
Abstract
The current gold standard diagnostic test for Johne’s disease (JD) is detecting Mycobacterium avium subsp. paratuberculosis (MAP) from fecal samples via culture and/or PCR. Other commercially available JD diagnostic tests focus on the detection of specific antibodies within the serum or milk of infected ruminants. These tests have a high specificity but low their sensitivity and usually fail to diagnose the disease until later stages of the disease. The ideal diagnostic test should detect infected animals already during the silent phase. Here, we evaluate the use of new and established approaches to define the silent phase of JD in experimentally infected goats. None of the established diagnostic tests or new approaches for the detection of humoral and cellular immune responses were positive during the first year of infection. Only the characterization of various subsets of peripheral blood leukocytes and the weight development gave some indication for the presence of a chronic, but silent, infection. Weight differences were present throughout the first year. In addition, some of the subsets of leukocytes (WC1+ γδ T cells, MHC class II+ leukocytes, CD1+ leukocytes, CD14+ granulocytes, and CD14+/MHC class II+ granulocytes) demonstrated significant differences, but only at certain time points.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
I. Introduction
Johne’s disease is a chronic intestinal disease in ruminants caused by Mycobacterium avium subspecies paratuberculosis (MAP) [1,2]. JD is commonly found in US dairy farms and at least one cattle with a positive MAP culture was detected in approximately 68 % of U.S. dairy cattle herds and in 95% of large herd with more than 500 dairy cows [3]. It shares with other mycobacterial diseases its chronic appearance [4,5,6,7,8,9]. While disease in wild ruminants is possible [10], JD is more often observed in domestic cattle, sheep, and goats. Most of these chronic mycobacterial infectious diseases have a late onset and Johne’s disease is no exception [11] with an average of two years but up to five years [12]. Animals usually acquire MAP early in life, in utero or from consumption of contaminated milk, feces, or water supplies. The progression of MAP infection, of which the last two are the actual Johne’s disease, is divided into four different distinct stages: silent, subclinical, clinical, advanced clinical [13]. In the silent stage there are no clinical or laboratory diagnostic indications of the infection. All diagnostic tests used so far are negative including culturing the pathogen from fecal samples. During the subclinical stage fecal shedding of the pathogen may be detected; this shedding is usually temporary and sporadic. Diagnostics tests are seldom indicative for the disease at this stage, and fecal culturing might be the only indication for JD. During the next two stages, clinical stage and advanced clinical stage, diagnostic tests are more often positive and animals show symptoms and signs for JD with weight loss and chronic diarrhea as the key characteristics. Even during these stages serological diagnostics are not always positive. Clinical symptoms during the later stages include a drastic decline in milk production and malabsorption of nutrients and osmotic diarrhea due to severe intestinal inflammation especially in dairy cattle, while in goats the clinical stages are dominated by anemia, emaciation and occasionally by diarrhea [14]. Most clinical animals shed large amounts of MAP in their feces and milk, spreading the disease to other members of the herd; however, some animals do not shed the bacteria at all. Death from malnourishment ensues.
The only effective control measure currently applied by most dairy farmers in the US is culling infected animals and/or instituting good herd management practices. While there are strong efforts to develop excellent vaccines [15,16], no successful efforts have been seen on the development of new diagnostic approaches to help in identify infected animals during the first two stages.
Economically, JD severely impacts dairy farmers [15,16]. Infected cows in the clinical stages do not produce as much milk as their uninfected counterparts or cows without clinical symptoms. Fecal shedding of MAP often occurs at or before the onset of clinical symptoms. Once an infected animal is symptomatic, it has most likely spread the bacteria to other animals in the herd. Diagnostic tests do exist for JD; however, while they do correctly diagnose animals at a frequency that allows for maintenance/decreased incidence of JD they only identify a portion of infected or diseased animals. The ‘gold standard’ diagnostic method for JD was culturing MAP from fecal samples, while currently the detection of the pathogen by PCR from fecal samples is replacing fecal culturing. While a positive result does indicate infection with nearly 100% certainty, fecal culturing only diagnoses animals that are already shedding MAP and consequently already infecting other animals. Furthermore, one has to consider the possibility of passive shedding [17]. Ideally, a diagnostic test for JD would positively identify MAP infected animals before they shed bacteria.
Eckstein et al (2006) recently discovered a MAP cell wall lipid not present in Mycobacterium avium subspecies avium, a bacterium that is physiologically similar to MAP and is common in the environment [18]. This lipid, termed Para-LP-01, reacts via ELISA to serum from JD-positive cows but not JD negative cows, indicating that it may be useful as a diagnostic tool. Since Para-LP-01 is, to the extent of our knowledge, unique to MAP, using it in diagnostic assays, such as the aforementioned ELISA, should significantly reduce the number of false positive results.
