chondrocyte phenotype, the maintenance of cell viability and ECM homeostasis are also necessary for large-scale chondrocyte expansion. Paradoxically, a higher expression of degradative enzymes such as matrix metalloproteinase 3 (MMP3) [42], improves the outcome of chondrocyte expansion. The mechanism is potentially associated with MMP-mediated remodeling of the ECM [43]. In contrast, the expression of MMPs is tightly linked with chondrocyte functional phenotype loss, contributing to dedifferentiation during in vitro culturing [44].
Due to the lack of basic knowledge on chondrocyte dedifferentiation, the ideal approach for effectively maintaining chondrocyte phenotype has not been determined. More in-depth investigations are required before practical application of small molecular compounds. Developing chemical compounds that facilitate chondrocyte function is important, as these potential drugs are likely to have a direct effect on chondrocytes, enabling their adoption in cell-free cartilage repair at joint surfaces in situ.
Small molecules facilitating reprogramming to generate chondrocytes
It has been shown that induced pluripotent stem cells (iPSCs) reprogrammed from patient somatic cells such as fibroblasts can form scaffoldless hyaline cartilaginous tissue in vivo, which can then be integrated into native cartilage [45]. This offers new options for regenerating cartilage tissue without ethical issues. Initially, the generation of iPSCs required virus-mediated expression of defined transcription factors (TFs) [46]. However, accumulating studies are reporting that chemical cocktails can improve reprogramming efficiency and even replace some of the defined factors [47]. Hou et al. used a cocktail of seven small molecule compounds (valproic acid (VPA), tranylcypromine, CHIR99021, 616,452, forskolin, 3-deazaneplanocin, and PD0325901) to achieve virus-free reprogramming of mouse fibroblasts into iPSCs in a step-wise manner [8]. Because the induction efficacy and the long period required for chemical reprogramming were still problematic for practical application [48], Zhao et al. significantly improved the efficiency of chemical reprogramming from somatic cells to chemically induced pluripotent stem cells (CiPSCs) and identified unique intermediate cells (XEN-like state) [49]. They induced mouse embryonic fibroblasts (MEFs) using an optimized protocol and chemical combinations were adopted as follows: VPA, CHIR99021, 616,452, tranylcypromine, forskolin, AM580, and EPZ004777 in stage 1; VPA, CHIR99021, 616,452, tranylcypromine, forskolin, AM580, DZNep, 5-aza-dC, and SGC0946 in stage 2, and CHIR99021 and PD0325901 in stage 3. In 2019, they enhanced the reprograming process by fine-tuning the chemical treatment and generated 500 CiPSC colonies from 100,000 re-plated cells from stage 1 [14], demonstrating that chemical reprogramming is a unique and promising strategy for generating functional iPSCs.
Somatic cells can be directly reprogrammed into other types of mature cells by bypassing pluripotency, meaning that chondrocytes can be generated through transdifferentiation. Chen et al. conducted a proof-of-concept study using a combination of five small molecular drugs (VPA, CHIR98014, 616,452, TTNPB, and Celecoxib) and induced MEFs into three-dimensional fibrocartilaginous tissues with in vivo mechanical function [50]. This approach avoids the pluripotent state, poses a lower risk of tumor development, and shortens the induction period, and may present a new opportunity for future pharmaceutically engineered cartilage. However, achieving cell-fate conversion of human somatic cells into functional hyaline chondrocytes via chemical reprogramming remains uncertain. Evidence suggests that more stringent chemical conditions are required for the reprogramming of human adult cells, but this will require a comprehensive understanding of human cell plasticity. More importantly, the low efficiency and long-term reprogramming period are technical concerns that still need to be addressed before any clinical application.
Several studies have demonstrated the convenience of using small molecular compounds for in vitro generation of functional chondrocytes. However, large gaps remain between the establishment of a well-recognized standard and the development of an in-house approach. First, most of the screening described above were conducted with reference to phenotypic measurements, not target-based drug design, and finding a hit was likely coincidental, although the targets could be identified using a deconvolution process [51]. In our opinion, the targets of chondrogenesis induction and reprogramming are highly relevant to major developmental signaling pathways. For example, the CBFβ-RUNX1 complex activates important genes involved in the differentiation of MSCs into cartilage. KGN can competitively bind to human cytosolic filamin A, a key binding partner of CBFβ, to displace CBFβ and accelerate its dimerization with transcription factor RUNX1 in the nucleus [52]. Similarly, TD-198946 strongly induces chondrogenic differentiation by regulating RUNX1 [24]. Cellular programs resembling cartilage development, including the activation of Bmp6, Tgfb2, and Wnt4, were demonstrated in a direct reprogramming study [50]. These compounds exert their effects by stimulating differentiation. SOX9 appears to be a common target in the modulation of the dedifferentiated phenotype [36, 38, 41], functioning as a master transcription factor determining chondrocyte differentiation and phenotype [53]. However, the main molecular events involved in chondrocyte expansion and dedifferentiation are still unclear, making it difficult to efficiently regulate the chondrocyte phenotype in long-term passaging.
Thus, boosting cartilage development may benefit chondrocyte generation. Nevertheless, there is a big caveat in developing a quality-control system for the cell source. Given that chondrocytes are not identical in different regions [54], a precise identification of the differentiation state or subtype is also urgently needed. Therefore, single-cell transcriptomic datasets are a powerful tool for dissecting differentiation trajectories and chondrocyte sub-identity [55,56,57]. The emergence of new techniques has made it more feasible to precisely evaluate cell quality and investigate the chemical condition of the medium used for differentiating cells into correct orientations.
Small molecules for manipulating chondrocyte pathological phenotype
Cell implantation is effective in the treatment of early focal cartilage defects [58]. However, OA is a more complex condition. Pharmaceutical interventions are usually administered to patients with OA during the painful process before disease end-stage as they contribute to symptom control, making them increasingly popular, with more than 1300 clinical trials registered worldwide in the last 10 years (https://clinicaltrials.gov). DMOADs are designed to slow OA deterioration and inhibit structural disease progression [59], but are still at the conceptualization stage.
Small molecular compounds may play a vital role in the development of DMOADs capable of controlling early OA in a more precise and defined manner. Here, we summarize a list of over 170 representative small molecular OA drugs previously or currently under clinical investigation (Table 1), according to the US National Institutes of Health database. Most of them are analgesics or anti-inflammatory agents. Understanding pharmacological activities that specifically block pathological processes and inhibit certain OA pathways is important for the manipulation of chondrocyte pathological phenotypes. In this section, we discuss potential OA targets and the corresponding small molecular drugs (Fig. 2).

Potential OA targets and representative developing small molecular drugs. BMSCs bone marrow mesenchymal stem cells, ECM extracellular matrix, IL-1β Interleukin 1β; TNF-α tumor necrosis factor α, MMP13 metalloproteinase 13, ADAMTS-5 a disintegrin and metalloproteinase with thrombospondin motifs 5
Small molecules inhibiting inflammation
OA is characterized by low-grade inflammation and chronic joint pain. Various inflammatory cytokines, including interleukin 1(IL-1) and tumor necrosis factor (TNF) α, may play a vital role in OA progression, and a direct inhibition of inflammation controls pain and reduces cartilage matrix degradation [60]. Here, several representative anti-inflammatory candidates are introduced based on inflammation-induced models or OA inflammatory pathways.
Diacerein is a symptomatic, slow-acting drug with anti-inflammatory, anti-catabolic, and pro-anabolic effects on cartilage in rat and dog models [61, 62]. The European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) confirmed that the effect of diacerein in clinical trials was similar to that of non-steroidal anti-inflammatory drugs (NSAIDs), but with fewer side effects [63]. The mechanism by which diacerein inhibits inflammation and prevents cartilage ECM degradation is potentially associated with the inhibition of IL-1β converting enzyme, the reduction of IL-1β receptors on chondrocytes, and the increase of IL-1 receptor antagonists. Diacerein was also shown to affect matrix biosynthesis and subchondral bone remodeling in an arthritis sheep model [63, 64].
Necrostatin-1(Nec-1) is an inhibitor of receptor-interacting protein kinase 1 (RIPK1) and specifically inhibits the phosphorylation of RIPK1, which regulates inflammation and cell death. Liang et al. found that Nec-1 dramatically suppressed the catabolic effect induced by IL-1β in mouse chondrocytes, including the downregulation of MMP and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS-5). Nec-1 also significantly reduced the destruction of OA cartilage in a destabilized medial meniscus (DMM) mouse model [65].
The transcription factor NF-κB is a key molecule in classical inflammatory pathways [66]. Salubrinal, a specific inhibitor of eIF2α phosphatase enzymes, attenuated cartilage degradation in mice with OA by inhibiting NF-κB [67]. NF-κB is also regulated by mechanical forces. Gremlin-1 can be elevated by mechanical stimulation in OA, which subsequently activates NF-κB signaling and promotes matrix degradation by MMP13 and ADAMTS-5 [68]. NSC23766 and EHT1864 are inhibitors of Rac1 (an upstream regulator of Gremlin-1) and can downregulate the expression of Gremlin-1 [68]. Indeed, Rac1 was found to be highly expressed in human and mouse OA cartilage, while its intrinsic inhibitor, OCRL, was inhibited [69, 70]. NSC23766 successfully suppressed chondrocyte hypertrophy and ADAMTS-5 expression in an OA mouse model [69].
Prostaglandin E2 (PGE2) is an important mediator of inflammation [71]. Otsuka et al. investigated the effect of PEG2 on the repair of chondral and osteochondral defects in rabbits using a selective agonist of the PEG2 receptor EP2 (ONO-8815Ly). They demonstrated that microspheres loaded with ONO-8815Ly could promote the regeneration of COL2-positive tissues, particularly at 12 weeks post-operation [72]. Similarly, the oral administration of KAG-308, a selective agonist of the PEG2 receptor EP4, suppressed the development of OA in mice by reducing chondrocyte hypertrophy and TNF secretion [73]. In addition, NSC117079, which targets Phlpp protein phosphatases, attenuated mechanical allodynia and slowed articular cartilage degradation in a murine OA model. It also increased the production of cartilage ECM in human OA cartilage explants [74].
