
Abstract
The discovery of iPSCs has led to the ex vivo production of differentiated cells for regenerative medicine. In the case of transfusion products, the derivation of platelets from iPSCs is expected to complement our current blood-donor supplied transfusion system through donor-independent production with complete pathogen-free assurance. This derivation can also overcome alloimmune platelet transfusion refractoriness by resulting in autologous, HLA-homologous or HLA-deficient products. Several developments were necessary to produce a massive number of platelets required for a single transfusion. First, expandable megakaryocytes were established from iPSCs through transgene expression. Second, a turbulent-type bioreactor with improved platelet yield and quality was developed. Third, novel drugs that enabled efficient feeder cell-free conditions were developed. Fourth, the platelet-containing suspension was purified and resuspended in an appropriate buffer. Finally, the platelet product needed to be assured for competency and safety including non-tumorigenicity through in vitro and in vivo preclinical tests. Based on these advancements, a clinical trial has started. The generation of human iPSC-derived platelets could evolve transfusion medicine to the next stage and assure a ubiquitous, safe supply of platelet products. Further, considering the feasibility of gene manipulations in iPSCs, other platelet products may bring forth novel therapeutic measures.
Similar content being viewed by others

Introduction
In 2006, induced pluripotent stem cells (iPSCs) were established from mouse fibroblasts by the introduction of four reprogramming factors, Oct3/4, Sox2, Klf4 and c-Myc [1]. In the next year, the establishment of human iPSCs was reported by the same or similar combination of reprogramming factors [2,3,4]. Analogous to embryonic stem cells (ESCs), iPSCs bear pluripotency and self-renewal capacity, in other words, they can proliferate indefinitely in vitro and have the capacity to differentiate into any cell type, recapitulating the development process in vivo (Fig. 1). Because iPSCs can be established from somatic cells such as skin fibroblasts or blood cells, they bear none of the ethical issues associated with embryo use and can be essentially established from any individual. In addition, iPSCs can be edited for their genes by CRISPR/Cas9 and other technologies. These characteristics have encouraged their application to disease remodeling in vitro and regenerative medicine [5, 6] (Fig. 1).

Applications of induced pluripotent stem cells (iPSCs). iPSCs are established from somatic cells such as skin fibroblasts and blood cells by the introduction of the reprogramming genes OCT3/4, SOX2, KLF4 and c-MYC or other combinations. iPSCs can be expanded indefinitely and induced to differentiate into any cell type in vitro. Cells differentiated from iPSCs are expected to contribute to disease remodeling in vitro, which can be used to clarify the pathogenesis and develop novel drugs and therapeutics, and to regenerative medicine as cell therapies
The versatility of iPSCs is applicable to the field of hematopoiesis. One way is to recapitulate hematopoietic diseases, ranging from monogenic congenital diseases or myeloproliferative diseases (MPD) to multifactorial hematopoietic malignancies and bone marrow failures, by using iPSCs established from patients [7, 8]. These models will contribute to uncovering the unknown pathogenesis or drug testing, thus leading to novel treatment modalities for the diseases. Another way is the clinical application of the hematopoietic cells differentiated from iPSCs, such as platelets, erythrocytes, lymphocytes and macrophages [5, 6]. This review will focus on the current status of the development, application and manipulation of iPSC-derived platelet-like particles (iPSC-PLTs), which has been leading the field of regenerative medicine using iPSC-derived blood cells and has reached the first-in-human clinical trial.
Expectations in manufacturing blood products from iPSCs
The primary treatment for patients with severe anemia or thrombocytopenia is the transfusion of blood products [9, 10]. Patients with primary hematopoietic failures and malignancies are those that require transfusion the most. Normal hematopoiesis is decreased while myelosuppressive treatments such as chemotherapy, radiotherapy and hematopoietic stem cell transplantation (HSCT) are applied. Non-hematopoietic cancers can also require transfusions due to chemotherapies or the invasion of malignant cells into the bone marrow. Other conditions that require transfusions include massive hemorrhage by trauma or surgery, the consumption and loss of platelets through extracorporeal circulation in cardiovascular surgeries using artificial heart and lung apparatuses, and respiratory failures using extracorporeal membrane oxygenation (ECMO).
Fortunately, many countries have well-established systems in which blood products manufactured from healthy blood donors are supplied to patients in need of blood transfusions (Table 1). However, the percentage of young blood donors is decreasing in developed countries due to declining birthrates and aging populations [11, 12]. Elderly people have higher incidences of hematological diseases such as leukemia and myelodysplastic syndromes (MDS) along with cancers and cardiovascular disease. Furthermore, owing to advances in medical care, the aforementioned aggressive treatments that require transfusions are being increasingly applied. Thus, there is concern that a supply shortage of blood products will occur in the future [11, 12].
In particular, platelet products are stored at room temperature to best maintain their function, but still have a short shelf life of only four or five days based on their loss of function and the risk of bacterial growth. Therefore, platelet products must be continuously supplied to meet demand. Moreover, supply can be significantly affected by extraneous factors such as holidays, inclement weather and pandemics, like the current COVID-19 pandemic [13]. The risk of supply shortages is especially higher for patients with rare blood types who require matched donors to avoid alloimmune rejection [12, 14]. In addition, although improved testing and collection procedures have made them rare, blood-borne infection and bacterial contamination cannot be completely eliminated partly due to the time window of the infection tests and emerging infections for which reliable tests are unavailable [15].
As an alternative to these donor-dependent blood product supply systems, biosynthesized blood products have been created by introducing molecules into liposomes and other platform materials to mimic the function of red blood cells or platelets [16,17,18]. These products are expected to have advantages in emergency situations with limited logistics, such as trauma at battlefields, owing to better preservation capacity, but it is especially difficult to recapitulate the delicate and complicated functional properties of native platelets.
Another approach is to derive the desired cells from iPSCs, which would realize the supply of bona fide blood cells independent of blood donors (Table 1). In addition, iPSC-derived blood products can cope with alloimmune reactions owing to the capability of establishing iPSCs from any desired person and are amenable to gene editing. In terms of safety, iPSC-derived blood products can be devoid of infection by aseptic manufacturing from the source cells, which are assured of being pathogen-free. From the perspective of iPSC-based regenerative medicine, since platelets and erythrocytes are non-nucleated cells, there is essentially no concern about their tumorigenesis, and the risk from nucleated cells that contaminate the product can also be dealt with by irradiation, which is generally performed for transfusion preparations. The differentiation of hematopoietic cells may be relatively easy compared with other cell types because cells can be cultured in suspension and do not require three-dimensional tissue construction. However, difficulties lie in the development of a production system that can provide 1011 platelets or 1012 red blood cells and a quality equivalent to current blood products. In addition, cost is a concern: currently, the cost for red blood cells is approximately US $150–450 per pack and for platelets is $400–1000 per pack in developed countries [19].
Hematopoiesis in vivo vs. hematopoiesis in vitro from iPSCs
Since the 1990s, several groups have achieved the hematopoietic differentiation of mouse ESCs [20,21,22] and human ESCs [23,24,25,26,27,28]. Soon after human iPSCs were established in 2007, the hematopoietic differentiation of iPSCs from healthy donors and patients were reported for many blood cell lineages [7, 29,30,31,32]. One issue with these pluripotent stem cells (PSCs), however, is that the differentiated hematopoietic cells tend to have primitive or early definitive of embryonic/fetal phenotype, not the definitive adult phenotype [33]. Primitive hematopoiesis occurs in the yolk sac and gives rise to primitive HSC-like cells, which differentiate into primitive erythroid cells, macrophages and megakaryocytes. Then, for a brief period, yolk sac gives rise to erythromyeloid progenitors (EMPs) which differentiate to both erythroid and myeloid cells, among which tissue-resident macrophages persist into adulthood [34]. Following these limited spectrums of hematopoiesis, definitive bona fide HSCs originate from the hemogenic endothelium in the aorta-gonad-mesonephros (AGM) during mid-gestation, then migrate to the fetal liver, spleen and finally the bone marrow, to differentiate to all adult-type hematopoietic cells throughout life [33]. Conventionally, HSCs are postulated to differentiate to multipotent progenitor cells that differentiate through a branching fashion to common myeloid progenitor cells (CMPs) and then to megakaryocyte/erythrocyte bipotent progenitor cells (MEPs) and finally megakaryocytes. However, megakaryocyte differentiation from HSCs has been recently revisited, with studies reporting megakaryocyte-biased HSCs and the direct differentiation of megakaryocyte progenitors from HSCs that bypasses CMPs and MEPs [35, 36].
The primitive or embryonic/fetal nature of iPSC-derived hematopoiesis may be the reason for the several limitations in differentiating certain hematopoietic cells. First, the induction of HSCs has not been successful unless the HSCs were collected from the bone marrow of mice in which teratomas were formed by transferring iPSCs [37, 38] or by the forced expression of seven transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1 and SPI1) in iPSC-derived hemogenic endothelial-like cells [39]. Second, the myeloid lineage is dominant, and lymphoid hematopoiesis have been less well reconstituted. Thus, most in vitro iPSC hematopoietic disease models are myeloid types, such as MPD, acute myeloid leukemia (AML), MDS and bone marrow failures [8]. Thirdly, the dominant types of hemoglobin in iPSC-derived erythrocytes are neonatal (HbE) or fetal (HbF), not adult-type HbA [33, 40,41,42]. Fourthly, the primitive or embryonic/fetal nature could also be the cause for the low yield of iPSC-PLTs from iPSC-derived megakaryocytes, which are currently differentiated from hematopoietic progenitor cells (HPCs), but not definite HSCs, since neonatal megakaryocytes have a higher capacity for expansion but lower polyploidy and platelet generation compared to the adult type [43]. In addition, fetal/neonatal platelets are suggested to be hyporeactive compared to adult-type platelets, although they survive longer, which may also become the nature of platelets derived in vitro from iPSCs [44, 45].