Cytokine levels could be measured to monitor cell-mediated immune responses to MAP. In experimentally infected cows and goats, elevated interferon-gamma (IFNγ), a TH1 stimulatory cytokine, in response to Johnin purified protein derivative (JPPD) occurred before fecal shedding [19]. Bovine macrophages infected with MAP secrete higher levels of Interleukin-10 (IL-10) than their uninfected counterparts [20]. Increased levels of IL-10 promote a TH2 response as opposed to a TH1 response, resulting in antibody production and decreased intracellular MAP killing. Monitoring IFNγ and IL-10 levels in infected animals could help determine when the TH1 to TH2 shift occurs and whether cytokine production would be a valid diagnostic tool [21].
All ruminants can develop JD, however, while the key animals of interest in the USA are dairy cattle because of the enormous losses associated with the disease [15,16], goats and sheep are the main focus in other countries. There are two key questions associated with the first two stages: Why is there a late onset of the clinical characteristics, and what could be done to improve detection of infected animals. Long-term studies with experimentally infected animals might provide more insights into the immune responses during the early stages. In addition, following peripheral blood leukocyte population changes might provide indication on what might happen at the local level in the intestine. In this study, we evaluated the use of various established and new disease-specific and none specific diagnostic approaches.
II. Materials and Methods
A. Animals
All research and housing procedures for the goats in this study were approved by Colorado State University IACUC and the approval number is #11-3120A. Twenty dairy goat kids of various breeds aged 3-5 days were purchased from CCI/Juniper Valley Products (Canon City, Colorado), a JD free goat dairy. The goat kids were housed on the Colorado State University Foothills Campus (Fort Collins, CO) in accordance with CSU animal ethics regulation. The goat kids were individually fed pasteurized cow milk three times daily for the first two months. Each goat kid had its own feeding bottle and nipple. All goat kids were housed in the same barn prior inoculation at week 7. The barn was clean and disinfected before used. Prior to weaning alfalfa hay was introduced to supplement the goats’ nutrition needs. Goats within the inoculated and non-inoculated groups were separated into different locations prior inoculation with MAP. Each group of goat kids was housed in non-adjacent corrals with open barns (fully covered, front wall open, all other walls closed) at the CSU Foothills Campus. Both groups had access to at least 4000 m2 pasture. All corrals at CSU Foothills Campus are not attached to other corrals and have significant space in between the corrals. Water and salt stones were provided ad libitum. Goats were fully milk fed for 7 weeks. Whole pasteurized cow milk was purchased from a local Walmart store (Walmart, Inc. Fort Collins, CO) in 1-gallon containers. Goats were fed with warm milk, individually by hand. Milk feeding was reduced to twice a day for 6 weeks and than reduced to once a day for additional 6 weeks. During this time alfalfa hay was supplemented. After 12 weeks post infection all goats received alfalfa hay as food supply. At week 6 post infection one goat died due to enterotoxaemia.
B. Goat infection and preparation of the inoculum
Goats were inoculated with MAP strain K-10 as described recently [22]. This strain is a bovine isolate from Nebraska and was provided to us by Dr. Vivek Kapur (University of Minnesota). The pathogen was grown on Middlebrook 7H11 supplemented with 10% OADC (oleic acid, albumin, dextrose, catalase) and 2 μg/ml mycobactin J. Cells were harvested and aliquots of 100 mg wet cell pellet in PBS (pH 7.2) were made for inoculation. 100 mg of wet cells equals roughly 109 cfu (cfu numbers were identified through serial dilutions of 1 μl of the cells used for inoculation) the required dose per inoculum. Cells were suspended in 20 ml warm pasteurized whole cow milk to a concentration of 109 cfu per inoculum and transferred to a 20 ml sterile syringe. Ten goats were inoculated three times on consecutive days with this suspension of 109 cfu, one day more than recommended by the International Committee of Johne’s Disease Researchers [23]. The inoculation was performed when the goat kids were 7 weeks old at a time point at which obtained all results from the day 0 data set. The ten goat kids within the negative control group received the same amount of milk but without the bacteria.
C. Blood collection and white blood cell preparation
Blood was collected prior the infection (termed day 0) and after inoculation at weeks 1, 3, and 8, and every four weeks thereafter. Characterization of samples collected on day 0 including flow cytometry data from different goats have been published previously [24]. For each of the blood draw 8 – 10 ml of blood were collected aseptically from the jugular vein into an EDTA solution for immediate analyses. Preparation of the peripheral blood leukocytes was performed as described previously [24]. Briefly, red blood cells were lysed with Gey’s solution (155 mM NH4Cl, 1 mM KHCO3). White blood cells were pelleted by centrifugation for 5 minutes at 259g at 4°C. The supernatant was discarded and the white cell pellet was washed twice with PBS. Clean pellets were resuspended in complemented RPMI 1640 medium (8.7% fetal bovine serum, 1 M HEPES buffer, 50x MEM without L-glutamine, 100 mM sodium pyruvate, 200 mM L-glutamine solution, penicillin [100 units/ml] and streptomycin [50 μg/ml] solution) (cRPMI). Four milliliters of blood were also drawn for serum collection, which was processed the same day and serum samples were stored at -80°C until serological testing.