Bradykinin (BK) is an intrinsic nonapeptide for vasodilation and has been identified as an inflammatory mediator in OA [75, 76]. BK mediates chronic inflammatory responses, inflammatory factor release, and NF-κB signaling pathway activation through the B2 receptor. Icatibant and fasitibant are two inhibitors of BK B2 receptors [75]. A clinical trial (NCT02205814) demonstrated that fasitibant had a moderate effect on OA.
The renin–angiotensin system (RAS) is associated with metabolic bone diseases and arthritis [77, 78]. RAS components were also expressed in hypertrophic or IL-1β/TNF-α stimulated chondrocytes [79], and Aliskiren, a RAS inhibitor, suppressed IL-1, TNF-α, and ECM degradation and chondrocyte hypertrophy in an OA rat model [80].
Transcriptional factor c-Fos/activator protein-1 (AP-1) has been shown to regulate inflammation and matrix degradation [81, 82]. In 2008, Aikawa et al. designed a de novo small molecular inhibitor of c-Fos/AP-1, T-5224, using three-dimensional (3D) pharmacophore modeling. Notably, administration of this compound prevented or resolved arthritis by reducing inflammatory cytokines and MMPs in mice [83]. In 2018, the same research group demonstrated that T-5224 prevented articular cartilage destruction by inhibiting MMP13 expression in a C57Black6/J OA model and prevented OA osteophyte formation [84].
Since rheumatoid arthritis (RA) is also characterized by inflammatory symptoms, RA drugs can potentially function like DMOADs and inhibit inflammation and pain. Methotrexate (MTX), a drug targeting synovitis in RA, was found to relieve pain and improve joint function in patients with OA after a 6-month treatment course. However, the outcome after 12 months of treatment was not significantly different from that of the placebo group [85]. Ketamine is an anesthetic clinically used to treat chronic pain [86]. It was recently shown to alleviate RA by antagonizing the N-methyl-D-aspartate (NMDA) receptor and inhibiting inflammatory and Th17 cellular responses [87,88,89]. In an OA rabbit model, ketamine protected articular cartilage from joint inflammation [88].
Although small molecules had great anti-inflammatory effects on OA, careful consideration must be given to the clinical heterogeneity of OA, as only parts of the patients are in the condition of acute high-level inflammation. Numerous studies have shown that simply blocking inflammation may not initiate the regenerative processes in cartilage [90]. Therefore, there is a need to increase our understanding of OA pathology and clarify the role of inflammation in tissue regeneration.
Small molecules for treating pain from OA
There is a strong association between inflammation and pain in patients with OA and several studies have suggested that pain from OA is driven by inflammation [91,92,93]. This theory has not been conclusively confirmed due to the complexity of measuring pain and OA pathogenesis [94]. However, as a major symptom of OA, pain is an important target and the primary outcome measure of OA disease management. There is an urgent need to develop analgesics with satisfactory clinical outcomes for many patients. Generally, both NSAIDs and glucocorticoids can target inflammation to treat OA pain. However, their efficacies are often short-lived and there are adverse effects associated with their long-term use [95,96,97,98,99]. Recent years have witnessed several promising candidates for pain control, including antibodies against nerve growth factor and some chemical small chemical molecules. These are discussed below based on their molecular targets.
G-protein coupled receptors (GPCRs) are highly druggable targets and are expressed in almost all cells that constitute the components of pain signaling in humans [100]. They include opioid receptors. Morphine is a strong analgesic that binds to opioid receptors and blocks the transmission of nociceptive signals, but its use is restricted due to safety concerns. Miller et al. provided a good summary of the drugs in this class [101]. Among them, CR845 has been shown to benefit OA. CR845 is a second, peripherally restricted, selective agonist of kappa opioid receptors [102]. In a phase 2B clinical trial (NCT02944448), oral CR845 (Cara Therapeutics) was well tolerated, with a statistically significant effect on pain. Another new drug, cebranopadol, targets both nociceptin/orphanin FQ peptide receptor and m opioid receptor. It showed more promising outcomes and better safety profiles in clinical trials compared with traditional opioids [103, 104]. However, results from clinical trials on OA are yet to be reported.
Apart from GPCRs, ion channels are also well-studied components in the pain signaling pathway [105], including OA chronic pain [106]. In this class, CNTX-4975, a highly purified, synthetic trans-capsaicin targeting the transient receptor potential vanilloid 1, has been tested in clinical trials. Based on results from a 24-week, randomized, double-blind study [107], a single injection of CNTX-4975 had analgesic effects on pain associated with walking, knee stiffness, and physical function. Additionally, NEO6860, an oral transient receptor potential vanilloid 1 blocker, showed positive clinical outcomes in a proof-of-concept study [101].
In conclusion, analgesics are the most commonly used OA drugs (Table 1), as pain relief remains the most direct need, considering patients’ perspectives during OA therapy. Unfortunately, even after decades of research in this field, drugs that can delay or prevent OA progression are still unavailable. One possible reason for this is that pain killers and disease-modifying drugs are often investigated separately in different fields of study [94, 108]. Importantly, we still lack evidence showing that analgesics can induce structural changes.
Small molecules regulating metabolism
Increasing evidence indicates that lipid metabolic disorders are relevant to the development of OA [109,110,111]. Statin drugs competitively bind to HMG-CoA reductase [112], thereby inhibiting cholesterol synthesis. In 2014, Yamashita et al. discovered that lovastatin treatment could rescue the FGFR3 skeletal dysplasia phenotype in a patient-derived iPSC chondrogenesis model [113]. Interestingly, recent studies have reported that statins have potential as chondroprotective reagents. Pathak et al. discovered that atorvastatin protects rat OA cartilage explants from IL-1β-stimulated degeneration in vitro [114]. Yudoh reported that, in a mouse model which spontaneously develops OA, statins could reduce articular degeneration [115]. However, clinical outcomes of statin therapy for OA remain controversial, and only partially indicate that statin use is associated with a lower risk of knee OA progression and knee pain [116,117,118]. The pharmacological mechanisms underlying these effects may be associated with the serum lipid control and anti-inflammatory ability of statins [118].
Estrogen-related receptors (ERRs) are involved in the metabolic activities of organs such as the liver, heart, and skeletal muscles, and are also associated with type II diabetes and metabolic syndrome [119, 120]. One of the ERR isoforms, ERRγ, was shown to be highly expressed in the cartilage of human patients with OA and in mouse models, and promoted OA development by upregulating MMP3 and MMP13. In the presence of the small molecule inhibitor GSK5182, ERRγ activation was inhibited and the OA phenotype was markedly ameliorated in a temporomandibular joint OA rat model [121].
Mitochondrial dysfunction has been observed as a typical feature of OA chondrocytes and is a promising pharmacological target for the maintenance of cartilage homeostasis [122, 123]. Inflammation is thought to be responsible for mitochondrial impairment, resulting in the increased production of inducible nitric oxide (iNO) by chondrocytes. Using an IL-1β-treated OA chondrocyte model, Eitner et al. showed that activation of cAMP by forskolin could prevent both iNO release and mitochondrial dysfunction [124]. However, the compound’s effect on the animal model was not confirmed.
Oxidative stress is a major factor that can cause mitochondrial dysfunction [125] and increase MMP, collagenase, and gelatinase levels in OA. Hence, antioxidation can be regarded as a therapeutic strategy for OA. Curcuminoids are strong, natural antioxidants. A randomized controlled trial showed that curcuminoids could increase serum superoxide dismutase (SOD) activity and decrease malondialdehyde (MDA) concentrations to relieve oxidative stress in OA, although its direct effect was not validated [126]. In 2019, Shi et al. discovered a potential DMOAD, BNTA, which stimulated ECM expression and suppressed inflammatory factors. In a trauma-induced rat OA model, intra-articular injection of BNTA delayed disease progression through superoxide dismutase 3 (SOD3) [127]. This study confirmed that SOD3 can act as a disease-modifying target. Withaferin A (WFA) is a plant extract that has been utilized as a drug in India. Choudhary et al. showed that treatment with WFA upregulated the expression of SOX9 and inhibited reactive oxygen species (ROS) synthesis, mitochondrial depolarization, and chondrocyte apoptosis induced by IL-1β or TNF-α. Daily oral administration of WFA for 28 days alleviated articular cartilage erosion and improved the subchondral bone structure in rats [128].
Mitochondria are widely distributed in cells and are in charge of complex processes required for cell survival, thus many endogenous protective pathways are involved in the regulation of mitochondrial function. Autophagy and mitophagy are, therefore, potential targets. Autophagy is a degradative process that breaks down subcellular content in response to various stimuli and is a protective behavior that prolongs cell survival [129]. Autophagy impairment has been associated with OA [130]. Notably, trehalose, a natural disaccharide, can exert cell protective effects under various stress conditions in many cells [131] by activating autophagy through the mammalian target of rapamycin (mTOR)-independent pathway. In a study of OA, trehalose promoted autophagic flux and attenuated mitochondrial dysfunction, and this was validated using a DMM mouse model [132]. Similarly, mitophagy is a quality-control process that governs basal autophagy by targeting and eliminating dysfunctional mitochondria [129]. Urolithin A is a natural dietary compound that induces mitophagy and thus prevents the accumulation of dysfunctional mitochondria [133]. Exploring its effect on a mechanical loading model using human chondrocytes in a hydrogel showed that Urolithin A protected chondrocytes from mechanical injury and supported cartilage homeostasis [134].
Despite the importance of mitochondria in tissue homeostasis, few mitochondria-targeting drugs are available in OA clinical trials [135]. This may be due to lack of non-invasive methods for measuring mitochondrial function in human cartilage in vivo. Interestingly, Harms et al. used the protoporphyrin IX triplet state lifetime technique to noninvasively measure mitochondrial oxygen tension (mitoPO2) in a rat model [136], providing a potential tool for clinical diagnosis. In addition, extensive investigations are needed to identify OA metabolic subtypes.
