
Abstract
The paracaspase MALT1 has a central role in the activation of lymphocytes and other immune cells including myeloid cells, mast cells and NK cells. MALT1 activity is required not only for the immune response, but also for the development of natural Treg cells that keep the immune response in check. Exaggerated MALT1 activity has been associated with the development of lymphoid malignancies, and recently developed MALT1 inhibitors show promising anti-tumor effects in xenograft models of diffuse large B cell lymphoma. In this review, we provide an overview of the present understanding of MALT1’s function, and discuss possibilities for its therapeutic targeting based on recently developed inhibitors and animal models.
Similar content being viewed by others


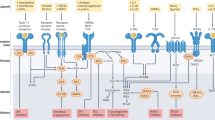
Introduction
The paracaspase MALT1 plays an essential role in the activation of immune cells by specific subtypes of immune receptors, which induce a common signaling pathway leading to the activation of the transcription factor NF-κB. NF-κB target genes include cytokines and anti-apoptotic proteins, which together promote the activation, proliferation and survival of the activated immune cells upon receptor triggering, and thereby allow the efficient generation of an immune response. MALT1 is also involved in the activation of non-immune cells, in which an NF-κB response can be induced by specific G protein-coupled receptors (GPCRs) or the epidermal growth factor (EGF) receptor (Fig. 1). In this review, we will focus mainly on the role of MALT1 in immune cells. Readers interested in the role of MALT1 in non-immune cells are referred to an excellent recent review covering this topic [1].

Receptor-induced signaling via MALT1. BCL10 and MALT1 act together to activate NF-κB downstream of receptors containing ITAMs and G protein-coupled receptors. The B- and T-cell receptor (BCR and TCR, respectively) and natural killer (NK)-cell receptors such as NKG2D, NK1.1, Ly49D and Ly49H, mediate signals through CARMA1. ITAM-containing receptors expressed by myeloid and mast cells, such as Dectins, FcγRs, OSCAR, TREM-1 and Mincle mediate signals through CARD9, while GPCRs signal through CARMA3 for the activation of the BCL10-MALT1 module. ITAM immunoreceptor tyrosine-based activation motif, FcRγ Fc receptor γ-chain; PKC protein kinase C, IKK inhibitor of NF-κB (IκB) kinase complex, OSCAR osteoclast associated, immunoglobulin-like receptor, TREM-1 triggering receptor expressed by myeloid cells 1
Molecular and biological functions of MALT1
MALT1 functions downstream of immunoreceptors with an ITAM sequence
The ligand-dependent activation of adaptive and innate immune cells via cell surface receptors that recognize various non-self features is an essential initiating step of the immune response. Not all immune receptors activate MALT1. Indeed, MALT1 specifically transmits signals from immune receptors with a so-called ITAM (immunoreceptor tyrosine-based activation motif), towards the activation of the transcription factor NF-κB (see Fig. 1 and specific references therein). Prototype members of these receptors are the B-cell and T-cell receptor (BCR and TCR), which associate with ITAM-containing CD79 or CD3 chains and recognize antigenic proteins or processed peptide antigens, respectively. Other types of immune cell receptors that signal via ITAM motifs are Fc receptors, which are expressed on myeloid and mast cells and recognize antibody-coated antigenic structures. The myeloid receptors Dectin-1, Mincle and TREM1, contain either an ITAM domain or associate with the ITAM-containing FcRγ chain, and activate innate immune cells upon recognition of microbial glycoproteins [2, 3]. Finally, activating natural killer (NK) cell receptors expressed on NK cells, which associate with ITAM-bearing signaling subunits such as DAP12 or CD3ζ, recognize cellular stress ligands or antigenic structures presented by non-classical MHC molecules [4]. A common consequence of the triggering of ITAM-containing receptors is the NF-κB-dependent expression of proliferation and inflammation-promoting genes, in particular of immune-stimulating cytokines, in the activated immune cells.
MALT1 has a central role in the immune response
Genetic studies using MALT1-deficient mice have revealed that MALT1 plays an essential role in immunoreceptor-induced activation events, since mice lacking functional MALT1 are immunodeficient [5, 6]. In particular, these mice show impaired B- and T-cell responses to immunization or viral infection, impaired Fc-receptor mediated cytokine responses of myeloid and mast cells, strongly reduced NK cell responses and impaired innate immunity to yeast infections [5–9]. Additionally, MALT1-deficient mice or mice expressing a catalytically inactive form of MALT1, have impaired development of specific B-cell subsets, such as B1 and marginal zone B cells, and of regulatory T cells [5, 6, 10–13], most likely as a consequence of impaired BCR and TCR signals during lymphocyte development (Fig. 2). MALT1-dependent TCR signaling is also strictly required for the development of effector T cells of the TH17 type [14, 15]. Recently, a small number of human patients with defects in MALT1 expression and/or function have been described [16–18]. A common feature of these patients is combined immunodeficiency, characterized by severe recurrent infections and impaired cellular and humoral immune responses despite normal numbers of circulating B- and T-cells. Collectively, these observations support an essential role for MALT1 in the immune response that is due to its essential signaling function downstream of ITAM-containing immunoreceptors.

Role of MALT1’s protease function in lymphocyte development and differentiation. MALT1 scaffold and protease functions are essential for the development of peritoneal B1 B cells, marginal zone (MZ) B cells and natural regulatory T cells (nTreg). Polarization of naïve CD4+ T cells into the TH17 subset of T helper cells is heavily dependent on MALT1 protease function
Immunoreceptors with ITAM motifs activate MALT1 via PKC and CARD proteins
How do ITAM-containing immunoreceptors activate MALT1? A common feature of these receptors is that their ITAM motif(s) become(s) rapidly phosphorylated by Src family kinases in response to binding of the antigenic ligand to the receptor. This is followed by the physical recruitment of the Syk family kinases ZAP70 or Syk to the doubly phosphorylated ITAM motif [19]. The signal is then relayed by Ser/Thr kinases of the PKC family, which in turn phosphorylate the scaffold protein CARMA1 (CARD-MAGUK1, also known as CARD11) [20, 21] or the CARMA1 homologue CARD9 [22], thereby promoting the physical assembly of oligomeric CARMA1/CARD9-BCL10-MALT1 (CBM) complexes (Fig. 1). The stoichiometry of these complexes is not well defined; recent findings suggest that the CBM signalosome is a filamentous assembly in which CARMA1 nucleates formation of BCL10 filaments that are decorated with MALT1 [23]. In non-immune cells, the CARMA1 homologue CARMA3 (also known as CARD10) is thought to play a similar role in promoting BCL10- and MALT1-dependent NF-κB activation downstream of G protein-coupled receptors or the EGFR [24–26]. The CARMA1-homologue CARMA2 (also known as CARD14) has been proposed to drive NF-κB activation in keratinocytes of the skin [27, 28], but the upstream signals triggering CARMA2 activation remain unknown. As a consequence of its BCL10-dependent physical recruitment into a CBM complex, MALT1 promotes NF-κB activation via both its scaffold and protease functions (Fig. 3). As a scaffold, MALT1 recruits ubiquitin ligases that promote the activation of the IκB kinase (IKK) complex [29–33]. IKK-dependent phosphorylation targets the NF-κB inhibitor, IκB, for proteasomal degradation and thereby allows NF-κB subunits to enter the nucleus and to initiate the transcription of NF-κB target genes [34]. The protease activity of MALT1, on the other hand is essential for NF-κB signal amplification and persistence, which is achieved mainly by the cleavage of two negative regulators of the NF-κB pathway, namely RelB and A20 [35, 36]. So far, the relevance of MALT1 protease activity for NF-κB activation has been formally demonstrated for receptors depending on CARMA1 or CARD9 [10–13, 35, 37]; whether it is also relevant for CARMA2- and CARMA3-dependent NF-κB signals remains uncertain [38]. Recently, several new substrates of MALT1 have been identified. These substrates control additional aspects of leukocyte activation such as activation of AP-1 transcription factors and regulation of transcript stability [39–41]. The molecular features of the MALT1 scaffold and protease functions and their biological relevance are further detailed below.