D. Antigen solution preparation
Antigens for cell stimulation included cRPMI media as a negative control, concanavalin A (ConA) as the positive control, bovine purified protein derivative (bPPD) (40 μg/ml), Para-LP-01 lipid (2.5 μg/ml), and Johnin purified protein derivative (JPPD) (20 μg/ml).
E. Cell stimulation for cytokine ELISA
The amount of 5x105 peripheral blood leukocytes were added per well to flat bottom, tissue culture 96-well plates (Corning Inc, NY). One hundred microliter of antigen dilutions were added to the appropriate wells. Plates were incubated 96 hours at 37°C, 5% CO2, 99% relative humidity, and then stored at -80°C until needed.
F. Cytokine ELISA
IFNγ monoclonal antibody bovine IFN-gamma-I (MabTech) and IL-10 capture antibody (AbD Serotec) were diluted in PBS (pH 7.2) (IFNγ: 25ul antibody per 1ml PBS; IL-10: 12.5ul antibody per 1ml PBS). One hundred microliter of primary antibody per well were added to the appropriate plates. Plates were incubated overnight at 4°C, then washed three times with 200ul PBS (pH 7.4) and blocked one hour at room temperature with 200 μl of a solution containing 3% bovine serum albumin and 5% Tween 20 in PBS, pH 7.4. Plates were washed again with 200 μl PBS (pH 7.4) per well. Supernatant from the antigen stimulated cells were added in a volume of 50 μl. Plates were incubated for one hour at room temperature.
IFNγ monoclonal biotinylated antibody (MabTech) and IL-10 mouse antibovine interleukin-10: biotin (AbD Serotec) were diluted in 10% fetal bovine serum in PBS, pH 7.4 (IFNγ: 2 μl antibody per 1 ml 2%FBS/PBS; IL-10: 2 μl antibody per 1ml 2%FBS/PBS). Plates were washed three times with PBS (pH 7.4). One hundred microliter per well of secondary antibody solution were added to the appropriate plates. Plates were incubated one hour at room temperature. HRP-streptavidin was diluted 1:1000 in 10% FBS/PBS. Plates were washed three times with 200 μl PBS (pH 7.2), 100 μl of HRP-streptavidin were added per well and plates were incubated 1 hour at room temperature in the dark. Plates were then washed three times with 200 μl PBS (pH 7.4) and 100 μl of 3,3’,5,5’-Tetramethylbenzidine liquid substrate (Sigma) were added per well. Plates were incubated at room temperature in the dark until color developed (approximately 10 minutes). To stop the reaction, 100 μl of 2N sulfuric acid were added per well. Plates were read using an iMark Microplate Reader (BioRad) at a wavelength of 450 nm.
G. ELISpot
Two days prior to blood collection, MultiScreen Filter Plates (0.45 μm Hydrophobic, Millipore) were pre-wet with 100 μl 35% ethanol per well. Plates were incubated for 1 minute before being washed three times with 150 μl PBS (pH 7.2) per well. IFNγ monoclonal antibody bovine IFN-gamma-I (MabTech) or IL-10 Capture Antibody (AbD Serotec) were diluted in PBS (pH 7.2) (IFNγ: 15 μl antibody per 1ml PBS; IL-10: 7.5 μl antibody per 1ml PBS) and 100 μl of appropriate antibody solution were added per well. Plates were incubated overnight at 4°C.
Cells obtained from the goat blood were added as follows: IFNγ (all antigens except Concanavalin A) – 5x105 cells/well; IFNγ (concanavalin A) – 1.25x105 cells/well; IL-10 (all antigens except concanavalin A) – 1.25 x 105 cells/well; IL-10 (concanavalin A) – 6.25 x 104 cells/well. 100 μl of diluted antigen solutions were added to the appropriate wells. All animal/antigen combinations were performed in triplicate. Plates were incubated overnight at 37°C, 5% CO2, 99% relative humidity.
IFNγ biotinylated monoclonal antibody (MabTech) and IL-10 mouse anti-bovine interleukin-10: biotin (AbD Serotec) were diluted in 2% Fish Skin Gelatin (FSG)/PBS (pH 7.2) (IFNγ: 1 μl antibody per 1 ml 2% FSG/PBS; IL-10: 0.5 μl antibody per 1ml PBS). Plates were washed three times with 150 μl PBS per well. 100 μl of appropriate antibody dilutions were added per well. Plates were incubated 2 hours at room temperature in the dark. HRP-Streptavidin was diluted 1:1000 in 2% FSG/PBS. Plates were washed three times with 200 μl PBS (pH 7.2) and 100 μl of HRP-Streptavidin solution was added per well. Plates were incubated 1 hour at room temperature in the dark.