Small molecules regulating epigenetic factors
Various physiological and pathological processes are epigenetically regulated, including skeletal development and OA occurrence [137,138,139,140,141], indicating that pharmaceutical interventions targeting epigenetics are promising as OA therapies. H3K27 methylation is generally gene repressive [142]. Enhancer of zeste homolog 2 (EZH2) is an H3K27 methylase that is highly expressed in some human patients with OA [143]. The small molecule EPZ005687 inhibited EZH2, downregulated ADAMTS-5 and MMP13 expression, and ameliorated chondrocyte hypertrophy in a murine model [139]. Consistently, the expression of H3K27 demethylase KDM6B was reduced in human patients with OA, and KDM6B knockout in mice accelerated OA progression [138]. The KDM6B (JMJD3) inhibitor, GSK-J4, prevented ex vivo cartilage destruction in humans and suppressed disease development in a DMM-induced OA mouse model [140, 144].
Thus, it is clear that epigenetic factors play an important role in OA, but knowledge on whether or how they exacerbate the disease is currently lacking. A continuous evolution of the technique in this field may provide high-resolution insights and a comprehensive understanding, enabling manipulation of specific targets at specific stages of OA.
Small molecules targeting other pathogenic pathways
Wnt signaling is one of the most essential pathways for chondrogenesis, osteogenesis, and OA progression [145,146,147]. Deshmukh et al. performed high-throughput screening and found that SM04690 significantly inhibited Wnt signaling, promoted chondrogenesis of human mesenchymal stem cells (MSCs), and suppressed the expression of members of the MMP family in chondrocytes [148]. A rodent model showed that this small molecule could improve cartilage regeneration, with increased cartilage thickness [103]. The underlying molecular mechanism was identified in 2019, and it suggested that SM04690 (lorecivivint) modulated the Wnt pathway by inhibiting CLK2 and DYRK1A [149]. The drug demonstrated general safety and tolerance without serious systemic adverse effects in phase I and phase II clinical trials. The study also showed clinical improvements in pain and joint function following a single intra-articular administration of 0.07 mg lorecivivint [150, 151]. No results are currently available for multiple rounds of administration of the drug. However, if the ongoing phase III trials exhibit good clinical efficacy profiles and marginal side effects [152], lorecivivint will become a promising DMOAD approved for knee OA therapy compared with intra-articular corticosteroid and hyaluronate injections [153]. Lietman et al. also reported that inhibiting Wnt/β-catenin signaling could ameliorate OA [154]. They successfully reduced cartilage degeneration and synovitis in a mouse model using a small molecular weight inhibitor, XAV-939 [154].
EGFR signaling is vital in cartilage development and OA [155,156,157,158]. EGFR was recently shown to be activated in a subpopulation of patients with OA. The specific inhibitor, Gefitinib, which has been used in the treatment of non-small cell lung cancer (NSCLC) for decades, inhibited EGFR activation and re-activated chondrocyte autophagy, restoring the balance between ECM synthesis and degradation. This was demonstrated and validated in a study of Col2a1-creERT2 conditional knockout mice [159]. However, another study illustrated that EGFR is important for articular superficial layer maintenance and inhibiting EGFR accelerated OA occurrence in mice lineages specifically overexpressing heparin-binding EGF-like growth factor (HBEGF) [160].
As these pathways are essential for cell survival and growth, simply inhibiting them may raise safety concerns for practical use. However, designing a delivery system to suppress a target in a specific chondrocyte subpopulation is doable. It also highlights the importance of single-cell analysis in OA pathology, which uncovers significant changes in cell cluster distribution.
Small molecules targeting bone remodeling
Subchondral bone undergoes structural remodeling as OA progresses [161] and is a potential a drug target. Strontium ranelate (SrR) regulates the balance between bone formation and resorption. Recently, SrR was shown to be capable of ameliorating OA with clinical improvement in joint structure and symptom control [162,163,164]. However, a separate study found that SrR did not improve cartilage in an OA porcine model and instead led to osteophyte overgrowth [165]. Odanacatib (MK0822) is a cathepsin K (CatK) inhibitor used to treat osteoporosis. High CatK expression in OA leads to cartilage matrix degradation, making it a promising therapeutic target for DMOADs [166]. Unfortunately, a 2006 clinical trial studying the effect of treating OA using odanacatib was terminated [167]. Other clinical profiles indicated that the development of odanacatib was stopped due to undesirable adverse effects such as higher incidence of cardio-cerebrovascular events [168]. Another cathepsin K inhibitor, MIV-711, was also tested in a clinical trial, but no significant effect was observed.
Increased attention has been paid on subchondral bone remodeling in OA programs. However, the complex interactions between cartilage and bone are still poorly understood. Future advancements in this field will rely mainly on the development of new methodologies for clinical diagnosis for identifying OA subtypes relevant to bone remodeling. The corresponding small molecules may work as potential DMOADs or be used in combination with agents targeting cartilage in OA pharmacological treatments.
Nevertheless, OA is not only caused by cartilage deterioration or bone remodeling, but also shows a complex and heterogeneous syndrome with multiple tissue involvement [169]. Therefore, it is difficult to design a “one fits all” molecule able to treat all tissues, including cartilage, bone, ligaments, synovium, and meniscus. Interestingly, a few such cases exist. For example, efforts have been made to explore the effect of KGN, a star molecule in cartilage regeneration, on other musculoskeletal tissues. As confirmed in various animal models, KGN serves as an efficacious therapy in the treatment of meniscus damage, intervertebral disc degeneration, and tendon-bone interface injury [170,171,172]. However, until meaningful clinical outcomes have been shown, designing a “one fits all” molecule suitable for the whole joint remains a considerable challenge.
Strategies for screening small molecules for cartilage regeneration
To efficiently develop new small molecular drugs, high-throughput and high-content screening have become important steps in the drug discovery workflow [173]. High-throughput systems allow automatic, rapid, sensitive, and specific screening of large-scale chemical libraries, taking advantage of advanced image equipment and algorithms. In this section, we briefly introduce the key elements in the primary screening of new drugs for cartilage regeneration (Fig. 3), including in vitro screening models, specific phenotype readout, and advanced methodologies that prospectively have an impact on future chemical cartilage regeneration.

Rational screening strategy of small molecular drug discovery for cartilage regeneration
In vitro screening models
Chondrogenesis models
Cells capable of chondrogenic differentiation may become an additional source for cell-based therapy in cartilage injury intervention. It is, therefore, reasonable to set up chondrogenesis models. Practically, easily obtained cells and simple models are widely used. For example, in the discovery of KGN [19], the authors used primary human bone marrow MSCs in their chondrogenesis model. In their image-based high-throughput screening, human bone marrow MSCs were seeded in 384-well plates to form chondrogenic nodules. Early cell condensation on day 3 was positively stained using rhodamine B. This fluorescent tag did not identify chondrocyte ECM, but distinctly labeled cell condensation in the imaging system. In this way, KGN was identified as a hit among 22,000 structurally diverse molecules. In other studies, the chondrocyte progenitor cell line, ATDC5, is the most extensively adopted cell source because it is proliferative and stable under expansion. The ATDC5 cell line is derived from mouse teratocarcinoma and can form cartilage-nodule-like cell aggregates in the early stages of chondrogenesis induction [174]. Shi et al. treated ATDC5 cells with 2320 natural and synthetic small compounds for 7 days [127]. The cartilage-specific matrix component, proteoglycan, was assessed using Alcian blue. Additionally, the initial identification of TD-198946, SNX19, and oxytetracycline all relied on the ATDC5 screening system [24, 34, 175]. iPSCs are promising cell sources for drug screening because of their genetically defined chondrogenic differentiation potential [176]. Willard et al. used high-density micromass culture to chondrogenically differentiate mouse iPSCs and establish an iPSC-derived cartilage OA model [177]. Using a high-throughput screen, they found that the NF-κB inhibitor, SC-514, effectively reduced cartilage damage in response to IL-1α.
Dedifferentiation model
In addition to stem/progenitor cells, primary chondrocytes are useful tools for exploring the molecular features of cartilage degeneration and regeneration, although chondrocyte dedifferentiation remains unsolved [178]. Thus, primary isolated chondrocytes were less likely to be used in high-throughput screening. However, Kobayashi et al. used induced chondrogenic cells (iChons), which were trans-differentiated from mouse dermal fibroblasts by defined factors [179]. They exploited doxycycline-treated Col11a1 EGFP iChons as a dedifferentiation model in the screening assay and identified a novel compound which inhibited SOX9 degradation [36].
3D organoids
Significant progress has been made in organoids research. Specifically, the concept of organ-on-a-chip has spread in a range of fields, including cancer therapies, as well as studies of the liver, blood vessels, and lungs [180]. Organoids are believed to be more sensitive to individual-specific drug screening. Moreover, organoid models are estimated to be cost-effective, simulate in vivo tissues, and respond more accurately to candidate drugs. Cartilage is susceptible to mechanical microenvironmental stimuli. Nevertheless, current drug screenings are often performed in 2D monolayer cultures because 3D cultures require a larger number of cells and longer Z-stack imaging. Tuan et al. [181] introduced an adult MSC-based 3D microsystem of the osteochondral complex for elucidating the pathogenesis of degenerative joint diseases and assessing the efficacy of potential therapeutics. The design of this 96-well microsystem involves a layered osteochondral tissue (bone, osteochondral interface, cartilage, or synovium) and mechanical loading modalities. The microtissue mimics tissue interactions and cellular responses to mechanical and inflammatory stimuli [181].