Development of iPSC-PLTs
Success and limitations in the direct differentiation of platelets from HSCs and PSCs in vitro
The derivations of erythrocytes and platelets from iPSCs have been both pursued, but the derivation of red blood cells is very much behind in terms of clinical application due to the scale and immature phenotype. As noted above, one magnitude more red blood cells is needed for transfusions compared with platelets (1012 vs. 1011]. In addition, only one red blood cell can be derived from one erythroblast through enucleation, while 800 to 2,000 platelets are fragmented from one megakaryocyte in vivo. Furthermore, with current methods, iPSC-derived erythroblasts do not complete enucleation or globin switching to adult-type HbA [40,41,42].
iPSC-PLTs have reached clinical trial, but to do so, several issues had to be overcome. The first was obtaining enough megakaryocytes, the mother cell of all platelets (Table 2). As megakaryocytes mature, they become large and polyploid through endomitosis, store platelet-specific granules and develop the demarcation membrane system (DMS) [46]. In steady state, mature megakaryocytes in the vascular niche of the bone marrow extend their proplatelets into the bone marrow sinusoid, where blood flow tears off proplatelets or smaller fragments, i.e. platelets, which are released into the systemic circulation [47].
Interestingly, the lung has been postulated to produce platelets or fragment to smaller platelets based on the observation that megakaryocytes reside in the lung and that the number of efferent platelets is more than of afferent platelets, whereas for megakaryocytes the opposite is true [48]. Although one report questions the lung as a platelet source [49], a recent in vivo imaging study of mouse bone marrow captured images of the vasculature with megakaryocytes trapped in the lung and spleen extending proplatelets and eventually releasing platelets as they do in the bone marrow [50]. The study further suggested that the lung also harbors HSCs and HPCs, which may migrate out of the lung to the bone marrow under thrombocytopenia and relative stem cell deficiency in the bone marrow. Meanwhile, our in vivo live imaging study of the mouse bone marrow observed that, under inflammatory conditions, megakaryocytes release platelets through a “rupture” fashion by an IL-1α- and caspase-3-dependent mechanism [51]. In the thrombopoietin (TPO)-dependent steady state, alpha and beta-1 tubulins are expressed in a regulated fashion to form proplatelets, but in the rupture state, the expression of tubulins is dysregulated, resulting in possibly diminished plasma membrane stability. Interestingly, our collaborator also reported that an activated form of tyrosyl-tRNA synthetase (YRSACT) functions to enhance megakaryopoiesis and platelet production in TPO-independent fashion, most likely under stress conditions [52].
Although megakaryocytes can be collected through bone marrow aspiration, they are not considered a source for the production of platelets ex vivo (Table 2). The collection procedure is invasive, and megakaryocytes are normally only about 50–150/μL, or less than 0.1% of nucleated cells in the bone marrow, which range from HSCs to fully differentiated hematopoietic-lineage cells [53, 54]. Because megakaryocytes cannot expand in vitro, it is unrealistic to prepare enough of them from the bone marrow for ex vivo production. HSCs, the origin of all adult-type hematopoietic cells, mostly reside in the bone marrow, but they can be isolated from cord blood without invasiveness or from peripheral blood under specific conditions such as mobilized conditions that reduce the invasiveness compared with the bone marrow (Table 2). Significantly owing to the discovery of the key cytokine TPO [55,56,57,58], HSCs can be differentiated into megakaryocytes in vitro [59,60,61]. However, the number of obtainable HSCs is small (106 order), and their complete expansion has not been achieved, thus they cannot provide a sufficient number of megakaryocytes for clinical application [62, 63].
PSCs, such as ESCs and iPSCs, offer a solution because they can expand limitlessly in vitro and have the potency to differentiate into essentially any kind of cell type, including hematopoietic lineages (Table 2). ESCs were successfully differentiated into megakaryocytes that could generate platelets in vitro [27, 64,65,66,67,68,69], an approach later replaced with iPSCs [30, 70] (Table 2). There are several ways to differentiate megakaryocytes from iPSCs. Mimicking embryogenesis, iPSCs are differentiated stepwise to mesodermal, HPC-like cells, and then to megakaryocytes by 2D cultivation using extracellular components [70] or embryoid bodies under 3D cultivation [71]. For the mesoderm and HPC differentiation, humoral factors such as BMP4, VEGF, bFGF, and WNT activators are added. For the megakaryocyte differentiation, TPO and SCF are indispensable but other humoral factors are also required. Feeder cells can also support HPC and megakaryocyte differentiation [30]. Based on these early successes, the amplification of iPSCs to large quantities and then their differentiation to megakaryocytes to derive platelets was considered [70]. However, the differentiation efficiency from iPSCs to megakaryocytes was low. We estimated that this technique would require hundreds or more liters of culture media supplemented with expensive cytokines and other substances for a long culture process, thus demanding high labor and unacceptable cost.
Establishment of expandable megakaryocytic cell lines through genetic modifications
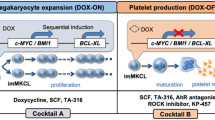
Genetic manipulation technology has enabled the induction of megakaryocytes through various ways (Fig. 2a). One approach is the genetic manipulation of iPSCs or HPCs to establish expandable megakaryocyte cell lines. Based on the finding that the transient activation of c-MYC is critical for efficient megakaryopoiesis from human iPSCs [30], we established immortalized megakaryocyte progenitor cell lines (imMKCLs) by the sequential introduction of c-MYC, BMI-1, and then BCL-XL during the differentiation process of iPSC-derived HPCs into megakaryocyte progenitors (Fig. 2b) [72]. Overexpressing c-MYC promotes proliferation, while BMI1 and BCL-XL suppress cell senescence and cell death, respectively. These three transgenes were incorporated into a doxycycline (DOX)-inducible gene expression vector to control their expression. In the presence of DOX, the expression of the transgenes is induced to endow imMKCLs to vigorously proliferate (DOX-ON). Then, when switched to DOX-free media, the expression of the three diminishes, which turns imMKCL from the proliferation state to maturation state, and platelets are released three days later (DOX-OFF) (Fig. 2b). We were able to expand imMKCLs effectively in static or rocking-motion type bioreactors [73]. Another group used a single-use bioreactor to expand commercial megakaryocytes through automated gas exchange and orbital shaking, which might be also useful [74].

Various types of induced megakaryocytes (MKs). a Cell sources and genetic manipulations to induce various types of MKs. b imMKCLs are established from iPSCs by introducing c-MYC and BMI at the hematopoietic progenitor cell stage and BCL-XL at the early megakaryocyte stage. The addition of doxycycline induces the expression of the transgenes, thereby forcing imMKCLs to self-replicate and expand (DOX-ON). The depletion of doxycycline ceases the expression of the transgenes, which allows imMKCLs to mature and release platelet-like particles (DOX-OFF)
Meanwhile, other groups have sought to activate transcription factors that determine hematopoiesis and megakaryopoiesis to induce megakaryocytes (Fig. 2a). Ono et al. reported the conversion of fibroblasts into megakaryocytes by introducing a set of three genes: NF-E2, MAFG and MAFK [75]. Later, Pulecio et al. reported similar transdifferentiation by a set of six genes: GATA2, RUNX1, GATA1, TAL-1, LMO2 and c-MYC [76]. Using another overexpression approach but with a different concept, Moreau et al. established an expandable megakaryocyte cell line, forward programmed MKs (fopMKs) [71]. Based on a screening of candidate megakaryocyte-specific transcription factor genes, GATA1, FLI1 and TAL1 were chosen to be introduced into PSCs two days before the differentiation into megakaryocytes. fopMKs can be cultivated in the absence of feeder cells and highly expanded in the presence of TPO and SCF. Then, by changing the cytokines to TPO and IL-1α, fopMKs mature to release platelets.
imMKCL and fopMKs can be cryopreserved, thawed upon demand, expanded and then induced to produce platelets upon switching to the maturation stage (Fig. 3). The issue of the low differentiation efficiency from iPSCs to megakaryocytes is removed, and the number of days required for the production is reduced. In principle, a clone that is confirmed for safety and high platelet productivity can be banked in advance for master cells, the source cells to start production. Then, after thawing, these cells would be subject to a standard operating procedure (SOP) that complies with good manufacturing practice (GMP) to stably produce platelets ex vivo with guaranteed clinical-grade quality (Fig. 3).