AEC substrate solution was prepared as in the eBioscience ELISpot Set Protocol. Plates were washed three times with 200 μl PBS (pH 7.2). 100 μl of AEC substrate solution were added per well. Plates were incubated at room temperature in the dark until spots developed (approximately 15-30 minutes). Spot development was stopped by rinsing plates in warm water. Dried plates were read using a CTL-ImmunoSpot S6 FluoroSpot equipped with Immunospot software version 5.0.9.
H. Para-LP-01 ELISA
Wells were coated with 100 ng Para-LP-01 dissolved in iso-propanol. Plates were incubated at room temperature until evaporated. Plates were blocked for one hour at room temperature with 100 μl 3%BSA (in PBS, pH 7.4) per well. ELISA was performed as described recently [18]. Briefly, 100 μl of each serum diluted 1:20 in 10% FBS/PBS (pH 7.4) was added to the wells and incubated for 30 minutes at room temperature. Plates were washed three times with PBS followed by adding secondary conjugated antibody (sheep anti-bovine IgG-h+l HRP conjugated antibody diluted 1:2000 in 10% FBS/PBS). 100 μl of secondary antibody solution were added per well and plates were incubated at room temperature for 30 minutes. Plates were washed as before and 100 μl of room temperature TMB were added per well. Plates were incubated at room temperature for 10 minutes. The reaction was stopped by adding 100 μl of 2N sulfuric acid per well. Plates were read at 450 nm using an iMark Microplate Reader (BioRad).
I. Antibodies for cell markers and staining of cell markers
Antibodies used for flow cytometry were the same as used in a previous study [24]. All antibodies were obtained from AbD Serotec (Raleigh, NC), except for CD14 monoclonal antibodies, which were purchased from BioLegend (San Diego, CA).
J. Flow cytometry
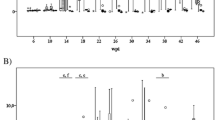
Flow cytometry was performed as described previously [24]. Briefly, samples were analyzed via flow cytometer (FACSCantoll, Becton Dickinson, USA) equipped with BD FACSDiva software (Becton Dickinson, USA). A minimum of 10,000 events was collected per sample. Profiles were analyzed with FlowJo software (TreeStar, Ashland, OR, USA). The following panels were used: CD4/CD8, CD4/WC1, CD1/CD14, and MHC class II/CD14. Gate strategies included selections of cell populations according to their appearance in side and forward scatter as shown in Figure 1 . Thereafter, the fluorescences of the positive cell signals were compared to their corresponding isotype-matched controls. The percentage of positive cells and intensity of fluorescence was recorded as percentage of positive cells and mean fluorescence channel (MFC), respectively. Specific cell populations (CD14+ granulocytes or CD14+ monocytes) were back gated to show the presence and amount of such population within the selected cell types.

Gating strategy of the flow cytometry of the various subsets of leukocytes and specific cell surface markers used in this study.
K. Statistical analyses for peripheral blood leukocyte population and for evaluation of diagnostic approaches
Repeated measures analysis was used for both the peripheral blood leukocyte populations and the ELISpot data. Analysis was done using SAS 9.3 (Cary, NC). A separate repeated measures analysis was done for each cell type using Proc Mixed. The within-subjects factor is time and the between-subjects factor is treatment group (infected or uninfected). A time*treatment interaction term was also included in the model. The arh(1) covariance structure was used, allowing for unequal variances at the different time points. Comparisons of means between treatment groups at each time point were considered. A Benjamini-Hochberg adjustment was applied to account for multiple testing across time points separately for each cell type [25]. Statistical significance was defined as a p-value of 0.05 or less. We note that a log transformation of the ELISpot data was considered to satisfy model assumptions. However, the conclusions were the same for both the original and log transformed data, so results on the original scale are presented here.
L. Fecal culturing of MAP and Serology
Fecal samples were obtained at the same time blood collection was performed. Culturing of MAP from fecal samples was performed at the Rocky Mountain Regional Animal Health Laboratory, Colorado Department of Agriculture Animal Industry Division. Serology for JD was performed by the Diagnostic Laboratory at the Veterinary Teaching Hospital at Colorado State University. Serology was performed using Paracheck (Prionics, Inc.) and an in-house developed AGID test.
M. Weight
Weights were obtained in pounds (lbs) with a commercially available scale until goats reached 50 pounds. The weight was determined by weighing the person holding the goat minus the weight of the person alone. After this period goat weights were determined with a hanging scale and a calf sling. Weights were obtained on a weekly basis during milk feeding and every month thereafter. Weights in pounds were later converted into kg (1 kg = 2.20462 lbs). Weights until the age of six months were already separately published [22].
III. Results
A. Fecal Cultures, Parachek ELISA, and AGID
All animals in the study were tested monthly via standard diagnostic techniques (fecal culturing, Parachek ELISA, and AGID). MAP could not be cultured at any time from fecal samples from any of the animals. Serology (Paracheck ELISA, AGID) for JD was negative for all goats (Data not shown).
B. Cell Counts
White blood cell counts were obtained from each goat throughout the study. While the average cell counts between the inoculated and negative goats were never significant different (p < 0.05), the infected goats consistently showed a higher average than the uninfected goats. In week #1 and week #8 post infection the p-value was less than 0.1 (Figure 2 ).