Cartilage explant models
Cartilage tissue explants are also an alternative model frequently used in cartilage regeneration [182,183,184,185]. Compared with isolated cells, they are estimated to simulate in vivo tissue and can serve as a “native cartilage” positive control [177]. When used in high-throughput screening, standardized ex vivo models provide a valuable platform. For instance, Schwab et al. established an ex vivo culture system for the long-term assessment of cartilage repair treatment strategies [186]. They isolated pig osteochondral biopsies and inserted them into an in-house developed six-well plate, where the cartilage and bone were cultured in separate compartments containing specific media. The viability, matrix content, and structural and mechanical properties of this model were maintained for at least 56 days, making the system a suitable tool for the characterization of cartilage regeneration treatment. Because the limited source of human joint tissue poses a challenge in large-scale experiments, Spinnen et al. developed a novel method that enables the preparation of 100 explant slices from a single human joint sample [187]. They used an orthopedic tissue punch to create an osteochondral cylinder punch (20 × 10 mm) from a tibial plateau and inserted the cylinder into a custom 3D-printed microtome. The cylinder was then divided into eight disc-shaped cuts with a thickness of 500–800 μm. The slices showed positive sensitivity to pharmacological testing after being cultured for three weeks. These findings indicate that ex vivo models can be applied in large-scale drug screening.
In vitro pathological models
OA inflammatory in vitro models are mostly based on the cellular responses of chondrocytes to inflammatory cytokines. When treated with IL-1β or TNF-α, chondrocytes exhibit an OA-like phenotype, with higher levels of expression of MMPs and ADAMTS4/5, and reduced expression of ECM genes. Chen et al. also showed that IL-1β substantially affected the mechanical properties of chondrocytes in vitro [188]. Alternatively, pathogenic inflammatory factors; IL-1α, IL-6, and TGF-α, have also been adopted in OA inflammatory models [189, 190].
Nevertheless, current inflammation-related models do not fully reflect OA conditions. In these models, chondrocytes were treated with one or two specific cytokines, for example IL-1β or TNF-α. However, the concentrations of other cytokines such as IL-8, vascular endothelial growth factor (VEGF), and monocyte chemoattractant protein-1 (MCP-1), are all elevated in OA joints [191], but these cytokines are rarely used. Moreover, cytokine-based models often use concentrations higher than those found in vivo [192]. To simulate the complex conditions associated with inflammation in OA, Hoff et al. added synovial fluid from patients with OA to a cell culture [193]. Synovial membranes isolated from patients with OA can also be used in co-culture models [194]. Mechanical injury is a major cause of OA. Jin et al. applied a mechanical pressure model with a self-designed mechanical pressure container that generated 10 MPa pressure on normal human chondrocytes for 60 min. In their study [195], mechanical pressure induced early apoptosis of chondrocytes. In response to mechanical injury, chondrocytes were forced to upregulate their expression of VEGF and downregulate their expression of Smad4. In 2013, Kim et al. [196] found that the accumulation of advanced glycation end-products (AGEs) in aged cartilages was the major cause of ECM stiffening. Treatment with ribose (one of the AGEs) significantly increased MMP expression and reduced ECM molecule expression in chondrocytes embedded in collagen matrices, but not in those in 2D culture. Supplementation with β-aminopropionitrile (BAPN), a specific inhibitor of the LOX family, reversed these changes. This study validated the feasibility of establishing mechanical-associated in vitro OA models.
Phenotype readout
Dyes for cartilage ECM
Reliable phenotypic indicators for image recognition are essential. Dyes for cartilage histological characterizations, including Alcian blue, Safranin O, and Toluidine blue, are regularly adopted in screening as they can positively mark cartilage ECM composites. Le et al. used a fluorescently labeled collagen-binding probe, CAN 35-Alexsa 488, to quantify the total collagen content of ATDC5 cultures [34]. These phenotypic indicators provide clues for drug selection but may not be completely specific to the cartilage lineage.
Reporter systems
Reporter systems make it easier to detect molecular phenotypes using imaging systems. The most widely used cartilage reporter genes are Col2a1 and Col11a2. Both are highly expressed in all cartilages and are essential for chondrocyte differentiation and skeletal morphogenesis [197]. A 1998 study indicated that Col11a2 chondrocyte-specific enhancer elements share many similarities with the Col2a1 48-bp enhancer, and Col11a2 and Col2a1 might coordinately regulate chondrocyte differentiation [197]. Yano et al. and Hojo et al. used a Col2GFP-ATDC5 reporter system for high-throughput screening of potential DMOADs [24, 33]. Similarly, Kan et al. built an ATDC5-C2ER system to optimize the detection and analysis of fluorescence [175]. Horton et al. generated a new mouse strain harboring a Col2-pd2EGFP reporter transgene. Because pd2EGF has a much shorter half-life than EGFP, it made a real-time reporter for Col2a1 expression in vivo and in vitro. They also demonstrated that this reporter has illustrated the decrease in Col2a1 in cultured articular chondrocytes [198]. Tsumaki et al. prepared chondrocytes from Col11a2-EGFP transgenic mice to provide a time-course of chondrocyte dedifferentiation [178]. They generated a Col11a2-EGFP human iPSC cell line using the piggyBac vector system and induced it to form hyaline chondrocytes [45]. They used the Col11a2 reporter cell line to screen 5822 compounds and found that A-674563 enhanced the expression of chondrocyte markers [36]. It is worth mentioning that the lineage-specificity of these biomarkers remains controversial and knowledge on hyaline chondrocyte biomarkers is still lacking. Generally, it is not advisable to identify small molecule drugs using a single-biomarker reporter in the primary screen. Therefore, efforts need to be made to identify more reliable cartilage biomarkers and establish multi-molecular reporter systems.
Mechanical property
As the mechanical microenvironment has been hypothesized to modulate chondrocyte function and fate [199], the biomechanical properties of cartilage explants or single chondrocytes are critical for cartilage regeneration tests. During the establishment of a post-traumatic OA model, Mohanraj et al. induced injury compression on engineered cartilage analogs and explants using a custom high-throughput mechanical screening device [200]. This device allowed for the compression of 48 samples in a standard 48-well plate and continuously recorded the load during injury. Although mechanical properties were not included as a phenotype readout, this case illustrated the possibility of applying mechanical stimulation and examination in high-throughput screening. In addition to cartilage tissue, chondrocyte mechanobiology has also been extensively studied [199, 201,202,203], and the atomic force microscope (AFM) has acted as a powerful tool for the characterization of single cells [201, 204]. The mechanical features of normal chondrocytes differed from those of OA chondrocytes, indicating that biomechanics could be a useful technique for investigating OA-related changes in human chondrocytes. However, as current measurements are time-consuming, a customer-friendly instrument optimal for large-scale testing, which supports easy and fast detection during high-throughput screening, is urgently required.
Advanced methodologies in other fields
Optimal reporter cell line
Ideas and methods for improving screening efficiency can be borrowed from other fields. Here, we introduce an interesting study that used a genetically modified reporter cell line for lung cancer drug discovery. Kang et al. developed a method for systematically identifying optimal reporter cell lines for annotating compound libraries (ORCALs) [205]. They trans-genetically built a library of reporter cell lines tagged with different fluorescence and allowed the algorithm to choose which ORCALs optimally classify drugs in a large compound library. They obtained an A549 reporter cell line with fluorescence-labeled nucleus, cell membrane, and a specific protein, XRCC5, which could accurately identify a training set of known drugs. This proof-of-concept study presented a new strategy for improving the efficiency, scale, and accuracy of phenotypic screening by optimizing reporter cell lines.
Bioinformatic-based screening
Bioinformatics is generally used in genome and proteome dissection, image/structure organization, and network analysis. It has also been adopted in multiple drug discovery to help predict drug targets and drug-target interactions using large-scale datasets, which can greatly improve screening efficiency. The Connectivity Map system (www.broadinstitute.org/cmap/), created by Lamb et al. in 2006, aims to create a database that links clinical drug effects with human genomics to provide more valuable information [206]. In their study, they used ~ 5000 small molecule compounds and ~ 3000 gene regulatory agents to process a variety of cell lines. Through the development of the L1000 gene rapid detection side system, they collected genome sequencing data from these cell lines following drug treatment. The effects and cell states were then ranked. As a result, their connectivity Map indicated connections among small molecules with similar chemical mechanisms or physiological processes. In 2016, Kidd et al. examined the responses of 221 immune cell types to 1309 drugs [207]. This network can effectively predict 69,995 drug-immune cell interactions and potentially predict immune responses or side effects in future clinical applications. In 2018, Brum et al. identified molecular targets that could positively regulate osteoblast differentiation using a connectivity map [208], which is a good demonstration of the applicability of this system for drug development.
Chemistry synthetic toolbox
Despite multiple advances in drug discovery, transforming a screening hit into a commercial drug is still slow and expensive. The cost of each drug launch was estimated to be in excess of 2.6 billion dollars [209]. Thus, increasing attention is being paid to compounds that can be easily translated. After the primary identification of candidates, the second step is to optimize them through design-make-test-analysis cycles [210]. The key mission for experts in medicinal chemistry is to design and synthesize bioactive compounds that have the potential for use as drugs. They get access to chemical databases with reaction data (such as SciFinder and Reaxys) to search for an exact structure for precedence. If the exact compound is not found, a substructure search is conducted. There are many cases where biological discoveries are made by synthetic chemists exploring various chemical routes, such as the development of benzodiazepines, penicillin, tetracyclines, and taxanes [211, 212]. The role of medicinal chemistry has changed considerably, and expanding the synthetic platforms and design toolbox should enhance the impact of new methodologies in future drug discovery. Advances in chemistry are expected to greatly accelerate the development of DMOADs.
Challenges and outlooks
Increasing numbers of small molecular drugs for cartilage regeneration or OA modification have been tested in clinical trials, but so far, none has had a significant long-term effect. In our opinion, several issues and challenges warrant further investigation.