Expandable megakaryocytes as a master cell line for the GMP grade production of iPSC-PLTs. Expandable megakaryocyte (MK) lines are established from iPSCs by the transduction of specified sets of genes. These cells are cryopreserved as a master cell stock, and upon requirement, thawed, expanded, matured and subjected to platelet production in good manufacturing practice (GMP) grade conditions
Besides genetic manipulation, Tozawa et al. reported that a human adipose tissue-derived mesenchymal stromal/stem cell line (ASCL) can produce platelets upon megakaryocyte induction (Fig. 2a) [77]. Interestingly, because of the unique ability of these ASCLs to produce endogenous TPO through transferrin stimulation, TPO does not need to be added to the media. Meanwhile, Patel et al. reported a scaled-up induction of cryopreservable megakaryocytes from human cord blood HSCs through an optimized culture condition [78]. While these induced megakaryocytes have not produced platelets at the clinically required quantity and quality, the findings may contribute to improved ex vivo platelet production.
Development of drugs that contribute to megakaryocyte maturation and platelet biogenesis
TPO and SCF are an indispensable combination of cytokines for the in vitro differentiation of iPSCs to megakaryocytes [70,71,72]. Concerns for using recombinant TPO are its high cost and immunogenicity. Indeed, a clinical trial of recombinant TPO for patients with thrombocytopenia was discontinued due to the formation of antibodies against TPO in general, leading to even lower platelet count than before the administration [79, 80]. In response, TPO mimetics have been developed for clinical use [81]. We developed TA-316, a low-molecular compound with effects higher than current TPO mimetics and comparable to recombinant TPO in vitro [82]. These TPO mimetics could be used as an alternative to recombinant TPO for platelet production ex vivo. Other cytokines such as interleukin-1 (IL-1) and IL-6 have also been used [83].
There have been many substances and conditions reported to enhance megakaryopoiesis and thrombopoiesis. Chemotactic factors secreted from endothelial and other cells in the bone marrow niche induce megakaryocyte migration and proplatelet formation. These include the secreted molecules stromal-derived factor 1 (SDF-1), sphingosine 1-phosphate (S1P) and chemokine ligand 5 (CCL5, or RANTES) [84,85,86]. WNT family proteins are also involved in megakaryocyte proliferation, maturation and proplatelet formation [87]. Nicotinamide increases the polyploidization and proplatelet formation by increasing TP53 activity [88]. Src-family kinases inhibit megakaryocyte proliferation and differentiation and either a Src inhibitor or a multi-kinase inhibitor induces polyploidization [89, 90]. Rho A and the downstream Rho-associated protein kinase (ROCK) contributes to actin function, and ROCK inhibitors have been reported to enhance megakaryocyte polyploidization and proplatelet formation [91,92,93,94]. A myosin inhibitor showed a similar effect [95]. A bromodomain and extraterminal motif protein (BET) inhibitor enhanced megakaryocyte maturation through the inhibition of c-MYC and enhancement of GATA1 expression [70]. Aurora B kinase is indispensable for megakaryocyte endomitosis, but its inhibitor has been reported to increase megakaryocyte ploidy, size and granularity [96, 97]. Aryl hydrocarbon receptor (AhR) antagonists are known to enhance HSC expansion [98,99,100] as well as increase the ploidy of megakaryocytes and the proportion of platelet-producing megakaryocyte precursors during in vitro differentiation [101]. In addition, hypercholesterolemia correlates with increased platelet biogenesis and function [102, 103]. Finally, hypoxia is postulated to exist in the bone marrow and reported to increase platelet production in vitro [104], and mild hyperthermia of 39 °C is also reported to enhance platelet production in vitro [105].
The combinations of different chemicals have been tested by many groups to enhance megakaryocyte maturation and eventual platelet production. For instance, Avanzi et al. found that the combination of nicotinamide, an Src inhibitor and Aurora B inhibitor was effective [96]. Feng et al. incorporated hypoxia, hyperthermia as well as a ROCK inhibitor and BET inhibitor [70]. In an in vivo megakaryocyte infusion setting, pre-infusion treatment with a Src inhibitor enhanced the platelet yield, but not a ROCK inhibitor or an Aurora kinase B inhibitor [97]. Through an assessment of relevant substances or drugs, we found that the combination of a ROCK inhibitor and an AhR antagonist enables platelet generation without feeder cells for imMKCL cultivation [73]. For further high-throughput screening of drugs that enhance megakaryocyte maturation, we developed an imMKCL with a fluorescent reporter that correlates with the expression of a maturation-specific protein [106].
During the in vitro generation of platelets, the metalloproteases ADAM17 and ADAM10 are activated at 37 ℃, the temperature at which cultivation is suitable. ADAM17 and ADAM10 cleave GPIbα (CD42b) and GPVI on the surface of platelets, respectively [107, 108], rendering the platelets to become like those in thrombasthenia, a series of diseases with significantly reduced platelet functionality due to mutations in genes including GPIbα and GPVI [107,108,109]. Matrix metalloproteinase (MMP) inhibitors have been utilized to inhibit these cleavages [110, 111], with an MMP8-specific inhibitor, MMP8-I, superior to a pan-MMP inhibitor, GM6001, in suppressing this effect [70]. We developed KP-457, which prevents the cleavage activity of ADAM17 and ADAM10, thus retaining the expression of GPIbα and GPVI on imMKCL-derived platelets [112].
The role of hydrodynamic forces in platelet generation from mature megakaryocytes and the development of bioreactors
Based on the live imaging analysis of proplatelet extension and platelet release in mouse bone marrow, Junt et al. concluded that shear stress caused by blood flow is the primary biophysical determinant for platelet release from megakaryocytes in vivo [47]. In fact, a simple study using flow chambers coated with vWF or extracellular matrix (ECM) showed that a high shear rate accelerated the in vitro platelet production from megakaryocytes derived from cord blood or bone marrow HSCs [113]. Accordingly, many groups have attempted to mimic the bone marrow environment for megakaryocytes in vitro through systems that are designed to have megakaryocytes extending proplatelets into artificial flow streams (Table 3).
Sullenbarger et al. created a 3D system in which cord blood cells were cultured in polyester fabric or hydrogel scaffolds [114]. In the material was TPO and/or fibronectin, and MK differentiating medium and gasses were continuously perfused. Compared to 2D systems, platelets were produced for a longer period of time and at a higher yield. We developed a microfluidic chip system in which PSC-derived megakaryocytes are trapped in slits [115]. Once trapped, the cells extend proplatelets from the pore to incur shear flow. Later, Martinez et al. designed slit-type bioreactors with optimal flow patterns and uniform shear stress based on extensive computational fluid dynamics analysis [116]. Thon et al. designed a microfluidic chip-based bioreactor that incorporates chemical and physical components of the bone marrow such as stiffness, the ECM, pore size, endothelial cells and shear stress flow [117]. Additionally, their bioreactor is capable of live-cell microscopy to observe the PSC-derived megakaryocytes extending proplatelets from the pores. To faithfully recapitulate the bone marrow niche environment, Di Buduo et al. created an artificial bone marrow niche system made from vascular tubes in natural silk protein sponges harboring cytokines, ECM components and endothelial-derived proteins [118]. Blin et al. built a different-type of a microfluidic device in which multiple vWF-coated micropillars stand within a wider channel to capture megakaryocytes. The cells extend proplatelets and release platelets depending on the controlled shear rates [119]. Avanzi et al. designed a bioreactor with a nanofiber membrane, through its’ loose texture megakaryocytes extend proplatelets into. They used this bioreactor to assemble a stepwise platelet production line from cord blood cell-derived CD34+ cells [120]. Fujiyama et al. used isolated porcine thighbone to create a closed culture circuit in a xenogeneic bone environment [121]. However, despite the many types of bioreactors, the yield of platelets is estimated to be no more than 20–30 per megakaryocyte. This number falls far short of the 800–2000 platelets per megakaryocyte in vivo [53, 54], and the quality of the produced platelets are inferior.
To readdress the hydrodynamic factors involved in platelet biogenesis, we performed in vivo live imaging analysis of mouse bone marrow megakaryocytes with flow dynamic visualization and observed that megakaryocytes actively generating platelets were exposed to turbulent flow [73]. We then tested a vertical reciprocal-motion stirrer, in which turbulent flow is generated, to cultivate imMKCLs at the maturation stage and succeeded in dramatically improving the yield and quality of iPSC-PLTs [73]. Simulation analysis identified shear stress and turbulent energy as key parameters of the turbulent flow conditions. By adjusting these two parameters, the same turbulent flow conditions were applied to an 8 L-scale bioreactor, which enabled the production of approximately 1011 competent iPSC-PLTs, half the number needed for a single transfusion [73]. Interestingly, the megakaryocytes in the new bioreactor released soluble factors that autonomously promoted the iPSC-PLT production [73]. Two of these, IGFBP2 and MIF, promoted the release of ECM proteins from the megakaryocytes. This release could create a scaffold-like condition for the megakaryocytes. Another soluble factor, NRDC, promoted the cleavage of platelet precursors from elongated proplatelets.
Alternatively, instead of pursuing bioreactors to produce platelets ex vivo, the intravenous infusion of megakaryocytes differentiated from cord blood HSCs has been proposed [122, 123]. Animal models have shown that the infused megakaryocytes can reach the lungs, where they are trapped by the capillary bed to produce platelets as endogenous megakaryocytes have been observed to do [50]. The potential disadvantages of this approach, however, are the delayed circulation of platelets compared with the direct infusion of platelets and the unknown safety and immunogenicity profiles of the infused nucleated megakaryocytes. Furthermore, it is difficult to continuously supply cells from the same source, because cord blood HSCs cannot be expanded like PSCs. However, this approach omits the necessity of producing platelets ex vivo, and the lung-generated platelets may be more physiological in nature.