Overview of MALT1-dependent lymphocyte activation via its scaffold and protease functions. Through its scaffold function, MALT1 promotes NF-κB activation via recruitment of the ubiquitin ligase TRAF6, the linear ubiquitin chain assembly complex LUBAC (composed of Sharpin/HOIL/HOIP) and the Ser/Thr kinase TAK1 to activate the IKK complex, which phosphorylates the NF-κB inhibitor IκB to target it for proteasomal degradation. MALT1 protease activity controls NF-κB activation, AP-1 activation, mRNA stability and cellular adhesion by the cleavage of various substrates (see text for details). Positions and sequences of MALT1 cleavage sites in the individual substrates are indicated
The scaffold function of MALT1 is defined by its protein binding motifs
Over the last few years, our understanding of the function of MALT1 has considerably been improved by molecular and biochemical analyses, which have dissected individual functions of specific structural subdomains present in the MALT1 protein (Fig. 4a). MALT1 contains a predicted N-terminal death domain of unknown function, followed by two immunoglobulin (Ig)-like domains that have been shown to be essential for its constitutive binding to the adaptor protein BCL10 [42, 43]. In addition, MALT1 contains a central catalytic domain that has homology with proteases of the caspase family [42]. Shared features include a conserved Cys and His residue required for catalysis and a typical three-dimensional fold of the protease domain, which can dimerize [42, 44, 45] (Fig. 4b). The protease domain is followed by a third Ig-like domain that contains important sites for mono- and polyubiquitination, and by a C-terminal extension of unknown structure. The capacity of MALT1 to recruit the ubiquitin ligase TRAF6 and to thereby activate the IKK complex has been attributed to two individual TRAF6 binding motifs located outside the protease domain [30, 31] (Fig. 4a). TRAF6 recruitment to MALT1 is thought to promote IKK activation by polyubiquitination of MALT1 together with its binding partner BCL10 and TRAF6 itself [29, 31, 46]. The resulting K63-linked polyubiquitin chains are thought to provide docking sites for the recruitment of the kinase TAK1 via TAB 2/3 adaptor proteins [47, 48], which allows TAK1 to phosphorylate and thereby activate the catalytic IKK subunit IKKβ [31, 48]. MALT1 also associates with the linear ubiquitination chain assembly complex LUBAC (also known as the Sharpin/HOIL/HOIP complex) [32, 33], but the consequences of this interaction for IKK activation remain incompletely understood. It is most likely that the linear ubiquitination of CBM- or associated components provides docking sites for the physical recruitment of the IKK complex through the linear ubiquitin chain-binding UBAN motif of the IKK subunit IKKγ (also known as NEMO) [47, 49]. Collectively, these findings support the idea that MALT1 can act as a scaffold to coordinate the recruitment and activation of TRAF6 and the kinases TAK1 and IKK for NF-κB activation.

Molecular structure and function of MALT1. a MALT1 contains an N-terminal death domain (DD), followed by two immunoglobulin-like domains (Ig), which are required for the interaction between MALT1 and BCL10. The central caspase-like domain, with the active site residues H415 and C464, is followed by a third Ig domain that contains K644, a monoubiquitination site that controls protease activity. MALT1 contains two binding motifs for the ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6). TRAF6 polyubiquitinates MALT1 on multiple C-terminal lysine residues, generating K63-linked ubiquitin chains that can in turn promote activation of the inhibitor of NF-κB kinase (IKK) complex through recruitment of the IKK-activating kinase TAK1 via the adaptor proteins TAB 2/3. Amino acid numbering in the figure refers to human MALT1. b Ribbon representation of the crystallographic structure of the MALT1 homodimer (figure reproduced from Yu et al., PNAS 108 (52), 21004–21009 (2011), with kind permission) [45]. The shown crystallized fragment comprises the paracaspase region (blue) and the adjacent C terminal Ig domain (orange). The covalently bound MALT1 peptide inhibitor is colored red and the subunit linker in the caspase-like fold is colored yellow. The sequences between the caspase-like domain and the Ig domain are disordered and represented as gray dotted lines
The protease activity of MALT1 is required for optimal NF-κB and AP-1 activation
Over the last years, it has become clear that MALT1 signaling downstream of ITAM-containing receptors depends not only on its scaffold function, but to a large extent on its protease activity. A specific feature of MALT1 activation that differentiates it from proteases of the caspase family is the requirement of the third Ig domain (Ig3), which forms several hydrophobic contacts with the adjacent protease domain that are released upon substrate binding (Fig. 3a, b) [44, 45]. This suggests a model in which the auto-inhibitory Ig3 domain contributes to MALT1 activation by an inducible conformational change in this region [44, 50]. Interestingly, MALT1 activity is tightly controlled by antigen receptor-induced monoubiquitination of MALT1 on residue K644 within the Ig3 domain (Fig. 3a), and this modification induces or stabilizes MALT1 dimerization via the protease domain [51, 52].
With the discovery of the proteolytic activity of MALT1 and of specific MALT1 protein substrates, our understanding of MALT1 function has been considerably enriched (Fig. 3c). The protease activity of MALT1 is thought to promote NF-κB activation through the cleavage of the deubiquitinating enzyme A20 and of the NF-κB subunit RelB. How exactly A20 cleavage affects NF-κB activation remains unclear. A20 can negatively regulate IKK-dependent NF-κB activation by deubiquitination of MALT1 [53], and A20 levels decrease shortly after T-cell stimulation [53]. However, MALT1 activity is neither required for A20 degradation [53] nor for IKK-dependent IκBα phosphorylation [10–12, 36, 53], and the proportion of inducible A20 cleavage is relatively small [35]. This suggests that MALT1-dependent A20 cleavage may serve to generate an A20 fragment that affects NF-κB activation in an IKK-independent manner that remains to be further explored. RelB, which is mainly known for its activating role in the alternative NF-κB pathway, acts as a negative regulator of canonical NF-κB activation by the antigen receptor [36, 54–56], most likely through the formation of transcriptionally inactive RelA/RelB heterodimers and/or competition at the DNA binding site [36, 57, 58]. MALT1-dependent RelB cleavage results in its proteasomal degradation, thereby lowering total levels of RelB in activated B- and T cells [36]. Through cleavage of RelB and A20, MALT1 thus expands the amplitude and duration of the NF-κB response in an IKK-independent manner. Interestingly, MALT1 also promotes NF-κB activation by its autoprocessing after Arg 149. The exact consequences of MALT1 autoprocessing remain unclear, but include an effect on NF-κB target gene expression downstream of nuclear NF-κB accumulation [59].