First, the induction efficacy and time required for phenotype control are major concerns. The fact that small molecules directly affect proteins, but not the genome, may become a double-edged sword for practical applications. It may cause induction failure, as chemical compounds are documented to only temporarily or reversibly alter cellular phenotypes. Chemical methods also require long-term step-wise induction. Second, individual patients can respond differently to the same chemical. Accumulating evidence supports the existence of OA subtypes [213,214,215]. Magnetic resonance imaging has been used to classify OA subtypes, but challenges still exist when it comes to diagnoses providing meaningful guidance for personalized therapeutic strategies. This necessitates a new diagnostic criterion allowing for early and individualized treatment, which not only relies on traditional imaging examinations, but also molecular detection. Third, a more in-depth understanding of cartilage biology and pathology is necessary to provide complete and informative data. This is important for the optimization of different chemical combinations to manipulate cell fate more precisely in a defined manner. However, since cartilage injury and OA are complex conditions, there are still obstacles when it comes to selecting compounds that work against multiple targets without excessive side effects, thus further investigations need to be conducted. Last, cartilage-regenerative drug screening systems are not satisfactory compared with those in other fields. Previous cell models merely simulated responses to one or two factors and the biomarkers for chondrocyte phenotype readout are limited in number. A continuous evolution in techniques is required to build a 3D cultured screening system allowing for multiple-factor analysis.
In this review, we have summarized the latest research on novel small molecular compounds which target cartilage regeneration. Advanced drug discovery strategies derived from new concepts of regenerative medicine contribute to the identification of potential small molecular drugs for cartilage repair. Small molecule drugs are convenient and cost-effective to design, modify, and synthesize for personalized pharmaceutical management, and will make a strong push for the development of new pharmaceutical therapies for cartilage regeneration.
Availability of data and material
The data and material that support this review are openly available.
References
Hinckel BB, Gomoll AH (2017) Autologous chondrocytes and next-generation matrix-based autologous chondrocyte implantation. Clin Sports Med 36(3):525–548
Engen CN, Årøen A, Engebretsen L (2015) Incidence of knee cartilage surgery in Norway, 2008–2011. BMJ Open 5(11):8423
Hunter DJ, Bierma-Zeinstra S (2019) Osteoarthritis. Lancet 393(10182):1745–1759
Medvedeva EV, Grebenik EA, Gornostaeva SN et al (2018) Repair of damaged articular cartilage: current approaches and future directions. Int J Mol Sci 19(8):2366
Luo Y, Sinkeviciute D, He Y et al (2017) The minor collagens in articular cartilage. Protein Cell 8(8):560–572
Caldwell KL, Wang J (2015) Cell-based articular cartilage repair: the link between development and regeneration. Osteoarthr Cartil 23(3):351–362
Chhabra M (2021) Biological therapeutic modalities. Transl Biotechnol
Hou P, Li Y, Zhang X et al (2013) Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341(6146):651–654
Cheng L, Hu W, Qiu B et al (2014) Generation of neural progenitor cells by chemical cocktails and hypoxia. Cell Res 24(6):665–679
Zhang Y, Cao N, Huang Y et al (2016) Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell 18(3):368–381
Ostrem JM, Shokat KM (2016) Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 15(11):771–785
Nanthakumar CB, Hatley RJ, Lemma S, Gauldie J, Marshall RP, Macdonald SJ (2015) Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov 14(10):693–720
Thomson NC (2017) New and developing non-adrenoreceptor small molecule drugs for the treatment of asthma. Expert Opin Pharmacother 18(3):283–293
Zhao T, Fu Y, Zhu J et al (2018) Single-cell RNA-Seq reveals dynamic early embryonic-like programs during chemical reprogramming. Cell Stem Cell 23(1):31-45.e7
Li W, Li K, Wei W, Ding S (2013) Chemical approaches to stem cell biology and therapeutics. Cell Stem Cell 13(3):270–283
Scott DE, Bayly AR, Abell C, Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 15(8):533–550
Bunpetch V, Wu H, Zhang S, Ouyang H (2017) From, “Bench to Bedside”: current advancement on large-scale production of mesenchymal stem cells. Stem Cells Dev 26(22):1662–1673
Hayek A, Kerstetter-Fogle AE, Sachlos E, Bollenbach T (2012) Kartogenin: a game-changer in regenerative medicine. Regen Med 7(4):475
Johnson K, Zhu S, Tremblay MS et al (2012) A stem cell-based approach to cartilage repair. Science 336(6082):717–721
Wang J, Wang Y, Sun X et al (2019) Biomimetic cartilage scaffold with orientated porous structure of two factors for cartilage repair of knee osteoarthritis. Artif Cells Nanomed Biotechnol 47(1):1710–1721
Kang ML, Kim JE, Im GI (2016) Thermoresponsive nanospheres with independent dual drug release profiles for the treatment of osteoarthritis. Acta Biomater 39:65–78
Kang ML, Jeong SY, Im GI (2017) Hyaluronic acid hydrogel functionalized with self-assembled micelles of amphiphilic PEGylated kartogenin for the treatment of osteoarthritis. Tissue Eng Part A 23(13–14):630–639
Fan W, Li J, Yuan L et al (2018) Intra-articular injection of kartogenin-conjugated polyurethane nanoparticles attenuates the progression of osteoarthritis. Drug Deliv 25(1):1004–1012
Yano F, Hojo H, Ohba S et al (2013) A novel disease-modifying osteoarthritis drug candidate targeting Runx1. Ann Rheum Dis 72(5):748–753
Yano F, Ohba S, Hosaka Y, Saito T, Chung UI (2014) Disease-modifying effects of TD-198946 on progressed osteoarthritis in a mouse model. Ann Rheum Dis 73(11):2062–2064
Chijimatsu R, Yano F, Saito T et al (2019) Effect of the small compound TD-198946 on glycosaminoglycan synthesis and transforming growth factor β3-associated chondrogenesis of human synovium-derived stem cells in vitro. J Tissue Eng Regen Med 13(3):446–458
Hwang KC, Kim JY, Chang W et al (2008) Chemicals that modulate stem cell differentiation. Proc Natl Acad Sci USA 105(21):7467–7471
Choi E, Lee J, Lee S et al (2016) Potential therapeutic application of small molecule with sulfonamide for chondrogenic differentiation and articular cartilage repair. Bioorg Med Chem Lett 26(20):5098–5102
Heck BE, Park JJ, Makani V, Kim EC, Kim DH (2017) PPAR-δ agonist with mesenchymal stem cells induces type II collagen-producing chondrocytes in human arthritic synovial fluid. Cell Transplant 26(8):1405–1417
Buhrmann C, Busch F, Shayan P, Shakibaei M (2014) Sirtuin-1 (SIRT1) is required for promoting chondrogenic differentiation of mesenchymal stem cells. J Biol Chem 289(32):22048–22062
Keshavarz G, Jalili C, Pazhouhi M, Khazaei M (2020) Resveratrol effect on adipose-derived stem cells differentiation to chondrocyte in three-dimensional culture. Adv Pharm Bull 10(1):88–96
Baghaban Eslaminejad M, Fallah N (2014) Small molecule-BIO accelerates and enhances marrow-derived mesenchymal stem cell in vitro chondrogenesis. Iran J Med Sci 39(2):107–116
Hojo H, Yano F, Ohba S et al (2010) Identification of oxytetracycline as a chondrogenic compound using a cell-based screening system. J Bone Miner Metab 28(6):627–633
Le BQ, Fernandes H, Bouten CV, Karperien M, van Blitterswijk C, de Boer J (2015) High-throughput screening assay for the identification of compounds enhancing collagenous extracellular matrix production by ATDC5 Cells. Tissue Eng Part C Methods 21(7):726–736
Duan L, Liang Y, Ma B et al (2017) DNA methylation profiling in chondrocyte dedifferentiation in vitro. J Cell Physiol 232(7):1708–1716
Kobayashi T, Fujita K, Kamatani T, Matsuda S, Tsumaki N (2018) A-674563 increases chondrocyte marker expression in cultured chondrocytes by inhibiting Sox9 degradation. Biochem Biophys Res Commun 495(1):1468–1475
Al-Rasheed NM, Al-Rasheed NM, Hasan IH et al (2017) Simvastatin ameliorates diabetic cardiomyopathy by attenuating oxidative stress and inflammation in rats. Oxid Med Cell Longev 2017:1092015
Terabe K, Takahashi N, Cobb M, Askew EB, Knudson CB, Knudson W (2019) Simvastatin promotes restoration of chondrocyte morphology and phenotype. Arch Biochem Biophys 665:1–11
Nardi A, Ventura L, Cozzi L, Tonini G (2016) Clodronate news of efficacy in osteoporosis. Clin Cases Miner Bone Metab 13(1):33–35
Rosa RG, Collavino K, Lakhani A et al (2014) Clodronate exerts an anabolic effect on articular chondrocytes mediated through the purinergic receptor pathway. Osteoarthr Cartil 22(9):1327–1336
Valenti MT, Mottes M, Biotti A, et al (2017) Clodronate as a therapeutic strategy against osteoarthritis. Int J Mol Sci 18(12)
Pelttari K, Lorenz H, Boeuf S et al (2008) Secretion of matrix metalloproteinase 3 by expanded articular chondrocytes as a predictor of ectopic cartilage formation capacity in vivo. Arthritis Rheum 58(2):467–474
Murphy G, Lee MH (2005) What are the roles of metalloproteinases in cartilage and bone damage. Ann Rheum Dis 64(Suppl 4):44–47
Francioli SE, Martin I, Sie CP et al (2007) Growth factors for clinical-scale expansion of human articular chondrocytes: relevance for automated bioreactor systems. Tissue Eng 13(6):1227–1234