Platelet purification and packaging
To prepare iPSC-PLTs into a transfusable format, the produced platelets must be washed, purified and resuspended in storage solution with appropriate conditions. Based on differences in size and specific gravity, filtrations and continuous centrifugation can be applied for the washing, purification and concentration before resuspension [73, 124]. Bicarbonate Ringer's solution added with ACD-A solution (BRS-A) is used for the washed platelet products [125] and could also be used as a resuspension solution for ex vivo platelets. Other solutions such as PAS solutions recently tested for blood products may also be applied [126]. Given the bacteria-free production procedure for iPSC-PLTs, the development of solutions that retain platelet function could contribute to significantly extending the shelf life of iPSC-PLTs, which is currently only 4 or 5 days, and reduce the difficulty in balancing the supply and demand.
Preclinical studies of the safety and efficacy of iPSC-PLTs
General overview and in vitro studies
Before clinical trials take place, rigorous preclinical tests to assess the safety and efficacy are required from regulation authorities (Fig. 4). A master cell bank, i.e. cell stock, that will be used as the original source for each batch of platelet production, whether cord blood HSCs, PSCs or megakaryocyte lines, will need to be thoroughly tested for viruses and any other infectious pathogens. A platelet production process in compliance with GMP grade from a certified pathogen-free master cell bank will assure products devoid of infection. Safety of the final platelet product needs to be tested in animals. The toxicity and mutagenicity of any additives used in the production process need to be tested in animals and in vitro unless previously proven safe or the final concentration of the additives are below the level determined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).

Preclinical tests for iPSC-PLTs at different stages. Megakaryocyte master cells, added substances and the final platelet product must each undergo thorough tests as listed above before the platelet product reaches clinical trials. PF4: platelet factor 4; βTG: β-thromboglobulin
A safety parameter always tested in regenerative medicine is tumorigenicity, which applies to iPSC-PLTs as well (Fig. 4). Platelets themselves are anucleate cells, which means they cannot proliferate. However, the batch may include contaminating nucleated cells. iPSCs form teratoma upon transfer in vivo [2,3,4], and expandable megakaryocytes may also proliferate after infusion [71, 72]. Chances are extremely low that PSCs remain in the long cultivation for megakaryocyte differentiation and maturation, but confirmation assays are still required. On the other hand, depending on the method, megakaryocytes with proliferation potential could substantially remain in the final platelet suspension [73]. These nucleated cells may also accumulate oncogenic mutations during the culture [127, 128]. Membrane filtration and centrifugation can reduce the number of nucleated cells, but not completely. To eliminate the risk of tumorigenicity, irradiation similar to that used to prevent GVHD after transfusion with current blood donation products [129] could be performed and should not affect the function of the platelets. Even if these procedures are taken, however, the risk of tumorigenicity still needs to be confirmed through in vitro culture and in vivo administration tests.
Platelets can be activated through various mechanisms, and thus are easy to deteriorate or aggregate by stimulation including low temperatures and physical pressures [126]. Therefore, it is crucial to evaluate the functional viability of the produced platelets (Fig. 4). Various in vitro measures include flow cytometry analysis of the CD42b and CD62P expressions, PAC-1 and Annexin V binding, as well as aggregation assays, clot retraction assays and electron microscopy observations [130, 131]. The high level of CD42b expression and low levels of CD62P expression and Annexin V binding at steady state indicate good quality. Although not fit for assessment before transfusion, observation of the intact platelet ultrastructure under electron microscopy is a reliable measure. A high level of CD62P expression, PAC-1 binding, aggregation and clot retraction upon stimulation with ligands like thrombin-activated receptor peptide (TRAP) and ADP suggest platelets with good functionality.
Animal models for in vivo studies of efficacy
For infused platelets to be effective in vivo, they need to circulate well and show hemostasis function at the bleeding site but at the same time not form unnecessary thrombi in the lungs or other organs. In humans, platelets are considered to have up 10 days of life, after which they are trapped and destroyed by splenic macrophages. However, in thrombocytopenia patients, the transfused platelets have a shorter lifetime, sometimes 3 days or less according to radiolabeling studies [132].
In vitro functionality tests are ultimately not sufficient to assess the in vivo functionality of platelets, therefore, to test the circulation and hemostasis function for the preclinical studies, animal models are adopted [Fig. 4). Using such animal models, the function of the platelets of interest could be compared with donor-derived product platelets. Immunodeficient mice have been used to assess circulation by flow cytometry and hemostasis through tail bleeding [73, 133, 134]. Radiation or treatment with anti-mouse CD41 antibodies is used to prepare thrombocytopenic conditions. In addition, mice with laser-induced vascular injury have been used to observe the incorporation of infused platelets by fluorescence intravital microscopy. [68, 72].
In addition to mice, rat, guinea pigs and dogs are also used for in vivo evaluations, but rabbit models are most commonly used, owing partly to the ease of assessment through the ear veins [135, 136]. Rabbits are pretreated with ethyl palmitate to suppress clearance by the reticuloendothelial system. Cytotoxic drugs such as busulfan are used to create thrombocytopenic conditions. Our collaborator found splenectomy improves the circulation [137]. Notably, rabbit and rat kidney or liver injury models have been used to assess hemostasis in massive visceral hemorrhaging for various platelet substitutes [130].
As mentioned above, iPSC-PLTs have a fetal/neonatal nature, which may give them longer survival, but also hyporeactivity compared with adult-type platelets [44, 45]. However, we observed iPSC-PLTs and blood donor-derived platelets have generally the same level of in vitro and in vivo functionality [73]. iPSC-PLTs are initially larger but become smaller after the transfusion and show a temporary increase in circulation number. We speculate that the younger, larger platelets become fragmented in the lung upon the circulation.
Applying iPSC-PLTs to alloimmune platelet transfusion refractoriness
Autologous product as a complete match source
As alloantigens, platelets express ABO antigens, human leukocyte antigen class I (HLA-I) and human platelet antigen (HPA). The production of alloantibodies against these antigens causes platelet transfusion refractoriness (allo-PTR), which appears as a poor increase of post-transfusion platelet count in transfused patients [14]. The contribution of ABO antigens is considered to play only a minor role in the platelet transfusion settings but may affect significantly platelets that highly express them [138, 139]. Alloantibodies against non-self HLA-I and HPA are produced through pregnancy or previous platelet transfusions or allotransplantations. Measures such as the leukoreduction of blood components or the use of single donor-derived products rather than multiple donor-derived pooled products have lowered the incidence of allo-PTR from about 15% to about 5% of patients receiving transfusions [14].
HLA-I is the major cause of allo-PTR, while HPA causes other forms of alloimmune responses, including post-transfusion purpura (PTP), in which the post-transfusion platelet count somehow becomes even lower several days after the transfusion, and fetal and neonatal alloimmune thrombocytopenia (FNAIT), in which platelets are cleared from infants by maternal anti-HPA alloantibodies [14, 140]. In patients with allo-PTR, only platelet products that match these alloantigens are effective, but such products may not be available for rare HLA-I or HPA or in emergencies.
iPSC-PLTs can cope with allo-PTR in several ways (Fig. 5). First, autologous iPSC-PLTs can be produced by establishing iPSCs from allo-PTR patients themselves. In 2019, we started a clinical trial of autologous iPSC-PLTs for an aplastic anemia patient with allo-PTR who had no matching blood donor in Japan [141]. Even from aplastic anemia patients, skin fibroblasts or blood lymphocytes can be used to establish iPSCs and produce iPSC-PLTs. This approach is also ethically highly compatible since it should not elicit an alloimmune response or blood-borne infections [142]. The limitation of autologous iPSC-PLTs, however, is that the iPSC production process is time-consuming, labor-intensive, and too costly for every patient. In addition, the production efficiency and quality of the platelets could vary depending on the source individual and iPSC line. Therefore, good autologous iPSC-PLTs cannot always be assured in a timely manner.

iPSC-PLTs for allo-PTR. Because iPSCs can be established from essentially anyone, autologous iPSC-PLTs can be produced from allo-PTR patients. These iPSC-PLTs are completely compatible for HLA-I and HPA, but require a long time for their production and have a high cost. Alternatively, allogeneic iPSC-PLTs with HLA-homozygous haplotype can be produced from stocked GMP-grade iPSCs. These cells act as an off-the-shelf uniform quality product and have a lower cost. Because it is relatively easy to genetically modify iPSCs, allogeneic iPSC-PLTs deleted for HLA-I can be produced, making them universally compatible for HLA-I. These iPSC-PLTs can be a single off-the-shelf product with low cost and thus are suitable as a platform for other platelet products
Allogeneic HLA-homozygous haplotype products as a stocked source
To overcome the limitations in the cost and availability of autologous iPSC-PLTs, premanufactured off-the-shelf allogeneic iPSC-PLTs from megakaryocyte cell lines that were confirmed to expand and produce platelets with high efficiency could be applied (Fig. 5). Allogeneic preparations are subject to strategies that overcome allogeneic immune responses. As for the ABO antigen, O-type donors could be selected. For HLA class I, good donors for the iPSCs are people with homozygous HLA. HLA genes are located as a cluster within chromosome 6p21, and two sets of HLA haplotypes exist in each person. HLA-homozygous grafts are compatible with a wide range of recipients because matching just one of the two haplotypes of the recipient evades rejection. In fact, blood donors with homozygous HLA are often registered and called upon when there is a demand for HLA-compatible platelets. Based on this wide compatibility of HLA-homologous grafts, iPSCs from HLA-homozygous subjects or cord blood are being stocked for regenerative medicine use [143, 144]. These iPSCs can be used to produce iPSC-PLTs. It is estimated that iPSC lines expressing the most frequent 10, 75 and 140 HLA would cover 50%, 80% and 90% of the Japanese population, respectively [144].