Total peripheral blood leukocyte cell count.
C. IFNγ ELISpot and IFNγ ELISA
The IFNγ ELISpots for all three stimulatory antigens (bPPD, Para-LP-01, JPPD) are shown in Figure 3 . With the exception of weeks 24 to 31 post infection for bovine (bPPD) and Johnin PPD (jPPD) as stimulatory antigens, there were no statistically significant (p-value < 0.05) differences between the two groups. The statistical significance was lost when the results of one goat within the non-inoculated group with a Corynebacterium pseudotuberculosis infection were removed (data not shown). While other goats within both groups had slightly elevated reactivities during this hot and dry summer period, this goat specifically reacted more robustly than any of the other goats. The IFNγ ELISAs for all three stimulatory antigens (bPPD, Para-LP-01, and jPPD) are depicted in

Interferon gamma ELISpot on peripheral blood leukocytes stimulated with bovine PPD, Johnin PPD, or Para-LP-01 of MAP-inoculated and non-inoculated goats.

Interferon gamma ELISA on peripheral blood leukocytes stimulated with bovine PPD, Johnin PPD, or Para-LP-01 of MAP-inoculated and non-inoculated goats.
D. IL-10 ELISpot and IL-10 ELISA
The IL-10 ELISpots for all three stimulatory antigens (bPPD, Para-LP-01, and jPPD) are presented in Figure 5 while those results for the IL-10 ELISA are shown in Figure 6 . No statistical significant differences were detected.
E. Para-LP-01 ELISA

IL-10 ELISpot on peripheral blood leukocytes stimulated with bovine PPD, Johnin PPD, and Para-LP-01 of MAP-inoculated and non-inoculated goats.

IL-10 ELISA on peripheral blood leukocytes stimulated with bovine PPD, Johnin PPD, or Para-LP-01 of MAP-inoculated and non-inoculated goats.
In addition to its use as a stimulatory antigen for cellular immune assays, Para-LP-01 was also used to determine humoral immune reactivity utilizing a lipid-based ELISA. The results for the Para-LP-01 ELISA are shown in Figure 7 , Panel A. The overall average O.D. showed only for the weeks #8 and #24 statistical significance for the infected group to have an increased O.D. However, beside the average O.D. numbers individual infected goats reacted strongly at certain weeks throughout the study, especially goats #2 and #6 (Figure 7 , Panel B). Another observation was that the goats had stronger reactivity during the first few weeks of life and that this reactivity was reduce to normal levels during the first few months.

Para-LP-01 based Lipid-ELISA on sera from MAP-inoculated and non-inoculated goats.
F. Key Populations of Leukocytes
The goat leukocyte samples were analyzed via flow cytometry to determine the abundance of the key populations: granulocytes, lymphocytes, and monocytes. The different cell types were defined as variable size and high granulation (SSC-Ahigh/FSC-Avariable: granulocytes), as small size and low granulation (SSC-Alow/FSC-Alow: lymphocytes), and as medium size and low granulation (SSC-Alow/FSC-Amedium: monocytes). Percentages of granulocytes, lymphocytes, and monocytes were determined in panels CD4/CD8, CD4/WC1, and CD14/MHC class II. No statistical differences were found for granulocytes, lymphocytes, or monocytes at any time point. Data are summarized in Figure 8 . Data from the other two panels showed very similar pattern (data not shown). The granulocyte populations did not appear different between the infected and uninfected goats. Monocytes in the infected goats seemed to have slightly increased throughout the first 31 weeks. A different observation was made for the lymphocyte populations. Lymphocytes were slightly higher in uninfected goats in the first few weeks, while the monocytes are slightly increased in inoculated goats.

Flow cytometry results of granulocytes (A), lymphocytes (B), and monocytes (C) in inoculated and uninfected goats.
G. Lymphocyte Subpopulations
The following goat lymphocyte populations were analyzed: CD4+ T cells, CD8+ T cells, CD4/CD8 double positive T cells, CD4/CD8 double negative T cells, and WC1+ γδ T cells. While CD4 and CD8 markers were analyzed in one panel, γδ T cells were analyzed in a separate panel with CD4 as the second marker. T cells analyzed for CD4 and CD8 did not show any statistical significant changes throughout the study. However, there was a clear trend for CD4 positive T cells during the silent stage with an increased CD4+ T cell population in infected goats (Figure 9 , Panel A). CD8+, CD4+/CD8+, and CD4-/CD8- T cell populations did not exhibit any trends or changes during the course of the study (Data not shown). Interestingly, WC1+ γδ T cells populations showed statistically significant differences during the study. Throughout the study, the WC1+ γδ T cells were decreased in the infected goats, and the decrease was statistically significant at week 24 (Figure 9 , Panel B). Thus the overall decreased in the lymphocyte population in the infected goats might have been due to decreased amounts of WC1+ γδ T cells.