Yamashita A, Morioka M, Yahara Y et al (2015) Generation of scaffoldless hyaline cartilaginous tissue from human iPSCs. Stem Cell Rep 4(3):404–418
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676
Chen S (2018) Screening-based chemical approaches to unravel stem cell biology. Stem Cell Rep 11(6):1312–1323
Niwa H (2015) A stepping stone to pluripotency. Cell 163(7):1570–1572
Zhao Y, Zhao T, Guan J et al (2015) A XEN-like State bridges somatic cells to pluripotency during chemical reprogramming. Cell 163(7):1678–1691
Chen Y, Wu B, Lin J et al (2020) High-resolution dissection of chemical reprogramming from mouse embryonic fibroblasts into fibrocartilaginous cells. Stem Cell Reports 14(3):478–492
Vandana JJ, Lacko LA, Chen S (2021) Phenotypic technologies in stem cell biology. Cell Chem Biol 28(3):257–270
Marini JC, Forlino A (2012) Replenishing cartilage from endogenous stem cells. N Engl J Med 366(26):2522–2524
Song H, Park KH (2020) Regulation and function of SOX9 during cartilage development and regeneration. Semin Cancer Biol 67(Pt 1):12–23
Hidaka C, Cheng C, Alexandre D, Bhargava M, Torzilli PA (2006) Maturational differences in superficial and deep zone articular chondrocytes. Cell Tissue Res 323(1):127–135
Wolock SL, Krishnan I, Tenen DE et al (2019) Mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep 28(2):302-311.e5
Mizuhashi K, Nagata M, Matsushita Y, Ono W, Ono N (2019) Growth plate borderline chondrocytes behave as transient mesenchymal precursor cells. J Bone Miner Res 34(8):1387–1392
He J, Yan J, Wang J et al (2021) Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res 31(7):742–757
Rakic R, Bourdon B, Hervieu M et al (2017) RNA Interference and BMP-2 stimulation allows equine chondrocytes redifferentiation in 3D-hypoxia cell culture model: application for matrix-induced autologous chondrocyte implantation. Int J Mol Sci 18(9)
Oo WM, Yu SP, Daniel MS, Hunter DJ (2018) Disease-modifying drugs in osteoarthritis: current understanding and future therapeutics. Expert Opin Emerg Drugs 23(4):331–347
Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H (2011) Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7(1):33–42
Kitadai HK, Takahashi HK, Straus AH et al (2006) Effect of oral diacerein (DAR) in an experimental hip chondrolysis model. J Orthop Res 24(6):1240–1248
Rezende MU, Gurgel HM, Vilaça Junior PR et al (2006) Diacerhein versus glucosamine in a rat model of osteoarthritis. Clinics (Sao Paulo) 61(5):461–466
Pavelka K, Bruyère O, Cooper C et al (2016) Diacerein: benefits, risks and place in the management of osteoarthritis an opinion-based report from the ESCEO. Drugs Aging 33(2):75–85
Ghosh P, Xu A, Hwa SY, Burkhardt D, Little C (1998) Evaluation of the effects of diacerhein in the sheep model of arthritis. Rev Prat 48(17 Suppl):S24-30
Liang S, Lv ZT, Zhang JM et al (2018) Necrostatin-1 attenuates trauma-induced mouse osteoarthritis and IL-1β induced apoptosis via HMGB1/TLR4/SDF-1 in primary mouse chondrocytes. Front Pharmacol 9:1378
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651
Hamamura K, Nishimura A, Iino T, Takigawa S, Sudo A, Yokota H (2015) Chondroprotective effects of Salubrinal in a mouse model of osteoarthritis. Bone Jt Res 4(5):84–92
Chang SH, Mori D, Kobayashi H et al (2019) Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat Commun 10(1):1442
Zhu S, Lu P, Liu H et al (2015) Inhibition of Rac1 activity by controlled release of NSC23766 from chitosan microspheres effectively ameliorates osteoarthritis development in vivo. Ann Rheum Dis 74(1):285–293
Zhu S, Dai J, Liu H et al (2015) Down-regulation of Rac GTPase-activating protein OCRL1 causes aberrant activation of Rac1 in osteoarthritis development. Arthritis Rheumatol 67(8):2154–2163
Nasry W, Martin CK (2021) Intersecting mechanisms of hypoxia and prostaglandin E2-mediated inflammation in the comparative biology of oral squamous cell carcinoma. Front Oncol 11:539361
Otsuka S, Aoyama T, Furu M et al (2009) PGE2 signal via EP2 receptors evoked by a selective agonist enhances regeneration of injured articular cartilage. Osteoarthr Cartil 17(4):529–538
Murahashi Y, Yano F, Chijimatsu R et al (2019) Oral administration of EP4-selective agonist KAG-308 suppresses mouse knee osteoarthritis development through reduction of chondrocyte hypertrophy and TNF secretion. Sci Rep 9(1):20329
Hwang SM, Feigenson M, Begun DL et al (2018) Phlpp inhibitors block pain and cartilage degradation associated with osteoarthritis. J Orthop Res 36(5):1487–1497
De Falco L, Fioravanti A, Galeazzi M, Tenti S (2013) Bradykinin and its role in osteoarthritis. Reumatismo 65(3):97–104
Meini S, Maggi CA (2008) Knee osteoarthritis: a role for bradykinin. Inflamm Res 57(8):351–361
Gebru Y, Diao TY, Pan H, Mukwaya E, Zhang Y (2013) Potential of RAS inhibition to improve metabolic bone disorders. Biomed Res Int 2013:932691
Cobankara V, Oztürk MA, Kiraz S et al (2005) Renin and angiotensin-converting enzyme (ACE) as active components of the local synovial renin-angiotensin system in rheumatoid arthritis. Rheumatol Int 25(4):285–291
Tsukamoto I, Akagi M, Inoue S, Yamagishi K, Mori S, Asada S (2014) Expressions of local renin-angiotensin system components in chondrocytes. Eur J Histochem 58(2):2387
Yan K, Shen Y (2017) Aliskiren has chondroprotective efficacy in a rat model of osteoarthritis through suppression of the local renin-angiotensin system. Mol Med Rep 16(4):3965–3973
Makino H, Seki S, Yahara Y et al (2017) A selective inhibition of c-Fos/activator protein-1 as a potential therapeutic target for intervertebral disc degeneration and associated pain. Sci Rep 7(1):16983
Lim H, Park H, Kim HP (2011) Effects of flavonoids on matrix metalloproteinase-13 expression of interleukin-1β-treated articular chondrocytes and their cellular mechanisms: inhibition of c-Fos/AP-1 and JAK/STAT signaling pathways. J Pharmacol Sci 116(2):221–231
Aikawa Y, Morimoto K, Yamamoto T et al (2008) Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol 26(7):817–823
Motomura H, Seki S, Shiozawa S, Aikawa Y, Nogami M, Kimura T (2018) A selective c-Fos/AP-1 inhibitor prevents cartilage destruction and subsequent osteophyte formation. Biochem Biophys Res Commun 497(2):756–761
Kingsbury SR, Tharmanathan P, Arden NK et al (2015) Pain reduction with oral methotrexate in knee osteoarthritis, a pragmatic phase iii trial of treatment effectiveness (PROMOTE): study protocol for a randomized controlled trial. Trials 16:77
Niesters M, Martini C, Dahan A (2014) Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol 77(2):357–367
Flood S, Parri R, Williams A, Duance V, Mason D (2007) Modulation of interleukin-6 and matrix metalloproteinase 2 expression in human fibroblast-like synoviocytes by functional ionotropic glutamate receptors. Arthritis Rheum 56(8):2523–2534
Lu W, Wang L, Wo C, Yao J (2016) Ketamine attenuates osteoarthritis of the knee via modulation of inflammatory responses in a rabbit model. Mol Med Rep 13(6):5013–5020
Lee JE, Lee JM, Park YJ, Kim BS, Jeon YT, Chung Y (2017) Inhibition of autoimmune Th17 cell responses by pain killer ketamine. Oncotarget 8(52):89475–89485
Yuan C, Pan Z, Zhao K et al (2020) Classification of four distinct osteoarthritis subtypes with a knee joint tissue transcriptome atlas. Bone Res 8(1):38
de Lange-Brokaar BJ, Ioan-Facsinay A, Yusuf E et al (2015) Association of pain in knee osteoarthritis with distinct patterns of synovitis. Arthritis Rheumatol 67(3):733–740
Yusup A, Kaneko H, Liu L et al (2015) Bone marrow lesions, subchondral bone cysts and subchondral bone attrition are associated with histological synovitis in patients with end-stage knee osteoarthritis: a cross-sectional study. Osteoarthr Cartil 23(11):1858–1864
Neogi T, Guermazi A, Roemer F et al (2016) Association of joint inflammation with pain sensitization in knee osteoarthritis: the multicenter osteoarthritis study. Arthritis Rheumatol 68(3):654–661
Conaghan PG, Cook AD, Hamilton JA, Tak PP (2019) Therapeutic options for targeting inflammatory osteoarthritis pain. Nat Rev Rheumatol 15(6):355–363
Jones IA, Togashi R, Wilson ML, Heckmann N, Vangsness CT Jr (2019) Intra-articular treatment options for knee osteoarthritis. Nat Rev Rheumatol 15(2):77–90
Jüni P, Hari R, Rutjes AW et al (2015) Intra-articular corticosteroid for knee osteoarthritis. Cochrane Database Syst Rev 10:CD005328
Osani MC, Vaysbrot EE, Zhou M, McAlindon TE, Bannuru RR (2020) Duration of symptom relief and early trajectory of adverse events for oral nonsteroidal antiinflammatory drugs in knee osteoarthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken) 72(5):641–651
Wenham CY, Hensor EM, Grainger AJ et al (2012) A randomized, double-blind, placebo-controlled trial of low-dose oral prednisolone for treating painful hand osteoarthritis. Rheumatol (Oxf) 51(12):2286–2294
McAlindon TE, LaValley MP, Harvey WF et al (2017) Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: a randomized clinical trial. JAMA 317(19):1967–1975
Roth BL, Kroeze WK (2015) Integrated approaches for genome-wide interrogation of the druggable non-olfactory g protein-coupled receptor superfamily. J Biol Chem 290(32):19471–19477
Miller RE, Block JA, Malfait AM (2018) What is new in pain modification in osteoarthritis. Rheumatol (Oxf) 57(Suppl_4):99–107
Cowan A, Kehner GB, Inan S (2015) Targeting itch with ligands selective for κ opioid receptors. Handb Exp Pharmacol 226:291–314