HLA-deficient products as universal products
With regards to HLA, platelets nullified of HLA class I could become a universal product [70, 145,146,147,148], and the mass production of one line would significantly lower the cost compared with the preparation of a product library (Fig. 5). HLA-I can be either depleted (knockout; KO) using CRISPR/Cas9 or other gene-editing technologies or downregulated (knockdown; KD) using RNA silencing technologies. One common KO and KD target has been beta2-microglobulin (B2M), a molecule composing the HLA-I heterodimer complex and required for the cell surface expression of all HLA-I family molecules [70, 145,146,147,148]. Another target would be HLA-ABC, because antigens of anti-HLA-I antibodies in allo-PTR are mostly HLA-A and HLA-B, with rare cases of HLA-C [149].
A general concern in HLA-I deficient products is that the total elimination of HLA-I could elicit rejection by natural killer (NK) cells because HLA-I molecules are inhibitory ligands of killer immunoglobulin-like receptors (KIRs) and CD94/NKG2 on NK cells [150, 151]. As means to avoid rejection by NK cells, KD, which can retain a low but not null expression level, is suggested. Other proposals include overexpressing HLA-E [152] or retaining the expression of HLA-C [153]. Interestingly, we found that B2M-KO iPSC-PLTs, which are completely devoid of HLA-I expression, do not elicit an NK cell response in vitro [147]. To confirm the circulation in vivo in the presence of NK cells, we developed humanized immune system (HIS) mice, which were reconstituted with a sufficient number of human NK cells to reject B2M-knockout iPSC-derived hematopoietic cells. B2M-KO iPSC-PLTs circulated comparably with wild-type iPSC-PLTs in the NK cell reconstituted HIS mice and also circulated in the presence of anti-HLA-I antibody [147]. While this observation removes the concern of NK cells rejecting HLA-KO platelets, the reason for the low immunogenicity of platelets remains unknown.
Low immunogenic O-type HLA-depleted iPSC-PLTs are suitable as a common platform for products with further genetic modifications or drug loading to endow an alternative or a new phenotype (Fig. 5). As for HPA incompatibility, the deletion strategy used for HLA-I cannot be applied, because HPA is a group of molecules essential for platelet function. Given that allo-PTR due to HPA frequently involves HLA-I as well, it would be appropriate to convert HPA on HLA-depleted iPSC-PLTs to the desired HPA type by gene modification [154].
Meanwhile, platelets are known to have functions besides hemostasis, such as contributing to the maintenance of vascular integrity, vascular and lung development in newborns, tissue regeneration, anti-bacterial immunity and anti-malarial immunity, and are also involved in the pathogenesis of rheumatoid arthritis and cancer [155]. Taking these into account, platelets have been studied as carriers to incorporate coagulation factors [156,157,158], anti-cancer drugs [159, 160], and oncolytic virus [161]. Platelets enable these factors to circulate in an encapsulated form and deliver to the sites they are attracted such as bleeding and cancer [162]. These substances could be loaded onto HLA-KO iPSC-PLTs through a defined process including genetic manipulation [163], providing a novel drug delivery system of standardized quality.
Conclusion
The discovery of iPSCs has brought new possibilities in regenerative medicine, but to reach clinical application, many qualitative and quantitative issues needed to be addressed. Platelets derived from iPSCs have reached a clinical trial owing to the development of expandable megakaryocyte lines, hydrodynamically designed bioreactors and novel drugs. Given that HLA-null universal iPSC-PLTs are suitable as a mass commercial product, transfusion medicine could evolve to a new generation, assuring a ubiquitous and safe supply of platelet products. Furthermore, iPSC-PLTs are amenable to other genetic manipulations, which may bring forth novel therapeutic measures.
References
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. https://doi.org/10.1016/j.cell.2006.07.024
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872
Yu J, Vodyanik MA, Smuga-Otto K et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858):1917–1920. https://doi.org/10.1126/science.1151526
Park IH, Zhao R, West JA et al (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451(7175):141–146. https://doi.org/10.1038/nature06534
Karagiannis P, Eto K (2016) Ten years of induced pluripotency: from basic mechanisms to therapeutic applications. Development 143:2039–2043
Blau HM, Daley GQ (2019) Stem cells in the treatment of disease. N Engl J Med 380(18):1748–1760
Park IH, Arora N, Huo H et al (2008) Disease-specific induced pluripotent stem cells. Cell 134(5):877–886
Kim H, Schaniel C (2018) Modeling hematological diseases and cancer with patient-specific induced pluripotent stem cells. Front Immunol 9:2243
Szczepiorkowski ZM, Dunbar NM (2013) Transfusion guidelines: when to transfuse. Hematol Am Soc Hematol Educ Program 2013:638–644
Estcourt LJ, Birchall J, Allard S, Bassey SJ, Hersey P, Kerr JP, Mumford AD, Stanworth SJ, Tinegate H (2017) Guidelines for the use of platelet transfusions. Br J Haematol 176:365–394
Whitaker BI, Hinkins S (2011) The 2011 National Blood Collection and Utilization Survey Report. The United States Department of Health and Human Services, Washington
Williamson LM, Devine DV (2013) Challenges in the management of the blood supply. Lancet 381:1866–1875
Stanworth SJ, New HV, Apelseth TO et al (2020) Effects of the COVID-19 pandemic on supply and use of blood for transfusion. Lancet Haematol S2352–3026(20):30186–30191. https://doi.org/10.1016/S2352-3026(20)30186-1
Stanworth SJ, Navarrete C, Estcourt L, Marsh J (2015) Platelet refractoriness–practical approaches and ongoing dilemmas in patient management. Br J Haematol 171(3):297–305
Kiely P, Gambhir M, Cheng AC, McQuilten ZK, Seed CR, Wood EM (2017) Emerging infectious diseases and blood safety: modeling the transfusion-transmission risk. Transfus Med. 312:154–164
Palmer AF, Intaglietta M (2014) Blood substitutes. Annu Rev Biomed Eng 16:77–101
Girish A, Sekhon U, Sen GA (2020) Bioinspired artificial platelets for transfusion applications in traumatic hemorrhage. Transfusion 60(2):229–231
Hagisawa K, Kinoshita M, Takikawa M, Takeoka S, Saitoh D, Seki S, Sakai H (2019) Combination therapy using fibrinogen γ-chain peptide-coated, ADP-encapsulated liposomes and hemoglobin vesicles for trauma-induced massive hemorrhage in thrombocytopenic rabbits. Transfusion 59(10):3186–3196
Toner RW, Pizzi L, Leas B, Ballas SK, Quigley A, Goldfarb NI (2011) Costs to hospitals of acquiring and processing blood in the US: a survey of hospital-based blood banks and transfusion services. Appl Health Econ Health Policy 9(1):29–37
Keller Keller G, Kennedy M, Papayannopoulou T, Wiles MV (1993) Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol 13(1):473–486
Nakano T, Kodama H, Honjo T (1994) Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science 265(5175):1098–1101
Palacios R, Golunski E, Samaridis J (1995) In vitro generation of hematopoietic stem cells from an embryonic stem cell line. Proc Natl Acad Sci U S A 92(16):7530–7534
Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA (2001) Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci USA 98:10716–10721
Eto K, Murphy R, Kerrigan SW et al (2002) Megakaryocytes derived from embryonic stem cells implicate CalDAG-GEFI in integrin signaling. Proc Natl Acad Sci USA 99(20):12819–12824
Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG (2005) Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood 106(5):1601–1603
Woll PS, Martin CH, Miller JS, Kaufman DS (2005) Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol 175(8):5095–5103
Gaur M, Kamata T, Wang S, Moran B, Shattil SJ, Leavitt AD (2006) Megakaryocytes derived from human embryonic stem cells: a genetically tractable system to study megakaryocytopoiesis and integrin function. J Thromb Haemost 4(2):436–442
Choi KD, Vodyanik MA, Slukvin II (2009) Generation of mature human myelomonocytic cells through expansion and differentiation of pluripotent stem cell-derived lin-CD34+CD43+CD45+ progenitors. J Clin Invest 119(9):2818–2829
Ye Z, Zhan H, Mali P et al (2009) Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 114(27):5473–5480
Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H et al (2010) Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med 207(13):2817–2830
Grigoriadis AE, Kennedy M, Bozec A, Brunton F, Stenbeck G, Park IH et al (2010) Directed differentiation of hematopoietic precursors and functional osteoclasts from human ES and iPS cells. Blood 115(14):2769–2776
Senju S, Haruta M, Matsumura K, Matsunaga Y, Fukushima S, Ikeda T et al (2011) Generation of dendritic cells and macrophages from human induced pluripotent stem cells aiming at cell therapy. Gene Ther 18(9):874–883
Ditadi A, Sturgeon CM, Keller G (2017) A view of human haematopoietic development from the Petri dish. Nat Rev Mol Cell Biol 18(1):56–67
Palis J (2016) Hematopoietic stem cell-independent hematopoiesis: emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett 590(22):3965–3974
Psaila B, Mead AJ (2019) Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 133(13):1427–1435
Sumide K, Matsuoka Y, Kawamura H, Nakatsuka R, Fujioka T, Asano H, Takihara Y, Sonoda Y (2018) A revised road map for the commitment of human cord blood CD34-negative hematopoietic stem cells. Nat Commun 9(1):2202
Amabile G, Welner RS, Nombela-Arrieta C, D’Alise AM, Di Ruscio A, Ebralidze AK, Kraytsberg Y, Ye M, Kocher O, Neuberg DS et al (2013) In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013(121):1255–1264
Suzuki N, Yamazaki S, Yamaguchi T, Okabe M, Masaki H, Takaki S, Otsu M, Nakauchi H (2013) Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther 2013(21):1424–1431
Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu Y-F, Goettel JA, Serrao E, Rowe RG, Malleshaiah M et al (2017) Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545:432–438
Lopez-Yrigoyen M, Yang CT, Fidanza A et al (2019) Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nat Commun 10(1):881
Vanuytsel K, Matte T, Leung A et al (2018) Induced pluripotent stem cell-based mapping of β-globin expression throughout human erythropoietic development. Blood Adv 2(15):1998–2011
Demirci S, Tisdale JF (2018) Definitive Erythropoiesis from Pluripotent Stem Cells: Recent Advances and Perspectives. Adv Exp Med Biol 1107:1–13
Elagib KE, Brock AT, Goldfarb AN (2018) Megakaryocyte ontogeny: Clinical and molecular significance. Exp Hematol 61:1–9
Margraf A, Nussbaum C, Sperandio M (2019) Ontogeny of platelet function. Blood Adv 3(4):692–703
Liu ZJ, Hoffmeister KM, Hu Z, Mager DE, Ait-Oudhia S, De- brincat MA, Pleines I, Josefsson EC, Kile BT, Italiano J Jr, Ramsey H, Grozovsky R, Veng-Pedersen P, Chavda C, Sola-Visner M. Expansion of the neonatal platelet mass is achieved via an extension of platelet lifespan. Blood 2014; 123: 3381–9.