Flow cytometry of selected T cell lymphocytes in infected and uninfected goats.
H. Monocyte Subpopulations
The overall amount of monocytes was very small when compared to those of granulocytes and lymphocytes and significant differences were difficult to detect. The amount of CD14+ monocytes fluctuated during the study without clear trends. During weeks 1 to 20, with the exception for weeks 3 and 8, there was an overall increase of CD14+ monocytes, however, without any statistical significance (Figure 10 , Panel A). No differences were obtained between the Monocyte/CD14+/MHC class II+ populations in infected and uninfected goats. Interestingly, however, the MHC class II+ cells within the monocyte population that were CD14- were increased in the infected goats between weeks 24 and 31. Week #28 differences were statistically significant (Figure 10 , Panel B). Furthermore, cells within the monocyte population without the markers CD14 and MHC class II had decreased numbers between the weeks 20 and 31 with statistically significant differences in week 28 (Figure 10 , Panel C). The other interesting marker for antigen-presenting cells in mycobacterial infections is CD1. No differences were seen for monocytes expressing both markers (CD14, CD1) (data not shown). However, cells within the monocyte population expressing CD1 but not CD14 demonstrated significant increase for weeks 28 and 31 (Figure 11 , Panel A). A slight increase was also seen early on from week 1 to week 8 with no statistical significance for any of the weeks in this period, while week 28 and week 31 demonstrated statistical significance. Cells within the monocyte population expressing neither CD14 not CD1 were reduced starting at week #20 with statistical significance for weeks 20 to 31 (Figure 11 , Panel B).

Flow cytometry of monocyte subset.

Flow cytometry of cells within the monocytes that do not express CD14.
I. Granulocytes Subpopulations
While the majority of CD14 positive cells are monocytes there is a significant population of CD14 positive cells that are granulocytes. During the first weeks, no trend was seen for the CD14 positive granulocytes. After week 12 there were differences in the percentage of CD14+ granulocytes between the infected and uninfected goats. While the CD14+ granulocytes of uninfected goats dropped the percentage of CD14+ granulocytes of the infected goats was the same as in earlier weeks post infection (Figure 12 , Panel A). Interestingly, a similar observation was made for a subset of those cells that also bear the marker MHC class II and were statistical significant in the same weeks as seen for CD14+ granulocytes (week 12, 16, and 28) (Figure 12 , Panel B).

Flow cytometry of granulocytes that are positive for CD14.
J. Weight & Standard Diagnostics (serology, fecal culture)
A brief analysis of the weight gain during the first 24 months after inoculation was previously demonstrated [22]. Here we extend our analysis of the total weight development on a 4-week basis. There is a clear separation of the average weights of inoculated versus non-inoculated goats with several time points demonstrating statistical significance with a p-value of less than 0.05 (weeks 22, 26, 34, and 42) (Figure 13 ). While an early significant difference in the weight gain was previously shown, we did not see a similar difference in 4-week weight gain during this time period.

Weight development of goats inoculated with MAP versus the negative control group.
All fecal cultures did not show any growth of Mycobacterium avium subsp. paratuberculosis. In addition, all serum samples had a negative serology using Paracheck.
IV. Discussion
Currently available diagnostics for JD are unable to consistently diagnose animals before they begin shedding MAP or exhibit clinical features. This is especially true for the silent and subclinical stages of this chronic disease. Since early diagnosis is key for controlling JD it is important to evaluate current diagnostics and new diagnostic tests during the pre-clinical stages as well as non-specific parameter such as weight gain, weight development, and peripheral blood leukocyte population changes. Thus, the overall scope of this study was to not only evaluate serology, cell-mediated immune assays, as well as the culturing of the pathogen from fecal samples during the silent stage, but also characterize immune cell population changes despite their non-specificity. In this study, we experimentally inoculated ten goats with MAP. Blood and fecal samples were collected on a monthly basis to determine not only the immune status during the early silent stage of Johne’s disease and efficacy of diagnostic approaches but also to monitor changes in the various population of the peripheral leukocytes. All results were compared to a control group of ten uninfected goats.
The results from the ELISpot, cytokine ELISA, and lipid ELISA do not clearly indicate if any of the goats are infected with MAP. There were no consistent significant differences between the two groups on any assay or with any antigen. Since the standard diagnostic assays (fecal culturing, Parachek ELISA, and AGID) have been negative for all goats thus far and we are testing new diagnostic approaches, we currently do not have a set “cut-off” value to determine whether an animal is positive or negative for Johne’s disease. While there are currently few differences between the two groups, the results from the Para-LP-01 Lipid-ELISA seem promising. Some of the infected goats have shown an increased antibody response to Para-LP-01 on the lipid ELISA (#2 and #6). However, those results were never consistent.