In addition to having defects in the NF-κB pathway, MALT1-deficient cells were also reported to have an impaired capacity to activate the JNK/AP-1 transcriptional pathway [5]. This may be attributed at least in part to the requirement of MALT1 for the cleavage of the deubiquitinating enzyme CYLD [39], which negatively regulates this pathway in lymphocytes through deubiquitination of the JNK upstream kinase TAK1 [60]. Expression of a non-cleavable form of CYLD inhibited activation of the JNK pathway and expression of AP-1 target genes in a T-cell line, although CYLD silencing only minimally increased JNK activation [39], possibly because of redundancy with other deubiquitinating enzymes. Of note, recent studies with protease-inactive MALT1 knock-in mice have shown that MALT1 protease activity is not required for JNK activation [10–12]. Thus, the exact mechanism of MALT1- and CYLD-dependent AP-1 activation and the role of JNK in this pathway remain to be further explored.
MALT1 controls transcript stability by cleavage of mRNA-destabilizing proteins
An entirely new aspect of MALT1’s biological function has been revealed with the recent discovery that Regnase-1 (also known as MCPIP1 or Zc3h12a) and the Roquin-1 and -2 proteins act as MALT1 substrates [40, 41]. Regnase-1 is an RNAse with an important function in mRNA degradation. Regnase-1 binds to specific mRNA stem loop structures and promotes mRNA degradation by its RNAse activity [61] and by recruitment of the RNA remodeling helicase UPF1 [62]. It was additionally suggested that Regnase-1 controls miRNA generation by processing of premiRNAs [63]. MALT1-dependent cleavage of Regnase-1 lowers its levels in activated T cells and thereby induces stabilization of a subset of pro-inflammatory transcripts, such as IL-6, IL-2, c-Rel and ICOS [40, 41]. As a consequence, MALT1-dependent Regnase-1 cleavage promotes T-cell activation [40]. Mice with a T-cell specific Regnase-1 deficiency have exaggerated T-cell responses, which account, to a large part, for the reported autoimmunity and systemic inflammation features of Regnase-1-deficient mice [61]. The MALT1 substrates Roquin-1 and Roquin-2 promote mRNA degradation by binding to 3’ located stem-loop structures in mRNA molecules known as conserved decay elements (CDE) [64, 65]. This allows Roquin proteins to promote mRNA deadenylation by recruitment of the cellular CCR4-CAF1-NOT deadenylase complex [64]. MALT1-dependent cleavage of Roquin proteins has been proposed to play a predominant role in the stabilization of specific transcripts, such as IL-6, ICOS, c-Rel and IRF4, which are also targets of Regnase-1 and collectively promote the differentiation of T effector cells of the TH17 subtype [41]. In addition to its roles in transcription and transcript stabilization, MALT1 has been suggested to have a role in promoting T-cell adhesion through cleavage of its binding partner BCL10, by mechanisms that are not yet understood [37]. Collectively, these findings suggest a broad role for MALT1, and in particular for its protease activity, in immune receptor-induced activation of transcription, RNA stabilization and cellular adhesion (Fig. 3).
Additional, less explored roles for MALT1 and its protease activity in lymphocytes
Beyond its established roles in activation of NF-κB and AP-1 and transcript regulation, MALT1 most likely controls other cellular activation pathways. It has for example been proposed that MALT1 controls NF-κB2 activation downstream of the BAFF-receptor in specific B-cell subsets, possibly via receptor-induced dissociation of MALT1 from the ubiquitin ligase TRAF3 [66] that negatively regulates BAFF-receptor-dependent survival signals [67]. Recently, MALT1 protease activity has been shown to be required for TCR/CD28-dependent induction of glutamine uptake and mTOR activation that controls metabolic changes required for T-cell activation [68]. The substrate responsible for this effect remains unknown.
MALT1 inhibition and its therapeutic potential for lymphomas
Different compounds can be used to inhibit MALT1 in vitro and in vivo
MALT1’s enzymatic activity has stimulated efforts to develop specific MALT1 inhibitors for research and therapeutic purposes. The first MALT1 inhibitor identified was z-VRPR-fmk, a modified tetrapeptide based on the optimal substrate Val-Arg-Pro-Arg of the Arabidopsis thaliana metacaspase AtmC9, conjugated to fluoromethyl ketone (fmk) [37]. This modified peptide irreversibly blocked MALT1 protease activity in an in vitro cleavage assay using recombinant MALT1 in a dose-dependent manner. It furthermore efficiently inhibited T cell activation and IL-2 secretion in Jurkat T cells and in human antigen specific CTLs [37]. An alternative version of this inhibitor, named z-LVSR-fmk, which is based on the LVSR substrate sequence in the MALT1 substrate RelB [36], also inhibits MALT1 efficiently [59]. Recently, two types of potential small molecule MALT1 inhibitors have been identified in high throughput screening approaches [69, 70], suggesting that it will be feasible to develop suitable MALT1 inhibitors for in vivo studies. Nagel and colleagues have identified three phenothiazine derivatives (mepazine, thioridazine, and promazine) as highly specific, noncompetitive and reversible MALT1 inhibitors [69]. A concurrent study by Fontan and colleagues has identified the compound MI-2 as a selective MALT1 inhibitor [70]. In contrast to phenothiazine derivatives, MI-2 engages and irreversibly binds the active site of MALT1 [70]. Co-crystallization of thioridazine with MALT1 has revealed that these compounds bind the interface between the protease domain and the Ig3 domain of MALT1, an allosteric site that is far from the active site of the enzyme [71]. Thus, thioridazine most likely affects MALT1 activity by preventing a conformational change in the protease-Ig3 interface that is essential for MALT1 activation [44, 45, 50]. Recently, two studies have reported the first generation of MALT1 activity-based probes derived from peptide- or phenothiazine inhibitors [72, 73]. While these are yet of limited sensitivity, improved probes may become useful in the future to detect MALT1 activity in pathological settings or for measuring patient responses to MALT1 inhibitor treatment.
The potential applications of MALT1 inhibitors in the fields of immunomodulation and the treatment of lymphomas are reviewed below and illustrated in Fig. 5.

Potential fields of application of clinical MALT1 inhibitors. Possible applications include treatment of lymphomas with constitutive MALT1 activity and immunomodulation in the context of transplantation tolerance, autoimmunity and various inflammatory disorders
MALT1 inhibition could be a strategy to target ABC DLBCL lymphomas
The first indication that Malt1 inhibition might be a promising strategy to treat human diseases came from two studies in 2009 which reported a preferential cytotoxicity of the MALT1 inhibitor z-VRPR-fmk on a subtype of cell lines derived from diffuse large B-cell lymphoma (DLBCL) [74, 75]. DLBCL can be genetically classified into molecularly distinct subtypes, including the germinal center B cell (GCB) and the activated B cell (ABC) subtype. Human cases of GCB DLBCL generally show a slow and chronic progression of the disease, whereas cases of ABC DLBCL have a faster course, worse 5 year survival rate and respond less well to chemotherapeutic treatment. Growth of ABC DLBCL is driven by constitutive NF-κB signaling that results from a variety of mechanisms [76]. These include activating mutations in the BCR-associated CD79A/B chains, (present in roughly 23 % of ABC DLBCL cases [77, 78]) the MALT1 activator CARMA1 (roughly 8 % of cases [77, 79]) or the TLR adaptor protein MyD88 (37 % of cases) [80], together with inactivating mutations in A20, a negative regulator of the NF-κB pathway (23 % of cases) [81]. Using an RNAi screen on ABC DLBCL cell lines, Ngo and colleagues demonstrated that these cell lines, which have combined mutations in MyD88 and either the CD79 or CARMA1 proteins, heavily depend on NF-κB activation via CARMA1, BCL10 and MALT1 for their survival and proliferation [82]. Inhibition of MALT1 by treatment with z-VRPR-fmk, or by expression of a catalytically inactive form of MALT1, decreased the expression of NF-κB target genes and dramatically reduced the viability and growth of cell lines derived from ABC DLBCL, but not from GCB DLBCL [74, 75]. Collectively, these findings suggested a key role for MALT1 activity in the growth of ABC DLBCL cells.