Machlus KR, Italiano JE Jr (2013) The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol 201(6):785–796
Junt T, Schulze H, Chen Z, Massberg S, Goerge T, Krueger A et al (2007) Dynamic visualization of thrombopoiesis within bone marrow. Science 317(5845):1767–1770
Borges I, Sena I, Azevedo P, Andreotti J, Almeida V, Paiva A, Santos G, Guerra D, Prazeres P, Mesquita LL, Silva LSB, Leonel C, Mintz A, Birbrair A (2017) Lung as a niche for hematopoietic progenitors. Stem Cell Rev Rep 13(5):567–574
Aliberti G, Proietta M, Pulignano I, Tritapepe L, Di Giovanni C, Schiappoli A, Vercillo G (2002) The lungs and platelet production. Clin Lab Haematol 24(3):161–164
Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM et al (2017) The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544(7648):105–109
Nishimura S, Nagasaki M, Kunishima S, Sawaguchi A, Sakata A, Sakaguchi H et al (2015) IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J Cell Biol 209(3):453–466
Kanaji T, Vo MN, Kanaji S, Zarpellon A, Shapiro R, Morodomi Y, Yuzuriha A, Eto K, Belani R, Do MH, Yang XL, Ruggeri ZM, Schimmel P (2018) Tyrosyl-tRNA synthetase stimulates thrombopoietin-independent hematopoiesis accelerating recovery from thrombocytopenia. Proc Natl Acad Sci USA 115(35):E8228–E8235
Kuter DJ. Megakaryocyte biology and the production of platelets. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com (Accessed on June 11, 2019.)
Michelson A, Cattaneo M, Frelinger A, Newman P (2019) Platelets, 4th edn. Academic Press, Cambridge
Lok S, Kaushansky K, Holly RD et al (1994) Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature 369:565–568
Kaushansky K, Lok S, Holly RD et al (1994) Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 369:568–571
de Sauvage FJ, Hass PE, Spencer SD et al (1994) Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 369:533–538
Wendling F, Maraskovsky E, Debili N et al (1994) cMpl ligand is a humoral regulator of megakaryocytopoiesis. Nature 369:571–574
Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P (1995) Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood 85(2):402–413
Matsunaga T, Tanaka I, Kobune M, Kawano Y, Tanaka M, Kuribayashi K, Iyama S, Sato T, Sato Y, Takimoto R, Takayama T, Kato J, Ninomiya T, Hamada H, Niitsu Y (2006) Ex vivo large-scale generation of human platelets from cord blood CD34+ cells. Stem Cells 24(12):2877–2887
Marini I, Rigoni F, Zlamal J, Pelzl L, Althaus K, Nowak-Harnau S, Rondina MT, Bakchoul T (2020) Blood donor-derived buffy coat to produce platelets in vitro. Vox Sang 115(1):94–102
Eaves CJ (2015) Hematopoietic stem cells: concepts, definitions, and the new reality. Blood 125:2605–2613
Kumar S, Geiger H (2017) HSC niche biology and HSC expansion ex vivo. Trends Mol Med 23:799–819
Fujimoto TT, Kohata S, Suzuki H, Miyazaki H, Fujimura K (2003) Production of functional platelets by differentiated embryonic stem (ES) cells in vitro. Blood 102(12):4044–4051
Wang L, Li L, Shojaei F, Levac K, Cerdan C, Menendez P, Martin T, Rouleau A, Bhatia M (2004) Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity 21(1):31–41
Takayama N, Nishikii H, Usui J, Tsukui H, Sawaguchi A, Hiroyama T et al (2008) Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 111(11):5298–5306
Klimchenko O, Mori M, Distefano A, Langlois T, Larbret F, Lecluse Y, Feraud O, Vainchenker W, Norol F, Debili N (2009) A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood 114(8):1506–1517
Lu SJ, Li F, Yin H, Feng Q, Kimbrel EA, Hahm E, Thon JN, Wang W, Italiano JE, Cho J, Lanza R (2011) Platelets generated from human embryonic stem cells are functional in vitro and in the microcirculation of living mice. Cell Res 21:530–545
Pick M, Azzola L, Osborne E, Stanley EG, Elefanty AG (2013) Generation of megakaryocytic progenitors from human embryonic stem cells in a feeder- and serum-free medium. PLoS ONE 8:e55530
Feng Q, Shabrani N, Thon JN, Huo H, Thiel A, Machlus KR et al (2014) Scalable generation of universal platelets from human induced pluripotent stem cells. Stem Cell Rep 3(5):817–831
Moreau T, Evans AL, Vasquez L, Tijssen MR, Yan Y, Trotter MW et al (2016) Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat Commun 7:11208
Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K et al (2014) Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell 14(4):535–548
Ito Y, Nakamura S, Sugimoto N, Shigemori T, Kato Y, Ohno M et al (2018) Turbulence activates platelet biogenesis to enable clinical scale ex vivo production. Cell 174:636–648
Nurhayati RW, Ojima Y, Dohda T, Kino-Oka M (2018) Large-scale culture of a megakaryocytic progenitor cell line with a single-use bioreactor system. Biotechnol Prog 34(2):362–369
Ono Y, Wang Y, Suzuki H et al (2012) Induction of functional platelets from mouse and human fibroblasts by p45NF-E2/Maf. Blood 120(18):3812–3821
Pulecio J, Alejo-Valle O, Capellera-Garcia S et al (2016) Direct conversion of fibroblasts to megakaryocyte progenitors. Cell Rep 17(3):671–683
Tozawa K, Ono-Uruga Y, Yazawa M et al (2019) Megakaryocytes and platelets from a novel human adipose tissue-derived mesenchymal stem cell line. Blood 133(7):633–643
Patel A, Clementelli CM, Jarocha D et al (2019) Pre-clinical development of a cryopreservable megakaryocytic cell product capable of sustained platelet production in mice. Transfusion 59(12):3698–3713
Li J, Yang C, Xia Y, Bertino A, Glaspy J, Roberts M et al (2001) Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 98:3241–3248
Basser RL, O’Flaherty E, Green M, Edmonds M, Nichol J, Menchaca DM et al (2002) Development of pancytopenia with neutralizing antibodies to thrombopoietin after multicycle chemotherapy supported by megakaryocyte growth and development factor. Blood 99:2599–2602
Ghanima W, Cooper N, Rodeghiero F, Godeau B, Bussel JB (2019) Thrombopoietin receptor agonists: ten years later. Haematologica 104(6):1112–1123
Aihara A, Koike T, Abe N, Nakamura S, Sawaguchi A, Nakamura T et al (2017) Novel TPO receptor agonist TA-316 contributes to platelet biogenesis from human iPS cells. Blood Adv 1(7):468–476
Di Buduo CA, Wray LS, Tozzi L et al (2015) Programmable 3D silk bone marrow niche for platelet generation ex vivo and modeling of megakaryopoiesis pathologies. Blood 125(14):2254–2264
Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, Jin DK, Dias S, Zhang F, Hartman TE, Hackett NR, Crystal RG, Witte L, Hicklin DJ, Bohlen P, Eaton D, Lyden D, de Sauvage F, Rafii S (2004) Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10:64–71
Zhang L, Urtz N, Gaertner F, Legate KR, Petzold T, Lorenz M, Mazharian A, Watson SP, Massberg S (2013) Sphingosine kinase 2 (Sphk2) regulates platelet biogenesis by providing intracellular sphingosine 1-phosphate (S1P). Blood 122:791–802
Machlus KR, Johnson KE, Kulenthirarajan R, Forward JA, Tippy MD, Soussou TS, El-Husayni SH, Wu SK, Wang S, Watnick RS, Italiano JE Jr, Battinelli EM (2016) CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood 127:921–926
Macaulay IC, Thon JN, Tijssen MR, Steele BM, MacDonald BT, Meade G, Burns P, Rendon A, Salunkhe V, Murphy RP, Bennett C, Watkins NA, He X, Fitzgerald DJ, Italiano JE Jr, Maguire PB (2013) Canonical Wnt signaling in megakaryocytes regulates proplatelet formation. Blood 121:188–196
Giammona LM, Panuganti S, Kemper JM et al (2009) Mechanistic studies on the effects of nicotinamide on megakaryocytic polyploidization and the roles of NAD+ levels and SIRT inhibition. Exp Hematol 37(1340–1352):e1343
Lannutti BJ, Blake N, Gandhi MJ, Reems JA, Drachman JG (2005) Induction of polyploidization in leukemic cell lines and primary bone marrow by Src kinase inhibitor SU6656. Blood 105(10):3875–3878