One goat in the uninfected group (goat #13) reacted strongly to bPPD and JPPD on the IFNγ ELISpot and ELISA. In these assays, he produced more IFNγ than the rest of the uninfected group and even of the infected group. He reacted barely to Para-LP-01 on these assays but did not exhibit any antibody response to Para-LP-01 on the lipid ELISA. While not reacting to Para-LP-0 the goat showed an increase of antibody response through Paracheck. Considering that goat #13 produced negative results on standard diagnostic assays, reacted weakly or not at all to Para-LP-01, was not exposed to MAP as well as was obtained from a JD-free goat dairy, and had similar results on both PPDs we do not think that his reactivity toward bPPD and jPDD is an indication of JD but rather an indicator of an infection due to a similar bacterial pathogen, such as Nocardia and Corynebacteria [26,27]. C. pseudotuberculosis, the causative agent of caseous lymphadenitis, is notorious for producing false positive results in JD diagnostics [30]. Thus, we hypothesized a subclinical C. pseudotuberculosis infection was responsible for goat #13’s strong reactivity toward bPPD and jPPD. Para-LP-01 may be a useful tool in diagnosing JD since it shows low to no cross-reactivity with animals infected with C. pseudotuberculosis.
In an experimental infection of angora goats, Stewart et al (2006) saw an initial IFNγ response to JPPD at 6 months post infection with a consistent IFNγ response beginning at 16 months post infection [29]. We did observe a peak in JPPD reactivity at 20 weeks post infection. Perhaps, this peak is analogous to that observed by Stewart et al; however, this peak was observed in both groups. Stewart et al. did not show IFNγ responses from their control animals; therefore, it is difficult to determine if the IFNγ response at 6 months post infection was due to JD. There is some debate over whether assays using components from the peripheral blood are effective in, or even capable of, diagnosing JD before fecal shedding occurs. Lybeck et al. (2010) discovered that goats naturally infected with MAP tend to shed the bacterium before immune responses to the disease are detectable [30].
Among the various subsets of monocytes analyzed in this study the most impressive results were obtained for monocytes expressing CD1 markers, which were more prominent in infected goats than in uninfected goats. CD1 molecules usually present lipid antigens and thus this is not a surprise since the majority of surface molecules in mycobacteria are lipids. Beard et al. (2000) investigated the role of γδ T cells and the presence of CD1 molecules [31]. While they found an increase of γδ T cells in the Peyer’s patches, no differences were found for the γδ T cells in the regional lymph nodes. They could not find any significant changes for the expression of CD1 in any of the analyzed tissues.
Lymphocytes are part of the early immune responses in mycobacterial infections. Among those CD4+, CD8+, and γδ T cells are the most studied immune cells. CD4+ T cells are associated with strong early immune responses on mycobacterial infections. Two classical studies on early cellular immune responses in experimentally infected cows demonstrated that the specific cellular immune responses led by activated CD4+ T cells is detectable no earlier than 6 months after inoculation. Most of these specific immune responses are significantly detectable around 9 months post infections. Our data were not in full accordance with those found by other studies on T cell populations during early infection in calves with JD [32,33]. The overall picture of CD4 T cells in the early infection is seen as a progressive increase of such cells with the expression of activation markers [32]. However, in our analyses of the CD4+ T cell populations we did not detect a steady increase on those cells in the peripheral blood. Although almost constantly higher numbers of those cells were found in infected goats, we did not observe a clear trend at all.
Another key element of the cellular immune responses in JD, especially in cattle, is the involvement of γδ T cells. While two phenotypes of γδ T cells were identified with respect to the presence of specific surface markers (CD8, CD2, and WC1) and the related cytokine profiles, we analyzed only the WC1+ population of γδ T cells. The γδ T cells that are WC1+ exhibit more pro-inflammatory characteristics while the other subset of γδ T cells shows anti-inflammatory properties [31]. The pro-inflammatory properties of the WC1+ subset are greatly related to their strong production of IFN-γ and IL-2 [31]. Badi et al. (2010) described an increase of such cells in non-seroconverted cattle with JD, while we did not observe such increase [34]. In our study this specific subset of γδ T cells was steadily declining and was always less present in infected goats when compared to uninfected goats. The differences in the observation might be due to the time point of evaluation with our goats being less than 1 year old and the cattle studied by Badi et al. (2010) are at least 2 years old [34].
The most surprising observation was the strong differences in weight gain during the first weeks after inoculation and the constant differences in the weight development between the inoculated and the uninfected goats. While reduced weight gain and reduced weight development are non-specific characteristics of any chronic disease, this still seems to be a surprise to us since we did not observe any clinical symptoms or positive results through specific diagnostics.
The goals of this study were (1) to test new diagnostic approaches using the Para-LP-01 lipid of MAP to potentially diagnose animals during the silent or sub-clinical stage of JD, and (2) to evaluate non-specific markers such as peripheral blood leukocytes. Thus far, we have no conclusive evidence of the efficacy of these approaches but have discovered that Para-LP-01 may be a useful tool in diagnosing JD in animals that may have been exposed to the similar bacterium C. pseudotuberculosis. Furthermore, we found that during the silent stage of JD changes occur in the various subsets of peripheral blood leukocytes. Finally, it seems that early weight gain differences as well as weight development during the silent phase are excellent non-specific characteristics pointing toward Johne’s disease in dairy farms that are suspected to have this chronic disease.