Kiguchi N, Ding H, Kishioka S, Ko MC (2020) Nociceptin/Orphanin FQ peptide receptor-related ligands as novel analgesics. Curr Top Med Chem 20(31):2878–2888
Koch ED, Kapanadze S, Eerdekens MH et al (2019) Cebranopadol, a novel first-in-class analgesic drug candidate: first experience with cancer-related pain for up to 26 weeks. J Pain Symptom Manage 58(3):390–399
Bagal SK, Brown AD, Cox PJ et al (2013) Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem 56(3):593–624
Malfait AM, Miller RJ (2016) Emerging targets for the management of osteoarthritis pain. Curr Osteoporos Rep 14(6):260–268
R Stevens DP, J Ervin JN, Nieves Y (2017) OP0167 efficacy and safety of CNTX-4975 in subjects with moderate to severe osteoarthritis knee pain: 24-week, randomized, double-blind, placebo-controlled, dose
Eitner A, Hofmann GO, Schaible HG (2017) Mechanisms of osteoarthritic pain studies in humans and experimental models. Front Mol Neurosci 10:349
Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U (2017) The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol 13(5):302–311
Zhuo Q, Yang W, Chen J, Wang Y (2012) Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol 8(12):729–737
Azamar-Llamas D, Hernández-Molina G, Ramos-Ávalos B, Furuzawa-Carballeda J (2017) Adipokine contribution to the pathogenesis of osteoarthritis. Mediators Inflamm 2017:5468023
Faubion SS, Kapoor E, Moyer AM, Hodis HN, Miller VM (2019) Statin therapy: does sex matter. Menopause 26(12):1425–1435
Yamashita A, Morioka M, Kishi H et al (2014) Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 513(7519):507–511
Pathak NN, Lingaraju MC, Balaganur V et al (2015) Anti-inflammatory and chondroprotective effects of atorvastatin in a cartilage explant model of osteoarthritis. Inflamm Res 64(3–4):161–169
Yudoh K, Karasawa R (2010) Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA). Aging (Albany NY) 2(12):990–998
Eymard F, Parsons C, Edwards MH et al (2018) Statin use and knee osteoarthritis progression: results from a post-hoc analysis of the SEKOIA trial. Jt Bone Spine 85(5):609–614
Veronese N, Koyanagi A, Stubbs B et al (2019) Statin use and knee osteoarthritis outcomes: a longitudinal cohort study. Arthritis Care Res (Hoboken) 71(8):1052–1058
Haj-Mirzaian A, Mohajer B, Guermazi A et al (2019) Statin use and knee osteoarthritis outcome measures according to the presence of heberden nodes: results from the osteoarthritis initiative. Radiology 293(2):396–404
Misra J, Kim DK, Choi HS (2017) ERRγ: a junior orphan with a senior role in metabolism. Trends Endocrinol Metab 28(4):261–272
Audet-Walsh É, Giguére V (2015) The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol Sin 36(1):51–61
Son YO, Park S, Kwak JS et al (2017) Estrogen-related receptor γ causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat Commun 8(1):2133
He Y, Makarczyk MJ, Lin H (2020) Role of mitochondria in mediating chondrocyte response to mechanical stimuli. Life Sci 263:118602
Zheng L, Zhang Z, Sheng P, Mobasheri A (2021) The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev 66:101249
Eitner A, Müller S, König C et al (2021) Inhibition of inducible nitric oxide synthase prevents IL-1β-induced mitochondrial dysfunction in human chondrocytes. Int J Mol Sci 22(5):2477
Ott M, Gogvadze V, Orrenius S, Zhivotovsky B (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12(5):913–922
Panahi Y, Alishiri GH, Parvin S, Sahebkar A (2016) Mitigation of systemic oxidative stress by curcuminoids in osteoarthritis: results of a randomized controlled trial. J Diet Suppl 13(2):209–220
Shi Y, Hu X, Cheng J et al (2019) A small molecule promotes cartilage extracellular matrix generation and inhibits osteoarthritis development. Nat Commun 10(1):1914
Choudhary D, Adhikary S, Ahmad N et al (2018) Prevention of articular cartilage degeneration in a rat model of monosodium iodoacetate induced osteoarthritis by oral treatment with Withaferin A. Biomed Pharmacother 99:151–161
Bloemberg D, Quadrilatero J (2019) Autophagy, apoptosis, and mitochondria: molecular integration and physiological relevance in skeletal muscle. Am J Physiol Cell Physiol 317(1):C111–C130
Valenti MT, Dalle Carbonare L, Zipeto D, Mottes M (2021) Control of the autophagy pathway in osteoarthritis: key regulators, therapeutic targets and therapeutic strategies. Int J Mol Sci 22(5):2700
Chen Q, Haddad GG (2004) Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol 207(Pt 18):3125–3129
Tang Q, Zheng G, Feng Z et al (2017) Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis 8(10):e3081
Ryu D, Mouchiroud L, Andreux PA et al (2016) Urolithin A induces mitophagy and prolongs lifespan in C elegans and increases muscle function in rodents. Nat Med 22(8):879–888
He Y, Yocum L, Alexander PG, Jurczak MJ, Lin H (2021) Urolithin A protects chondrocytes from mechanical overloading-induced injuries. Front Pharmacol 12:703847
Zhang L, Zhang Z, Khan A, Zheng H, Yuan C, Jiang H (2020) Advances in drug therapy for mitochondrial diseases. Ann Transl Med 8(1):17
Harms FA, Bodmer SI, Raat NJ, Mik EG (2015) Non-invasive monitoring of mitochondrial oxygenation and respiration in critical illness using a novel technique. Crit Care 19(1):343
Kim KI, Park YS, Im GI (2013) Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J Bone Miner Res 28(5):1050–1060
Dai J, Yu D, Wang Y et al (2017) Kdm6b regulates cartilage development and homeostasis through anabolic metabolism. Ann Rheum Dis 76(7):1295–1303
Chen L, Wu Y, Wu Y, Wang Y, Sun L, Li F (2016) The inhibition of EZH2 ameliorates osteoarthritis development through the Wnt/β-catenin pathway. Sci Rep 6:29176
Yapp C, Carr AJ, Price A, Oppermann U, Snelling SJ (2016) H3K27me3 demethylases regulate in vitro chondrogenesis and chondrocyte activity in osteoarthritis. Arthritis Res Ther 18(1):158
Monteagudo S, Cornelis F, Aznar-Lopez C et al (2017) DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat Commun 8:15889
Jambhekar A, Dhall A, Shi Y (2019) Roles and regulation of histone methylation in animal development. Nat Rev Mol Cell Biol 20(10):625–641
Du X, Chen Y, Zhang Q et al (2020) Ezh2 ameliorates osteoarthritis by activating TNFSF13B. J Bone Miner Res 35(5):956–965
Jun Z, Xinmeng J, Yue L et al (2020) Jumonji domain containing-3 (JMJD3) inhibition attenuates IL-1β-induced chondrocytes damage in vitro and protects osteoarthritis cartilage in vivo. Inflamm Res 69(7):657–666
De Santis M, Di Matteo B, Chisari E et al (2018) The role of Wnt pathway in the pathogenesis of OA and its potential therapeutic implications in the field of regenerative medicine. Biomed Res Int 2018:7402947
Stampella A, Monteagudo S, Lories R (2017) Wnt signaling as target for the treatment of osteoarthritis. Best Pract Res Clin Rheumatol 31(5):721–729
Day TF, Guo X, Garrett-Beal L, Yang Y (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8(5):739–750
Deshmukh V, Hu H, Barroga C et al (2018) A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr Cartil 26(1):18–27
Deshmukh VO, Green AL et al (2019) Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr Cartil 27(9):1347–1360
Tambiah J, Kennedy S, Swearingen CJ et al (2021) Individual participant symptom responses to intra-articular lorecivivint in knee osteoarthritis: post hoc analysis of a phase 2B trial. Rheumatol Ther 8(2):973–985
Yazici Y, McAlindon TE, Gibofsky A et al (2020) Lorecivivint, a novel intraarticular CDC-like kinase 2 and dual-specificity tyrosine phosphorylation-regulated kinase 1A inhibitor and wnt pathway modulator for the treatment of knee osteoarthritis: a phase II randomized trial. Arthritis Rheumatol 72(10):1694–1706
Goswami VG, Patel BD (2021) Recent updates on Wnt signaling modulators: a patent review (2014–2020). Expert Opin Ther Pat 1–35
Sabha M, Siaton BC, Hochberg MC (2020) Lorecivivint, an intra-articular potential disease-modifying osteoarthritis drug. Expert Opin Investig Drugs 29(12):1339–1346
Lietman C, Wu B, Lechner S et al (2018) Inhibition of Wnt/β-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight 3(3)
Shepard JB, Jeong JW, Maihle NJ, O’Brien S, Dealy CN (2013) Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Arthritis Res Ther 15(3):R60
Shin SY, Pozzi A, Boyd SK, Clark AL (2016) Integrin α1β1 protects against signs of post-traumatic osteoarthritis in the female murine knee partially via regulation of epidermal growth factor receptor signalling. Osteoarthr Cartil 24(10):1795–1806
Staal B, Williams BO, Beier F, Vande Woude GF, Zhang YW (2014) Cartilage-specific deletion of Mig-6 results in osteoarthritis-like disorder with excessive articular chondrocyte proliferation. Proc Natl Acad Sci U S A 111(7):2590–2595
Zhang X, Siclari VA, Lan S et al (2011) The critical role of the epidermal growth factor receptor in endochondral ossification. J Bone Miner Res 26(11):2622–2633
Sun H, Wu Y, Pan Z et al (2018) Gefitinib for epidermal growth factor receptor activated osteoarthritis subpopulation treatment. EBioMedicine 32:223–233
Jia H, Ma X, Tong W et al (2016) EGFR signaling is critical for maintaining the superficial layer of articular cartilage and preventing osteoarthritis initiation. Proc Natl Acad Sci USA 113(50):14360–14365
Zhen G, Wen C, Jia X et al (2013) Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med 19(6):704–712
Han W, Fan S, Bai X, Ding C (2017) Strontium ranelate, a promising disease modifying osteoarthritis drug. Expert Opin Investig Drugs 26(3):375–380