Recently, small molecule inhibitors of MALT1 have been shown to be effective against ABC DLBCL in xenograft models [69, 70]. The phenothiazine derivatives identified by Nagel et al., which act as allosteric MALT1 inhibitors, showed selective activity against ABC DLBCL cell lines in vitro and xenotransplanted ABC DLBCL tumors in vivo [69]. The compound MI-2, an active site MALT1 inhibitor developed by Fontan et al., also successfully suppressed the growth of human ABC DLBCL xenografts in mice without notable toxicity [70]. Thus, MALT1 inhibitors may have potential in the treatment of ABC DLBCL, in particular for those cases that have oncogenic CARMA1 mutations that will render them insensitive to compounds inhibiting BCR signaling upstream of CARMA1, such as the Btk inhibitor Ibrutinib [76].
Additional types of lymphomas may be targeted by MALT1 inhibitors
Another type of lymphoma with constitutive MALT1 activity is the MALT lymphoma. A large proportion of cases with advanced stages of this disease are characterized by the presence of a chromosomal translocation (t(11;18)(q21;q21) that encodes the oncogenic IAP2-MALT1 fusion protein, composed of the N terminus of IAP2 linked to the C terminus of MALT1. The IAP2 moiety mediates auto-oligomerization of the fusion protein [83], rendering IAP2-MALT1 constitutively active and able to stimulate NF-κB independently of upstream signals. The IAP2–MALT1 fusion recruits the ubiquitin ligase TRAF6 to promote canonical, IKK-dependent NF-κB1 activation [30]. In addition, it has constitutive protease activity as a consequence of its monoubiquitination [51], and can cleave natural MALT1 substrates such as A20 to promote NF-κB1 activation [35]. Aside from activating the canonical NF-κB pathway, the IAP2–MALT1 fusion protein specifically triggers NF-κB2 activation by the cleavage of the Ser/Thr kinase NIK, which is detectable in MALT lymphoma samples [84]. NIK promotes the activation of the NF-κB2 pathway through phosphorylation and subsequent proteolytic maturation of the NF-κB2 precursor p100 into its transcriptionally active form p52 [85]. Normally, the cellular activity of NIK is limited by its very short half-life, but MALT1-dependent cleavage of NIK generates a NIK fragment that is stable and active, thereby promoting constitutive NF-κB2 activation to drive cellular adhesion and protection from apoptosis of the malignant cells [84]. In addition to cleaving NIK, the IAP2-MALT1 fusion protein has recently been shown to cleave the tumor suppressor protein LIMA1, and LIMA1 cleavage products are present in MALT lymphoma samples expressing IAP2-MALT1 [86]. LIMA1 cleavage abolishes its tumor suppressor function and generates a novel fragment with oncogenic properties that contributes to cellular transformation [86]. The IAP2-MALT1 fusion protein thus seems to drive cellular transformation by multiple means, suggesting that inhibition of the MALT1 protease activity could be an attractive new treatment approach for t(11;18)-positive MALT lymphoma.
Finally, a recent study by Rahal and collegues [87] identified a subset of MCL lines with chronic activation of the BCR mediated CBM–NF-κB signaling pathway and constitutive RelB cleavage, suggesting that a subset of MCL may also be responsive to MALT1 inhibition. While MALT1 activity has not yet been assessed in other lymphoma types, it seems likely that additional types of B- or T-cell lymphomas with constitutive antigen receptor signaling are sensitive to MALT1 inhibition.
Use of MALT1 inhibition for immunomodulation
MALT1 may control transplant rejection
The potential relevance of MALT1-dependent signaling has thus far only been addressed using mice deficient for the MALT1 upstream regulator CARMA1 and pancreatic islet allografts [88]. Transplanted CARMA1-deficient mice displayed a lack of T cell priming, showed very few mononuclear cell infiltrates in the grafts and accepted fully allogeneic islet allografts long-term. These results hold promise that MALT1 inhibition might be beneficial in tissue transplantation, especially for the transplantation of less immunogenic tissues. However, since MALT1 protease inhibition is less efficient in preventing T cell activation than complete CARMA1-deficiency, the benefits will have to be determined experimentally. In addition, it is of concern that CBM deficient mice as well as mice expressing a catalytically inactive form of MALT1 display a developmental lack of regulatory T (Treg) cells. Considering that Treg cells play a critical role in preventing graft rejection [15], this lack of Treg cells will complicate the interpretation of grafting experiments using these mouse models. Whether prolonged treatment of mice with a MALT1 inhibitor could adversely affect engraftment by reducing Treg numbers remains to be determined.
MALT1 may be relevant for inflammatory lung and skin disorders
CARMA1 has been shown to play an important role in the induction of allergic asthma [89]. Immunization and challenge of wild type mice with an allergen leads to eosinophilic airway inflammation, with production of Th2 cytokines and IgE, mucus cell hypertrophy and airway hyperresponsiveness, whilst CARMA1-deficient mice are protected from disease induction. A follow up study by the same group [90] revealed that CARMA1 is also important in recall responses when the initial priming has occurred in the presence of CARMA1, which more closely resembles the natural conditions of a therapeutic intervention. The CARMA1 homologue CARMA3 (also known as CARD10) that is expressed by airway epithelial cells is implicated in allergic airway inflammation by mediating proinflammatory cytokine production, DC maturation and migration, antigen processing and T cell proliferation [91]. MALT1, which acts downstream of both CARMA1 and CARMA3, may thus bridge innate and adaptive immune responses in the context of allergic asthma. Interestingly, mutations in the CARMA1 homologue CARMA2 (also known as CARD14) have been recently associated with the development of psoriasis in human patients [27, 28]. Some of these mutants were found to be hyperactive and to promote expression of psoriasis-associated chemokines such as CCL20 and IL-8 in keratinocytes [28]. Collectively, these findings suggest that inhibition of CARMA1/2/3-dependent signaling with a MALT1 inhibitor may have potential in the treatment of various inflammatory disorders through effects on immune and non-immune cells.
MALT1 may play an important role in GPCR- and EGFR-driven pathologies
To date, little is known about the requirement of MALT1 protease activity for signaling by GPCR or the EGFR, which both depend on MALT1 and the upstream regulator CARMA3 (also known as CARD10) for NF-κB signaling (see Fig. 1 and references therein). EGFR signaling does not appear to depend on MALT1 protease activity [38], while the requirement for MALT1 protease activity for signaling by GPCRs has not yet been thoroughly investigated. Signaling by GPCRs depending on CARMA3 and MALT1 regulates various inflammatory processes, including lysophosphatidic acid (LPA)-induced lung inflammation [92], platelet-activating factor (PAF)-induced intestinal inflammation [93] and angiotensin II-, thrombin- and IL-8-mediated vascular inflammation [24, 94, 95]. Moreover, GPCR-signaling via CARMA3 and MALT1 may be important for LPA- or SDF-1-induced carcinogenesis [96, 97]. Thus, whether MALT1 inhibition might positively affect these pathological conditions merits further investigation.