Nurhayati RW, Ojima Y, Taya M (2015) BMS-777607 promotes megakaryocytic differentiation and induces polyploidization in the CHRF-288-11 cells. Hum Cell 28(2):65–72
Chang Y, Auradé F, Larbret F, Zhang Y, Le Couedic JP, Momeux L, Larghero J, Bertoglio J, Louache F, Cramer E, Vainchenker W, Debili N (2007) Proplatelet formation is regulated by the Rho/ROCK pathway. Blood 109(10):4229–4236
Lordier L, Jalil A, Aurade F, Larbret F, Larghero J, Debili N, Vainchenker W, Chang Y (2008) Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood 112:3164–3174
Avanzi MP, Goldberg F, Davila J, Langhi D, Chiattone C, Mitchell WB (2014) Rho kinase inhibition drives megakaryocyte polyploidization and proplatelet formation through MYC and NFE2 downregulation. Br J Haematol 164:867–876
Gobbi G, Mirandola P, Carubbi C, Masselli E, Sykes SM, Ferraro F, Nouvenne A, Thon JN, Italiano JE Jr, Vitale M (2013) Proplatelet generation in the mouse requires PKCε-dependent RhoA inhibition. Blood 122:1305–1311
Shin JW, Swift J, Spinler KR, Discher DE (2011) Myosin-II inhibition and soft 2D matrix maximize multinucleation and cellular projections typical of platelet-producing megakaryocytes. Proc Natl Acad Sci USA 108(28):11458–11463
Avanzi MP, Chen A, He W, Mitchell WB (2012) Optimizing megakaryocyte polyploidization by targeting multiple pathways of cytokinesis. Transfusion 52(11):2406–2413
Jarocha D, Vo KK, Lyde RB, Hayes V, Camire RM, Poncz M (2018) Enhancing functional platelet release in vivo from in vitro-grown megakaryocytes using small molecule inhibitors. Blood Adv 2(6):597–606
Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, Schultz PG, Cooke MP (2010) Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329:1345–1348
Smith BW, Rozelle SS, Leung A, Ubellacker J, Parks A, Nah SK, French D, Gadue P, Monti S, Chui DH, Steinberg MH, Frelinger AL, Michelson AD, Theberge R, McComb ME, Costello CE, Kotton DN, Mostoslavsky G, Sherr DH, Murphy GJ (2013) The aryl hydrocarbon receptor directs hematopoietic progenitor cell expansion and differentiation. Blood 122:376–385
Wagner JE Jr, Brunstein CG, Boitano AE, DeFor TE, McKenna D, Sumstad D, Blazar BR, Tolar J, Le C, Jones J, Cooke MP, Bleul CC (2016) Phase I/II trial of stemregenin-1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell 18:144–155
Strassel C, Brouard N, Mallo L, Receveur N, Mangin P, Eckly A, Bieche I, Tarte K, Gachet C, Lanza F (2016) Aryl hydrocarbon receptor-dependent enrichment of a megakaryocytic precursor with a high potential to produce proplatelets. Blood 127:2231–2240
Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, Wang Y, Shaw JA, Levine RL, Ni H, Tall AR, Wang N (2013) Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med 19:586–594
Wang N, Tall AR (2016) Cholesterol in platelet biogenesis and activation. Blood 127:1949–1953
Lasky LC, Sullenbarger B (2011) Manipulation of oxygenation and flow-induced shear stress can increase the in vitro yield of platelets from cord blood. Tissue Eng Part C Methods 17(1081–8):10
Pineault N, Boucher JF, Cayer MP, Palmqvist L, Boyer L, Lemieux R, Proulx C (2008) Characterization of the effects and potential mechanisms leading to increased megakaryocytic differentiation under mild hyperthermia. Stem Cells Dev 17:483–493
Seo H, Chen SJ, Hashimoto K et al (2018) A β1-tubulin-based megakaryocyte maturation reporter system identifies novel drugs that promote platelet production. Blood Adv 2(17):2262–2272
Bergmeier W, Piffath CL, Cheng G et al (2004) Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res 95:677–683
Gardiner EE, Karunakaran D, Shen Y et al (2007) Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost 5:1530–1537
Dumont B, Lasne D, Rothschild C et al (2009) Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood 114:1900–1903
Bergmeier W, Burger PC, Piffath CL et al (2003) Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro- aged or -injured mouse platelets. Blood 102:4229–4235
Nishikii H, Eto K, Tamura N et al (2008) Metalloproteinase regulation improves in vitro generation of efficacious platelets from mouse embryonic stem cells. J Exp Med 205:1917–1927
Hirata S, Murata T, Suzuki D, Nakamura S, Jono-Ohnishi R, Hirose H et al (2016) Selective inhibition of ADAM17 efficiently mediates glycoprotein Ibalpha retention during ex vivo generation of human induced pluripotent stem cell-derived platelets. Stem Cells Transl Med. 6:720–730
Dunois-Lardé C, Capron C, Fichelson S, Bauer T, Cramer-Bordé E, Baruch D (2009) Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood 114(9):1875–1883
Sullenbarger B, Bahng JH, Gruner R, Kotov N, Lasky LC (2009) Prolonged continuous in vitro human platelet production using three-dimensional scaffolds. Exp Hematol 37(1):101–110
Nakagawa Y, Nakamura S, Nakajima M, Endo H, Dohda T, Takayama N et al (2013) Two differential flows in a bioreactor promoted platelet generation from human pluripotent stem cell-derived megakaryocytes. Exp Hematol 41(8):742–748
Martinez AF, McMahon RD, Horner M, Miller WM (2017) A uniform-shear rate microfluidic bioreactor for real-time study of proplatelet formation and rapidly-released platelets. Biotechnol Prog 33(6):1614–1629
Thon JN, Mazutis L, Wu S, Sylman JL, Ehrlicher A, Machlus KR, Feng Q, Lu S, Lanza R, Neeves KB, Weitz DA, Italiano JE Jr (2014) Platelet bioreactor-on-a-chip. Blood 124:1857–1867
Di Buduo CA, Wray LS, Tozzi L, Malara A, Chen Y, Ghezzi CE, Smoot D, Sfara C, Antonelli A, Spedden E, Bruni G, Staii C, De Marco L, Magnani M, Kaplan DL, Balduini A (2015) Programmable 3D silk bone marrow niche for platelet generation ex vivo and modeling of megakaryopoiesis pathologies. Blood 125:2254–2264
Blin A, Le Goff A, Magniez A, Poirault-Chassac S, Teste B, Sicot G, Nguyen KA, Hamdi FS, Reyssat M, Baruch D (2016) Microfluidic model of the platelet-generating organ: beyond bone marrow biomimetics. Sci Rep 6:21700
Avanzi MP, Oluwadara OE, Cushing MM, Mitchell ML, Fischer S, Mitchell WB (2016) A novel bioreactor and culture method drives high yields of platelets from stem cells. Transfusion 56:170–178
Fujiyama S, Hori N, Sato T, Enosawa S, Murata M, Kobayashi E (2020) Development of an ex vivo xenogeneic bone environment producing human platelet-like cells. PLoS ONE 15(4):e0230507
Xi J, Zhu H, Liu D, Nan X, Zheng W, Liu K, Shi W, Chen L, Lv Y, Yan F, Li Y, Xie X, Wang Y, Yue W, Xu X, Wei X, Zhu J, Huang X, Pei X (2013) Infusion of megakaryocytic progenitor products generated from cord blood hematopoietic stem/progenitor cells: results of the phase 1 study. PLoS ONE 8:e54941
Wang Y, Hayes V, Jarocha D, Sim X, Harper DC, Fuentes R, Sullivan SK, Gadue P, Chou ST, Torok-Storb BJ, Marks MS, French DL, Poncz M (2015) Comparative analysis of human ex vivo-generated platelets vs megakaryocyte-generated platelets in mice: a cautionary tale. Blood 125:3627–3636
Schlinker AC, Radwanski K, Wegener C, Min K, Miller WM (2015) Separation of in-vitro-derived megakaryocytes and platelets using spinning-membrane filtration. Biotechnol Bioeng 112(4):788–800
Oikawa S, Taguchi T, Endo K, Hoshi T, Kawashima W, Horibe Y et al (2015) Storage of washed platelets in BRS-A platelet additive solutions based on two types of clinically available bicarbonated Ringer’s solutions with different electrolyte concentrations. Transfus Apheres Sci 53(2):233–237
Hegde S, Akbar H, Zheng Y, Cancelas JA (2018) Towards increasing shelf life and haemostatic potency of stored platelet concentrates. Curr Opin Hematol 25(6):500–508