Lafeber FP, van Laar JM (2013) Strontium ranelate: ready for clinical use as disease-modifying osteoarthritis drug. Ann Rheum Dis 72(2):157–161
Reginster JY, Badurski J, Bellamy N et al (2013) Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: results of a double-blind, randomised placebo-controlled trial. Ann Rheum Dis 72(2):179–186
Chu JG, Dai MW, Wang Y et al (2017) Strontium ranelate causes osteophytes overgrowth in a model of early phase osteoarthritis. BMC Musculoskelet Disord 18(1):78
Kozawa E, Cheng XW, Urakawa H et al (2016) Increased expression and activation of cathepsin K in human osteoarthritic cartilage and synovial tissues. J Orthop Res 34(1):127–134
Lewiecki EM (2009) Odanacatib, a cathepsin K inhibitor for the treatment of osteoporosis and other skeletal disorders associated with excessive bone remodeling. IDrugs 12(12):799–809
Dai R, Wu Z, Chu HY et al (2020) Cathepsin K: the action in and beyond bone. Front Cell Dev Biol 8:433
Loeser RF, Goldring SR, Scanzello CR, Goldring MB (2012) Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 64(6):1697–1707
Li H, Liao Z, Yang Z et al (2021) 3D printed poly(ε-Caprolactone)/meniscus extracellular matrix composite scaffold functionalized with kartogenin-releasing PLGA microspheres for meniscus tissue engineering. Front Bioeng Biotechnol 9:662381
Yu C, Li D, Wang C et al (2021) Injectable kartogenin and apocynin loaded micelle enhances the alleviation of intervertebral disc degeneration by adipose-derived stem cell. Bioact Mater 6(10):3568–3579
Zhu J, Shao J, Chen Y et al (2021) Fibrin glue-kartogenin complex promotes the regeneration of the tendon-bone interface in rotator cuff injury. Stem Cells Int 2021:6640424
Gerry CJ, Schreiber SL (2018) Chemical probes and drug leads from advances in synthetic planning and methodology. Nat Rev Drug Discov 17(5):333–352
Yao Y, Wang Y (2013) ATDC5: an excellent in vitro model cell line for skeletal development. J Cell Biochem 114(6):1223–1229
Kan A, Ikeda T, Saito T et al (2009) Screening of chondrogenic factors with a real-time fluorescence-monitoring cell line ATDC5-C2ER: identification of sorting nexin 19 as a novel factor. Arthritis Rheum 60(11):3314–3323
Yu J, Vodyanik MA, Smuga-Otto K et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858):1917–1920
Willard VP, Diekman BO, Sanchez-Adams J, Christoforou N, Leong KW, Guilak F (2014) Use of cartilage derived from murine induced pluripotent stem cells for osteoarthritis drug screening. Arthritis Rheumatol 66(11):3062–3072
Minegishi Y, Hosokawa K, Tsumaki N (2013) Time-lapse observation of the dedifferentiation process in mouse chondrocytes using chondrocyte-specific reporters. Osteoarthr Cartil 21(12):1968–1975
Hiramatsu K, Sasagawa S, Outani H, Nakagawa K, Yoshikawa H, Tsumaki N (2011) Generation of hyaline cartilaginous tissue from mouse adult dermal fibroblast culture by defined factors. J Clin Invest 121(2):640–657
Skardal A, Shupe T, Atala A (2016) Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov Today 21(9):1399–1411
Lozito TP, Alexander PG, Lin H, Gottardi R, Cheng AW, Tuan RS (2013) Three-dimensional osteochondral microtissue to model pathogenesis of osteoarthritis. Stem Cell Res Ther 4(Suppl 1):S6
Choi CH, Kim TH, Sung YK et al (2014) SKI306X inhibition of glycosaminoglycan degradation in human cartilage involves down-regulation of cytokine-induced catabolic genes. Korean J Intern Med 29(5):647–655
Mixon A, Savage A, Bahar-Moni AS, Adouni M, Faisal T (2021) An in vitro investigation to understand the synergistic role of MMPs-1 and 9 on articular cartilage biomechanical properties. Sci Rep 11(1):14409
Black RM, Wang Y, Struglics A et al (2020) Proteomic analysis reveals dexamethasone rescues matrix breakdown but not anabolic dysregulation in a cartilage injury model. Osteoarthr Cartil Open. 2(4):100099
Wardale J, Mullen L, Howard D, Ghose S, Rushton N (2015) An ex vivo model using human osteoarthritic cartilage demonstrates the release of bioactive insulin-like growth factor-1 from a collagen-glycosaminoglycan scaffold. Cell Biochem Funct 33(5):277–284
Schwab A, Meeuwsen A, Ehlicke F et al (2017) Ex vivo culture platform for assessment of cartilage repair treatment strategies. Altex 34(2):267–277
Spinnen J, Shopperly LK, Rendenbach C et al (2021) A novel method facilitating the simple and low-cost preparation of human osteochondral slice explants for large-scale native tissue analysis. Int J Mol Sci 22(12):6394
Chen C, Xie J, Deng L, Yang L (2014) Substrate stiffness together with soluble factors affects chondrocyte mechanoresponses. ACS Appl Mater Interfaces 6(18):16106–16116
Sokolove J, Lepus CM (2013) Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis 5(2):77–94
Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS (2011) Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 63(9):2732–2743
Sohn DH, Sokolove J, Sharpe O et al (2012) Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther 14(1):R7
Johnson CI, Argyle DJ, Clements DN (2016) In vitro models for the study of osteoarthritis. Vet J 209:40–49
Hoff P, Buttgereit F, Burmester GR et al (2013) Osteoarthritis synovial fluid activates pro-inflammatory cytokines in primary human chondrocytes. Int Orthop 37(1):145–151
Ushiyama T, Chano T, Inoue K, Matsusue Y (2003) Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann Rheum Dis 62(2):108–112
Jin L, Zhao J, Jing W et al (2014) Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int J Mol Med 34(2):451–463
Kim JH, Lee G, Won Y et al (2015) Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc Natl Acad Sci USA 112(30):9424–9429
Bridgewater LC, Lefebvre V, de Crombrugghe B (1998) Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem 273(24):14998–15006
Tryfonidou MA, Lunstrum GP, Hendriks K et al (2011) Novel type II collagen reporter mice: new tool for assessing collagen 2α1 expression in vivo and in vitro. Dev Dyn 240(3):663–673
Anderson DE, Johnstone B (2017) Dynamic mechanical compression of chondrocytes for tissue engineering: a critical review. Front Bioeng Biotechnol 5:76
Mohanraj B, Meloni GR, Mauck RL, Dodge GR (2014) A high-throughput model of post-traumatic osteoarthritis using engineered cartilage tissue analogs. Osteoarthr Cartil 22(9):1282–1290
Hsieh CH, Lin YH, Lin S, Tsai-Wu JJ, Herbert Wu CH, Jiang CC (2008) Surface ultrastructure and mechanical property of human chondrocyte revealed by atomic force microscopy. Osteoarthr Cartil 16(4):480–488
Oftadeh R, Perez-Viloria M, Villa-Camacho JC, Vaziri A, Nazarian A (2015) Biomechanics and mechanobiology of trabecular bone: a review. J Biomech Eng 137(1):0108021–01080215
Das RH, Jahr H, Verhaar JA, van der Linden JC, van Osch GJ, Weinans H (2008) In vitro expansion affects the response of chondrocytes to mechanical stimulation. Osteoarthr Cartil 16(3):385–391
Darling EM, Zauscher S, Guilak F (2006) Viscoelastic properties of zonal articular chondrocytes measured by atomic force microscopy. Osteoarthr Cartil 14(6):571–579
Kang J, Hsu CH, Wu Q et al (2016) Improving drug discovery with high-content phenotypic screens by systematic selection of reporter cell lines. Nat Biotechnol 34(1):70–77
Lamb J, Crawford ED, Peck D et al (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313(5795):1929–1935
Kidd BA, Wroblewska A, Boland MR et al (2016) Mapping the effects of drugs on the immune system. Nat Biotechnol 34(1):47–54
Brum AM, van de Peppel J, Nguyen L et al (2018) Using the Connectivity Map to discover compounds influencing human osteoblast differentiation. J Cell Physiol 233(6):4895–4906
DiMasi JA, Grabowski HG, Hansen RW (2016) Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 47:20–33
Davis AM, Plowright AT, Valeur E (2017) Directing evolution: the next revolution in drug discovery. Nat Rev Drug Discov 16(10):681–698
Boström J, Brown DG, Young RJ, Keserü GM (2018) Expanding the medicinal chemistry synthetic toolbox. Nat Rev Drug Discov 17(10):709–727
Wright PM, Seiple IB, Myers AG (2014) The evolving role of chemical synthesis in antibacterial drug discovery. Angew Chem Int Ed Engl 53(34):8840–8869
Zheng S, Hunter DJ, Xu J, Ding C (2016) Monoclonal antibodies for the treatment of osteoarthritis. Expert Opin Biol Ther 16(12):1529–1540
Arden N, Richette P, Cooper C et al (2015) Can we identify patients with high risk of osteoarthritis progression who will respond to treatment? A focus on biomarkers and frailty. Drugs Aging 32(7):525–535
Waarsing JH, Bierma-Zeinstra SM, Weinans H (2015) Distinct subtypes of knee osteoarthritis: data from the osteoarthritis initiative. Rheumatology (Oxford) 54(9):1650–1658
Funding
This work was supported by the National Key R&D Program of China (2017YFA0104900), the Natural Sciences Foundation of China (81630065, 82002319), and the China Postdoctoral Science Foundation (2020M681892).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Consent for publication was obtained from all participants.
Conflict of interest
The authors declare that they have no conflicts of interest to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, Y., Sun, H., Yao, X. et al. Pharmaceutical therapeutics for articular regeneration and restoration: state-of-the-art technology for screening small molecular drugs. Cell. Mol. Life Sci. 78, 8127–8155 (2021). https://doi.org/10.1007/s00018-021-03983-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03983-8