MALT1 protease-deficient mice reveal contradictory roles for MALT1 in autoimmunity
To assess the biological relevance of the MALT1 protease activity in vivo, several groups have recently generated mice expressing a catalytically inactive form of the MALT1 protease (MALT1 protease dead, MALT1-PD) [10–13]. CARMA1- or MALT1-deficient mice are fully protected from induction of experimental autoimmune encephalomyelitis (EAE) by immunization with myelin oligodendrocyte glycoprotein (MOG) [14, 98, 99], and treatment of mice with the MALT1 inhibitor mepazine attenuates onset and progression of the disease [100]. Interestingly, MALT1-PD mice are completely protected from induction of EAE [10, 11], suggesting an important role of MALT1 in the generation and activation of autoreactive T cells. TH17 cells play a major role in the pathology of EAE, and both MALT1-deficient and MALT1-PD mice show an impaired differentiation of naïve T cells into TH17 cells in vitro [11, 14]. Interestingly, MOG-immunized MALT1-PD mice also have reduced IL-17 positive T cells in the CNS [10], suggesting that MALT1 protease-mediated signals are required for the differentiation of autoreactive TH17 cells.
In a second autoimmune setting, using a T cell transfer model of colitis, we demonstrated that naïve T cells from these mice less efficiently induce colon inflammation than wild-type T-cells [10]. Unfortunately, the analysis of other models of autoimmune diseases in MALT1-PD mice has been severely hampered by the fact that these mice spontaneously develop a severe form of autoimmune disease (Table 1). Indeed, MALT1-PD mice fail to thrive, show neurological defects, but most intriguingly, exhibit a very pronounced lymphadenopathy with a strong increase in B- and T cell numbers in the lymph nodes, the presence of T cells with an effector/memory phenotype and lymphocyte infiltration into various organs, including the stomach, glands, lungs, peripheral nerves and other organs [10–13]. Similar to MALT1-deficient mice, MALT1-PD mice have a defect in regulatory T cell development, which leads to strongly reduced regulatory T cell numbers in the thymus, spleen and lymph nodes [10–12]. In contrast to T cells from MALT1 knock-out mice, which are refractory to stimulation, T cells from MALT1-PD mice retain some residual activation potential, most likely though MALT1 scaffold-dependent IKK and JNK activation [10–13]. The imbalance between the residual partial T cell activation potential on the one hand, and the lack of efficient counter-regulation by regulatory T cells on the other hand, most likely leads to the observed fatal autoimmune disease in the otherwise immune-compromised animals. Interestingly, some patients with MALT1 mutations also show combined signs of immunodeficiency and autoimmune features (such as inflammatory bowel disease) that correlate with reduced peripheral Treg numbers [16, 17].
The compromised Treg function in MALT1-deficient or -mutant humans and mice raises the concern that prolonged treatment of individuals with MALT1 inhibitors may favor the development of autoimmunity. It is important to note, however, that the in vitro differentiation of naïve T cells from MALT1 PD mice into regulatory T cells was possible [10, 11], indicating that this process would not be severely hampered by a therapeutic MALT1 inhibitor. Along these lines, mepazine did not change the percentages of regulatory T cells in mice that were immunized with MOG peptide and subsequently treated daily with the inhibitor for up to 17 days [100]. Furthermore, the presence of mepazine did not impede the in vitro polarization of naïve mouse T cells into regulatory T cells [100]. Thus, MALT1 is strictly required for the development of natural Treg cells, while MALT1-independent mechanisms seem to be important for the generation of inducible Treg cells. Further experiments are however required to assess potential adverse effects of MALT1 inhibition on Treg cell development and function.
Conclusions and perspectives
MALT1 has an essential role in the immune response and the growth of lymphoma cells with constitutive MALT1 activity. Studies with MALT1-deficient mice and MALT1 inhibitors have suggested that MALT1 could be a rational drug target for immunomodulation and lymphoma treatment. The recent development of mice expressing catalytically inactive MALT1 confirms not only its essential role in the immune response, but also highlights the fact that MALT1 activity is required for the development of natural Treg cells that prevent autoimmunity. Importantly, MALT1 inhibition does not prevent the differentiation of naïve T cells into Treg cells. Additional studies are thus required to assess whether the long-term use of MALT1 inhibitors may affect Treg functions in the adult, which is of obvious concern for immunomodulation, but maybe less evident for the treatment of lymphomas.
An important perspective for future research on MALT1 will be to further explore its molecular function and to identify the full spectrum of its substrates, which may open additional windows for therapeutic applications. Another major challenge will be to better understand MALT1’s physiological function in non-immune cells, and its relevance not only for immune but also for non-immune pathologies.
Abbreviations
- ABC:
-
Activated B cell
- BCL10:
-
B-cell lymphoma-10
- CARD:
-
Caspase recruitment domain
- CARMA1:
-
CARD-containing membrane-associated guanylate kinase-1
- DLBCL:
-
Diffuse large B-cell lymphoma
- EAE:
-
Experimental autoimmune encephalomyelitis
- GCB:
-
Germinal center B cell
- MALT1:
-
Mucosa-associated lymphoid tissue protein-1
- MCL:
-
Mantle cell lymphoma
References
Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM (2011) From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle 10(15):2485–2496
Kingeter LM, Lin X (2012) C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell Mol Immunol 9(2):105–112. doi:10.1038/cmi.2011.58
Tessarz AS, Cerwenka A (2008) The TREM-1/DAP12 pathway. Immunol Lett 116(2):111–116. doi:10.1016/j.imlet.2007.11.021
Lanier LL (2008) Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9(5):495–502. doi:10.1038/ni1581
Ruland J, Duncan GS, Wakeham A, Mak TW (2003) Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity 19(5):749–758
Ruefli-Brasse AA, French DM, Dixit VM (2003) Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science 302(5650):1581–1584
Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J (2006) Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442(7103):651–656
Gross O, Grupp C, Steinberg C, Zimmermann S, Strasser D, Hannesschlager N, Reindl W, Jonsson H, Huo H, Littman DR, Peschel C, Yokoyama WM, Krug A, Ruland J (2008) Multiple ITAM-coupled NK cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-{kappa}B and MAPK activation to selectively control cytokine production. Blood 112(6):2421–2428
Klemm S, Gutermuth J, Hultner L, Sparwasser T, Behrendt H, Peschel C, Mak TW, Jakob T, Ruland J (2006) The Bcl10-Malt1 complex segregates Fc epsilon RI-mediated nuclear factor kappa B activation and cytokine production from mast cell degranulation. J Exp Med 203(2):337–347
Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, Perroud M, Golshayan D, Gaide O, Thome M (2014) Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J 33(23):2765–2781. doi:10.15252/embj.201488987
Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil-Allaoui I, Rush JS, Smith PA, Bigaud M, Junker-Walker U, Burkhart C, Dawson J, Niwa S, Katopodis A, Nuesslein-Hildesheim B, Weckbecker G, Zenke G, Kinzel B, Traggiai E, Brenner D, Brustle A, St Paul M, Zamurovic N, McCoy KD, Rolink A, Regnier CH, Mak TW, Ohashi PS, Patel DD, Calzascia T (2015) Deficiency of MALT1 Paracaspase Activity Results in Unbalanced Regulatory and Effector T and B Cell Responses Leading to Multiorgan Inflammation. J Immunol 194(8):3723–3734. doi:10.4049/jimmunol.1402254
Gewies A, Gorka O, Bergmann H, Pechloff K, Petermann F, Jeltsch KM, Rudelius M, Kriegsmann M, Weichert W, Horsch M, Beckers J, Wurst W, Heikenwalder M, Korn T, Heissmeyer V, Ruland J (2014) Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep 9(4):1292–1305. doi:10.1016/j.celrep.2014.10.044
Yu JW, Hoffman S, Beal AM, Dykon A, Ringenberg MA, Hughes AC, Dare L, Anderson AD, Finger J, Kasparcova V, Rickard D, Berger SB, Ramanjulu J, Emery JG, Gough PJ, Bertin J, Foley KP (2015) MALT1 Protease Activity Is Required for Innate and Adaptive Immune Responses. PLoS One 10(5):e0127083. doi:10.1371/journal.pone.0127083
Brustle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, Reardon C, Lacher SM, Ruland J, Ohashi PS, Mak TW (2012) The NF-kappaB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest 122(12):4698–4709. doi:10.1172/JCI63528
Wood KJ, Bushell A, Hester J (2012) Regulatory immune cells in transplantation. Nat Rev Immunol 12(6):417–430. doi:10.1038/nri3227
Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, Mollenauer M, Srinivasan R, Brenner SE, Mulder A, Claas FH, Weiss A, Puck JM (2015) Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol 35(2):135–146. doi:10.1007/s10875-014-0125-1
McKinnon ML, Rozmus J, Fung SY, Hirschfeld AF, Del Bel KL, Thomas L, Marr N, Martin SD, Marwaha AK, Priatel JJ, Tan R, Senger C, Tsang A, Prendiville J, Junker AK, Seear M, Schultz KR, Sly LM, Holt RA, Patel MS, Friedman JM, Turvey SE (2014) Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol 133 (5):1458–1462, 1462 e1451–1457. doi:10.1016/j.jaci.2013.10.045
Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, Chouery E, Mikhael R, Gorka O, Gewies A, Portales P, Nakayama T, Hosokawa H, Revy P, Herrod H, Le Deist F, Lefranc G, Ruland J, Geha RS (2013) A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol 132(1):151–158. doi:10.1016/j.jaci.2013.04.047
Bezbradica JS, Medzhitov R (2012) Role of ITAM signaling module in signal integration. Curr Opin Immunol 24(1):58–66. doi:10.1016/j.coi.2011.12.010
Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ (2005) Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23(6):561–574
Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Lin X (2005) Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity 23(6):575–585
Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP, Brombacher F, Urlaub H, Baier G, Brown GD, Leitges M, Ruland J (2012) Syk kinase-coupled C-type lectin receptors engage protein kinase C-sigma to elicit Card9 adaptor-mediated innate immunity. Immunity 36(1):32–42. doi:10.1016/j.immuni.2011.11.015
Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, Bracken C, Rosen M, Melnick A, Egelman EH, Wu H (2013) Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell 51(6):766–779. doi:10.1016/j.molcel.2013.08.032
McAllister-Lucas LM, Ruland J, Siu K, Jin X, Gu S, Kim DS, Kuffa P, Kohrt D, Mak TW, Nunez G, Lucas PC (2007) CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci USA 104(1):139–144. doi:10.1073/pnas.0601947103
Grabiner BC, Blonska M, Lin PC, You Y, Wang D, Sun J, Darnay BG, Dong C, Lin X (2007) CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev 21(8):984–996. doi:10.1101/gad.1502507
Jiang T, Grabiner B, Zhu Y, Jiang C, Li H, You Y, Lang J, Hung MC, Lin X (2011) CARMA3 is crucial for EGFR-Induced activation of NF-kappaB and tumor progression. Cancer Res 71(6):2183–2192. doi:10.1158/0008-5472.CAN-10-3626
Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, Duffin KC, Stuart PE, Goldgar D, Hayashi G, Olfson EH, Feng BJ, Pullinger CR, Kane JP, Wise CA, Goldbach-Mansky R, Lowes MA, Peddle L, Chandran V, Liao W, Rahman P, Krueger GG, Gladman D, Elder JT, Menter A, Bowcock AM (2012) Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet 90(5):796–808. doi:10.1016/j.ajhg.2012.03.013
Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, Ryan C, Duan S, Helms CA, Liu Y, Chen Y, McBride AA, Hwu WL, Wu JY, Chen YT, Menter A, Goldbach-Mansky R, Lowes MA, Bowcock AM (2012) PSORS2 is due to mutations in CARD14. Am J Hum Genet 90(5):784–795. doi:10.1016/j.ajhg.2012.03.012
Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SC, Ruland J, Scheidereit C, Krappmann D (2007) Malt1 ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J 26(22):4634–4645
Noels H, van Loo G, Hagens S, Broeckx V, Beyaert R, Marynen P, Baens M (2007) A Novel TRAF6 Binding Site in MALT1 Defines Distinct Mechanisms of NF-kappaB Activation by API2-MALT1 Fusions. J Biol Chem 282(14):10180–10189
Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 14(3):289–301
Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, Wright GW, Zhao H, Yang Y, Xu W, Rosenwald A, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Wiestner A, Kruhlak MJ, Iwai K, Bernal F, Staudt LM (2014) Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov 4(4):480–493. doi:10.1158/2159-8290.CD-13-0915
Dubois SM, Alexia C, Wu Y, Leclair HM, Leveau C, Schol E, Fest T, Tarte K, Chen ZJ, Gavard J, Bidere N (2014) A catalytic-independent role for the LUBAC in NF-kappaB activation upon antigen receptor engagement and in lymphoma cells. Blood 123(14):2199–2203. doi:10.1182/blood-2013-05-504019
Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663. doi:10.1146/annurev.immunol.18.1.621
Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P, Beyaert R (2008) T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol 9(3):263–271
Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton JE, Guzzardi M, Decaillet C, Grau M, Dorken B, Lenz P, Lenz G, Thome M (2011) Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA 108(35):14596–14601
Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, Gaide O, Guzzardi M, Iancu EM, Rufer N, Fasel N, Thome M (2008) The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 9:272–281
Pan D, Jiang C, Ma Z, Blonska M, You MJ, Lin X (2015) MALT1 is required for EGFR-induced NF-kappaB activation and contributes to EGFR-driven lung cancer progression. Oncogene. doi:10.1038/onc.2015.146
Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, Gevaert K, Beyaert R (2011) T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J 30(9):1742–1752. doi:10.1038/emboj.2011.85
Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, Satoh T, Mino T, Suzuki Y, Standley DM, Tsujimura T, Rakugi H, Isaka Y, Takeuchi O, Akira S (2013) Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell 153(5):1036–1049
Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, Vogel KU, Rehage N, Warth SC, Edelmann SL, Gloury R, Martin N, Lohs C, Lech M, Stehklein JE, Geerlof A, Kremmer E, Weber A, Anders HJ, Schmitz I, Schmidt-Supprian M, Fu M, Holtmann H, Krappmann D, Ruland J, Kallies A, Heikenwalder M, Heissmeyer V (2014) Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol 15(11):1079–1089. doi:10.1038/ni.3008
Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM (2000) Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6(4):961–967
Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, Nunez G (2001) Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem 276(22):19012–19019
Wiesmann C, Leder L, Blank J, Bernardi A, Melkko S, Decock A, D’Arcy A, Villard F, Erbel P, Hughes N, Freuler F, Nikolay R, Alves J, Bornancin F, Renatus M (2012) Structural Determinants of MALT1 Protease Activity. J Mol Biol 419(1–2):4–21. doi:10.1016/j.jmb.2012.02.018
Yu JW, Jeffrey PD, Ha JY, Yang X, Shi Y (2011) Crystal structure of the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase region. Proc Natl Acad Sci USA 108(52):21004–21009. doi:10.1073/pnas.1111708108
Wu CJ, Ashwell JD (2008) NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-{kappa}B activation. Proc Natl Acad Sci U S A 105:3023–3028
Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D (2009) Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep 10(5):466–473. doi:10.1038/embor.2009.55
Bhoj VG, Chen ZJ (2009) Ubiquitylation in innate and adaptive immunity. Nature 458(7237):430–437. doi:10.1038/nature07959
Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I (2009) Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136(6):1098–1109. doi:10.1016/j.cell.2009.03.007
Staal J, Beyaert R (2012) A two-step activation mechanism of MALT1 paracaspase. J Mol Biol 419(1–2):1–3. doi:10.1016/j.jmb.2012.03.006
Pelzer C, Cabalzar K, Wolf A, Gonzalez M, Lenz G, Thome M (2013) MALT1 protease activity is controlled by monoubiquitination. Nature Immunol 14:337–345. doi:10.1038/ni.2540
Cabalzar K, Pelzer C, Wolf A, Lenz G, Iwaszkiewicz J, Zoete V, Hailfinger S, Thome M (2013) Monoubiquitination and Activity of the Paracaspase MALT1 Requires Glutamate 549 in the Dimerization Interface. PLoS One 8(8):e72051. doi:10.1371/journal.pone.0072051
Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, Krappmann D (2009) A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol 182(12):7718–7728. doi:10.4049/jimmunol.0803313
Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R (1995) Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell 80(2):331–340
Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R (1996) Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J Immunol 157(9):3974–3979
Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, Neumann M (2001) Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-kappaB control. Oncogene 20(56):8142–8147. doi:10.1038/sj.onc.1204884
Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M (2003) RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem 278(22):19852–19860. doi:10.1074/jbc.M301945200
Saccani S, Pantano S, Natoli G (2003) Modulation of NF-kappaB activity by exchange of dimers. Mol Cell 11(6):1563–1574
Baens M, Bonsignore L, Somers R, Vanderheydt C, Weeks SD, Gunnarsson J, Nilsson E, Roth RG, Thome M, Marynen P (2014) MALT1 auto-proteolysis is essential for NF-kappaB-dependent gene transcription in activated lymphocytes. PLoS One 9(8):e103774. doi:10.1371/journal.pone.0103774
Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, Sun SC (2007) Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 204(6):1475–1485. doi:10.1084/jem.20062694
Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S (2009) Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458(7242):1185–1190. doi:10.1038/nature07924
Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, Vinuesa CG, Ohler U, Standley DM, Landthaler M, Fujiwara T, Takeuchi O (2015) Regnase-1 and roquin regulate a common element in inflammatory mrnas by spatiotemporally distinct mechanisms. Cell 161(5):1058–1073
Suzuki HI, Arase M, Matsuyama H, Choi YL, Ueno T, Mano H, Sugimoto K, Miyazono K (2011) MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol Cell 44(3):424–436. doi:10.1016/j.molcel.2011.09.012
Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G (2013) Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 153(4):869–881. doi:10.1016/j.cell.2013.04.016
Schlundt A, Heinz GA, Janowski R, Geerlof A, Stehle R, Heissmeyer V, Niessing D, Sattler M (2014) Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat Struct Mol Biol 21(8):671–678. doi:10.1038/nsmb.2855
Tusche MW, Ward LA, Vu F, McCarthy D, Quintela-Fandino M, Ruland J, Gommerman JL, Mak TW (2009) Differential requirement of MALT1 for BAFF-induced outcomes in B cell subsets. J Exp Med 206(12):2671–2683. doi:10.1084/jem.20091802
Gardam S, Sierro F, Basten A, Mackay F, Brink R (2008) TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28(3):391–401. doi:10.1016/j.immuni.2008.01.009
Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, Sun SC (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40(5):692–705. doi:10.1016/j.immuni.2014.04.007
Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, Hlahla D, Neuenschwander M, Peter von Kries J, Hadian K, Dorken B, Lenz P, Lenz G, Schendel DJ, Krappmann D (2012) Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 22(6):825–837. doi:10.1016/j.ccr.2012.11.002
Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, Garcia M, Cerchietti L, Shaknovich R, Yang SN, Fang F, Gascoyne RD, Martinez-Climent JA, Glickman JF, Borden K, Wu H, Melnick A (2012) MALT1 Small Molecule Inhibitors Specifically Suppress ABC-DLBCL In Vitro and In Vivo. Cancer Cell 22(6):812–824. doi:10.1016/j.ccr.2012.11.003
Schlauderer F, Lammens K, Nagel D, Vincendeau M, Eitelhuber AC, Verhelst SH, Kling D, Chrusciel A, Ruland J, Krappmann D, Hopfner KP (2013) Structural Analysis of Phenothiazine Derivatives as Allosteric Inhibitors of the MALT1 Paracaspase. Angew Chem Int Ed Engl 52(39):10384–10387. doi:10.1002/anie.201304290
Hachmann J, Edgington-Mitchell LE, Poreba M, Sanman LE, Drag M, Bogyo M, Salvesen GS (2015) Probes to monitor activity of the paracaspase MALT1. Chem Biol 22(1):139–147. doi:10.1016/j.chembiol.2014.11.011
Eitelhuber AC, Vosyka O, Nagel D, Bognar M, Lenze D, Lammens K, Schlauderer F, Hlahla D, Hopfner KP, Lenz G, Hummel M, Verhelst SH, Krappmann D (2015) Activity-based probes for detection of active MALT1 paracaspase in immune cells and lymphomas. Chem Biol 22(1):129–138. doi:10.1016/j.chembiol.2014.10.021
Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, Penas EM, Dierlamm J, Chan WC, Staudt LM, Thome M (2009) Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 106(47):19946–19951
Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C, Krappmann D, Ruland J (2009) Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 206(11):2313–2320. doi:10.1084/jem.20091167
Roschewski M, Staudt LM, Wilson WH (2014) Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol 11(1):12–23. doi:10.1038/nrclinonc.2013.197
Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R (2011) Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 43(9):830–837. doi:10.1038/ng.892
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, Xiao W, Powell J, Jiang JK, Thomas CJ, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Johnson NA, Rimsza LM, Campo E, Jaffe ES, Wilson WH, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pierce SK, Staudt LM (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463(7277):88–92. doi:10.1038/nature08638
Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM (2008) Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science 319(5870):1676–1679. doi:10.1126/science.1153629