Merkle FT, Ghosh S, Kamitaki N et al (2017) Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545(7653):229–233
Rohani L, Johnson AA, Naghsh P, Rancourt DE, Ulrich H, Holland H (2018) Concise review: molecular cytogenetics and quality control: clinical guardians for pluripotent stem cells. Stem Cells Transl Med 7(12):867–875
Treleaven J, Gennery A, Marsh J, Norfolk D, Page L, Parker A et al (2011) Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 152(1):35–51
Cardigan R, Turner C, Harrison P (2005) Current methods of assessing platelet function: relevance to transfusion medicine. Vox Sang 88(3):153–163
Escolar G, McCullough J (2019) Platelet in vitro assays: their correspondence with their in vivo hemostatic potential. Transfusion 59(12):3783–3793
Hanson SR, Slichter SJ (1985) Platelet kinetics in patients with bone marrow hypoplasia: evidence for a fixed platelet requirement. Blood 66(5):1105–1109
Newman PJ, Aster R, Boylan B (2007) Human platelets circulating in mice: applications for interrogating platelet function and survival, the efficacy of antiplatelet therapeutics, and the molecular basis of platelet immunological disorders. J Thromb Haemost 5(Suppl 1):305–309
Gelderman MP, Cheng C, Xu F, Skripchenko A, Ryan J, Li Y, Whitley P, Wagner SJ, Vostal JG (2020) Validation of a SCID mouse model for transfusion by concurrent comparison of circulation kinetics of human platelets, stored under various temperature conditions, between human volunteers and mice. Transfusion. https://doi.org/10.1111/trf.15953 (Epub ahead of print)
Blajchman MA, Lee DH (1997) The thrombocytopenic rabbit bleeding time model to evaluate the in vivo hemostatic efficacy of platelets and platelet substitutes. Transfus Med Rev 11:95–105
Rothwell SW, Maglasang P, Krishnamurti C (1998) Survival of fresh human platelets in a rabbit model as traced by flow cytometry. Transfusion 38:550–556
Watanabe N, Nogawa M, Ishiguro M et al (2017) Refined methods to evaluate the in vivo hemostatic function and viability of transfused human platelets in rabbit models. Transfusion 57(8):2035–2044
Ogasawara K, Ueki J, Takenaka M, Furihata K (1993) Study on the expression of ABH antigens on platelets. Blood 82(3):993–999
Dunbar NM, Ornstein DL, Dumont LJ (2012) ABO incompatible platelets: risks versus benefit. Curr Opin Hematol 19(6):475–479
Curtis BR (2015) Recent progress in understanding the pathogenesis of fetal and neonatal alloimmune thrombocytopenia. Br J Haematol 171(5):671–682
jRCTa050190117; Clinical study of autologous transfusion of iPS cell-derived platelets for thrombocytopenia (iPLAT1) https://jrct.niph.go.jp/en-latest-detail/jRCTa050190117 (Accessed on September 6, 2020.)
Akabayashi A, Nakazawa E, Jecker NS (2019) The world’s first clinical trial for an aplastic anemia patient with thrombocytopenia administering platelets generated from autologous iPS cells. Int J Hematol 109(2):239–240
Turner M, Leslie S, Martin NG, Peschanski M, Rao M, Taylor CJ, Trounson A, Turner D, Yamanaka S, Wilmut I (2013) Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 13:382–384
Umekage M, Sato Y, Takasu N (2019) Overview: an iPS cell stock at CiRA. Inflamm Regen 2(39):17
Gras C, Schulze K, Goudeva L, Guzman CA, Blasczyk R, Figueiredo C (2013) HLA-universal platelet transfusions prevent platelet refractoriness in a mouse model. Hum Gene Ther 24:1018–1028
Borger AK, Eicke D, Wolf C, Gras C, Aufderbeck S, Schulze K et al (2016) Generation of HLA-universal iPSCs-derived megakaryocytes and platelets for survival under refractoriness conditions. Mol Med 22:274–285
Suzuki D, Flahou C, Yoshikawa N, Stirblyte I, Hayashi Y, Sawaguchi A, Akasaka M, Nakamura S, Higashi N, Xu H, Matsumoto T, Fujio K, Manz MG, Hotta A, Takizawa H, Eto K, Sugimoto N (2020) iPSC-derived platelets depleted of HLA class I are inert to anti-HLA class I and natural killer cell immunity. Stem Cell Rep 14(1):49–59
Norbnop P, Ingrungruanglert P, Israsena N et al (2020) Generation and characterization of HLA-universal platelets derived from induced pluripotent stem cells. Sci Rep 10:8472
Saito S, Ota S, Seshimo H, Yamazaki Y, Nomura S, Ito T, Miki J, Ota M, Fukushima H, Maeda H (2002) Platelet transfusion refractoriness caused by a mismatch in HLA-C antigens. Transfusion 42:302–308
Lanier LL (2008) Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9:495–502
Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S (2013) Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 31:227–258
Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, Prunkard D, Colunga AG, Hanafi L-A, Clegg DO, Turtle C, Russell DW (2017) HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 35:765–772
Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, Kitaoka F, Takahashi T, Okita K, Yoshida Y, Kaneko S, Hotta A (2019) Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 24:1–13
Zhang N, Zhi H, Curtis BR, Rao S, Jobaliya C, Poncz M et al (2015) CRISPR/Cas9-mediated conversion of human platelet alloantigen allotypes. Blood 127(6):675–680
Leslie M (2010) Cell biology. Beyond clotting: the powers of platelets. Science 328(5978):562–564
Zhang N, Newman PJ (2019) Packaging functionally important plasma proteins into the α-granules of human-induced pluripotent stem cell-derived megakaryocytes. J Tissue Eng Regen Med 13(2):244–252
Gao C, Schroeder JA, Xue F et al (2019) Nongenotoxic antibody-drug conjugate conditioning enables safe and effective platelet gene therapy of hemophilia A mice. Blood Adv 3(18):2700–2711
Du LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, Haberichter SL, Merricks E, Raymer RA, Fang J, Koukouritaki SB, Jacobi PM, Hawkins TB, Cornetta K, Shi Q, Wilcox DA (2013) Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun 4:2773
Zhang X, Wang J, Chen Z et al (2018) Engineering PD-1-presenting platelets for cancer immunotherapy. Nano Lett 18(9):5716–5725
Xu P, Jiang Y, Zuo H et al (2019) Vincristine-loaded platelets coated with anti-CD41 mAbs: a new macrophage targeting proposal for the treatment of immune thrombocytopenia. Biomater Sci 7(11):4568–4577
Nishikawa T, Tung LY, Kaneda Y (2014) Systemic administration of platelets incorporating inactivated Sendai virus eradicates melanoma in mice. Mol Ther 22(12):2046–2055
Xu P, Wang R, Wang X, Ouyang J (2016) Recent advancements in erythrocytes, platelets, and albumin as delivery systems. Onco Targets Ther 9:2873–2884
Wilcox DA (2016) Megakaryocyte- and megakaryocyte precursor-related gene therapies. Blood 127(10):1260–1268
Acknowledgements
The authors thank Dr. Peter Karagiannis for critical reading of the manuscript and Misaki Ouchida for assistance on the figures. This work was supported in part by Core Center for iPS Cell Research (JP19bm0104001, N.S. and K.E.), Practical Applications of Regenerative Medicine (JP19bk0104003h, K.E.), Projects for Technological Development (JP19bm0404037, N.S. and K.E.) from the Japan Agency for Medical Research and Development (AMED), and by Grant-in-Aid for scientific research (18H04164, K.E.) from the Japan Society for the Promotion of Science (JSPS). N.S. and K.E. have applied for patents related to this manuscript. K.E. is a founder of Megakaryon and a member of its scientific advisory board without salary; the interests of K.E. were reviewed and are managed by Kyoto University in accordance with its conflict-of-interest policies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sugimoto, N., Eto, K. Generation and manipulation of human iPSC-derived platelets. Cell. Mol. Life Sci. 78, 3385–3401 (2021). https://doi.org/10.1007/s00018-020-03749-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03749-8