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470(7332):115–119. doi:10.1038/nature09671
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L (2009) Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459(7247):717–721. doi:10.1038/nature07968
Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM (2006) A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441(7089):106–110
Zhou H, Du MQ, Dixit VM (2005) Constitutive NF-kappaB activation by the t(11;18)(q21;q21) product in MALT lymphoma is linked to deregulated ubiquitin ligase activity. Cancer Cell 7(5):425–431
Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, Hamoudi RA, Noels H, Sagaert X, Van Loo P, Baens M, Du MQ, Lucas PC, McAllister-Lucas LM (2011) Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science 331(6016):468–472. doi:10.1126/science.1198946
Sun SC (2012) The noncanonical NF-kappaB pathway. Immunol Rev 246(1):125–140. doi:10.1111/j.1600-065X.2011.01088.x
Nie Z, Du MQ, McAllister-Lucas LM, Lucas PC, Bailey NG, Hogaboam CM, Lim MS, Elenitoba-Johnson KS (2015) Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun 6:5908. doi:10.1038/ncomms6908
Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chun Chan F, Meissner B, Bhang HE, Ruddy D, Kauffmann A, Farsidjani A, Derti A, Rakiec D, Naylor T, Pfister E, Kovats S, Kim S, Dietze K, Dorken B, Steidl C, Tzankov A, Hummel M, Monahan J, Morrissey MP, Fritsch C, Sellers WR, Cooke VG, Gascoyne RD, Lenz G, Stegmeier F (2014) Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med 20(1):87–92. doi:10.1038/nm.3435
Porras DL, Wang Y, Zhou P, Molinero LL, Alegre ML (2012) Role of T-cell-specific nuclear factor kappaB in islet allograft rejection. Transplantation 93(10):976–982. doi:10.1097/TP.0b013e31824d11d7
Medoff BD, Seed B, Jackobek R, Zora J, Yang Y, Luster AD, Xavier R (2006) CARMA1 is critical for the development of allergic airway inflammation in a murine model of asthma. J Immunol 176(12):7272–7277
Ramadas RA, Roche MI, Moon JJ, Ludwig T, Xavier RJ, Medoff BD (2011) CARMA1 is necessary for optimal T cell responses in a murine model of allergic asthma. J Immunol 187(12):6197–6207. doi:10.4049/jimmunol.1101348
Causton B, Ramadas RA, Cho JL, Jones K, Pardo-Saganta A, Rajagopal J, Xavier RJ, Medoff BD (2015) CARMA3 is critical for the initiation of allergic airway inflammation. J Immunol. doi:10.4049/jimmunol.1402983
Medoff BD, Landry AL, Wittbold KA, Sandall BP, Derby MC, Cao Z, Adams JC, Xavier RJ (2009) CARMA3 mediates lysophosphatidic acid-stimulated cytokine secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol 40(3):286–294. doi:10.1165/rcmb.2008-0129OC
Borthakur A, Bhattacharyya S, Alrefai WA, Tobacman JK, Ramaswamy K, Dudeja PK (2010) Platelet-activating factor-induced NF-kappaB activation and IL-8 production in intestinal epithelial cells are Bcl10-dependent. Inflamm Bowel Dis 16(4):593–603. doi:10.1002/ibd.21092
Delekta PC, Apel IJ, Gu S, Siu K, Hattori Y, McAllister-Lucas LM, Lucas PC (2010) Thrombin-dependent NF-{kappa}B activation and monocyte/endothelial adhesion are mediated by the CARMA3.Bcl10.MALT1 signalosome. J Biol Chem 285(53):41432–41442. doi:10.1074/jbc.M110.158949
Martin D, Galisteo R, Gutkind JS (2009) CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem 284(10):6038–6042. doi:10.1074/jbc.C800207200
Mahanivong C, Chen HM, Yee SW, Pan ZK, Dong Z, Huang S (2008) Protein kinase C alpha-CARMA3 signaling axis links Ras to NF-kappa B for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. Oncogene 27(9):1273–1280. doi:10.1038/sj.onc.1210746
Rehman AO, Wang CY (2009) CXCL12/SDF-1 alpha activates NF-kappaB and promotes oral cancer invasion through the Carma3/Bcl10/Malt1 complex. Int J Oral Sci 1(3):105–118. doi:10.4248/IJOS.09059
Mc Guire C, Wieghofer P, Elton L, Muylaert D, Prinz M, Beyaert R, van Loo G (2013) Paracaspase MALT1 deficiency protects mice from autoimmune-mediated demyelination. J Immunol 190(6):2896–2903. doi:10.4049/jimmunol.1201351
Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre ML (2012) T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci USA 109(45):18529–18534. doi:10.1073/pnas.1204557109
Mc Guire C, Elton L, Wieghofer P, Staal J, Voet S, Demeyer A, Nagel D, Krappmann D, Prinz M, Beyaert R, van Loo G (2014) Pharmacological inhibition of MALT1 protease activity protects mice in a mouse model of multiple sclerosis. J Neuroinflammation 11:124. doi:10.1186/1742-2094-11-124
Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, Mak TW (2001) Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell 104(1):33–42
Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, Wang D (2003) Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol 4:857–865
Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, Cook MC, Kucharska EM, Hara H, Penninger JM, Domashenz H, Hong NA, Glynne RJ, Nelms KA, Goodnow CC (2003) Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity 18(6):751–762
Newton K, Dixit VM (2003) Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr Biol 13(14):1247–1251
Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, Thome M, Littman DR (2003) Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr Biol 13(14):1252–1258
Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, D’Acquisto F, Ghosh S, Yeh WC, Baier G, Rottapel R, Penninger JM (2003) The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity 18(6):763–775
Gringhuis SI, Wevers BA, Kaptein TM, van Capel TM, Theelen B, Boekhout T, de Jong EC, Geijtenbeek TB (2011) Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog 7(1):e1001259. doi:10.1371/journal.ppat.1001259
Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T (2007) The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nature Immunol 8(6):619–629
Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, Oland S, Hieny S, Caspar P, Yamasaki S, Lin X, Ting JP, Trinchieri G, Besra GS, Cerundolo V, Sher A (2013) Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol 190(11):5722–5730. doi:10.4049/jimmunol.1203343
Zhao XQ, Zhu LL, Chang Q, Jiang C, You Y, Luo T, Jia XM, Lin X (2014) C-type lectin receptor dectin-3 mediates trehalose 6,6′-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. J Biol Chem 289(43):30052–30062. doi:10.1074/jbc.M114.588574
McAllister-Lucas LM, Jin X, Gu S, Siu K, McDonnell S, Ruland J, Delekta PC, Van Beek M, Lucas PC (2010) The CARMA3-Bcl10-MALT1 signalosome promotes angiotensin II-dependent vascular inflammation and atherogenesis. J Biol Chem 285(34):25880–25884. doi:10.1074/jbc.C110.109421
Pan D, Lin X (2013) Epithelial growth factor receptor-activated nuclear factor kappaB signaling and its role in epithelial growth factor receptor-associated tumors. Cancer J 19(6):461–467. doi:10.1097/PPO.0000000000000001
Acknowledgments
We would like to thank Zala Jevnikar and Ming Zhang for critical reading of the manuscript, and Slavica Masina for grammatical revisions. M.T. acknowledges support by the Leenaards Foundation, the Swiss Cancer League (Oncosuisse) and the Swiss National Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jaworski, M., Thome, M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell. Mol. Life Sci. 73, 459–473 (2016). https://doi.org/10.1007/s00018-015-2059-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2059-z