References
E. J. Manning, and M. T. Collins, “Mycobacterium avium subsp. paratuberculosis: pathogen, pathogenesis and diagnosis,” Rev. Sci. Tech., vol. 20, pp. 133–50, April 2001.
O. Chacon, L. E. Bermudez, and R. G. Barletta, “Johne’s disease, inflammatory bowel disease, and Mycobacterium paratuberculosis,” Annu. Rev. Microbiol., vol. 58, pp. 329–363, October 2004.
USDA, APHIS, Info Sheet, Veterinary Services, Center for Epidemiology and Animal Health. April 2008. Johne’s Disease on U.S. Dairies, 1991–2007.
M. W. Borgdorff, M. Sebek, R. B. Geskus, K. Kremer, N. Kalisvaart, and D. van Soolingen, “The incubation period distribution of tuberculosis estimated with a molecular epidemiological approach,” Int. J. Epidemiol., vol. 40, pp. 964–970, August 2011.
D. A. Enarson, A. Seita, and P. Fujiwara, “Global elimination of tuberculosis: implementation, innovation, investigation,” Int. J. Tuberc. Lung. Dis., vol. 7(Suppl 3), pp. S328-32, December 2003.
P. K. Murthy, “Clinical manifestations, diagnosis and classification of leprosy,” J. Indian Med. Assoc., vol. 102, pp. 678–679, December 2004.
M. Tsukamura and S. Ichiyama, “Correlation of minimal inhibitory concentration values of antituberculosis agents against Mycobacterium avium complex strains with incubation period,” Kekkaku, vol. 63, pp. 107–110, February 1988.
J. A. Trubiano, C. J. Lavender, J. A. Fyfe, S. Bittmann, and P. D. Johnson, “The incubation period of Buruli ulcer (Mycobacterium ulcerans infection),” PLoS Negl. Trop. Dis., vol. 7, pp. e246–3, October 2013.
J. A. Jernigan and B. M. Farr, “Incubation period and sources of exposure for cutaneous Mycobacterium marinum infection: case report and review of the literature,” Clin. Infect. Dis., vol. 31, pp. 439–43, August 2000.
A. S. Motiwala, A. Amonsin, M. Strother, E. J. Manning, V. Kapur V, et al., “Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis isolates recovered from wild animal species,” J. Clin. Microbiol., vol. 42, pp. 1703–1712, April 2004.
R. W. Sweeney, “Pathogenesis of paratuberculosis,” Vet. Clin. North Am. Food Anim. Pract., vol. 27, pp. 537–546, November 2011.
C. J. Clarke, “The pathology and pathogenesis of paratuberculosis in ruminants and other species,” J. Comp. Pathol., vol. 116, pp. 217–261, April 1997.
A. Tiwari, J. A. VanLeeuwen, S. L. McKenna, G. P. Keefe, and H. W. Barkema, “Johne’s disease in Canada Part I: clinical symptoms, pathophysiology, diagnosis, and prevalence in dairy herds,” Can. Vet. J., vol. 47, pp. 874–882, September 2006.
R. F. Slocombe, “Combined Streptomycin-Isoniazid-Rifampin Therapy in the Treatment of Johne’s Disease in a Goat,” Can. Vet. J., vol. 23, pp. 160–163, May 1982.
F. Bastida and R. A. Juste, “Paratuberculosis control: a review with a focus on vaccination,” J. Immune Based Ther. Vaccines, vol. 9, pp. 8, October 2011.
J. Cho, L. W. Tauer, Y. H. Schukken, M. I. Gomez, R. L. Smith, et al., “Economic analysis of Mycobacterium avium subspecies paratuberculosis vaccines in dairy herds,” J. Dairy Sci., vol. 95, pp. 1855–1872, April 2012.
P. Kralik, R. Pribylova-Dziedzinska, A. Kralova, K. Kovarcik, and I. Slana, “Evidence of passive faecal shedding of Mycobacterium avium subsp. paratuberculosis in a Limousin cattle herd,” Vet. J., vol. 201, pp. 91–94, July 2014.
T. M. Eckstein, S. Chandrasekaran, S. Mahapatra, M. R. McNeil, D. Chatterjee, et al., “A major cell wall lipopeptide of Mycobacterium avium subspecies paratuberculosis,” J. Biol. Chem., vol. 281, pp. 5209–5215, February 2006.
H. Mikkelsen, C. Aagaard, S. S. Nielsen, and G. Jungersen, “Correlation of antigen-specific IFN-γ responses of fresh blood samples from Mycobacterium avium subsp. paratuberculosis infected heifers with responses of day-old samples co-cultured with IL-12 or anti-IL-10 antibodies,” Vet. Immunol. Immunopathol., vol. 147, pp. 69–76, June 2012.
N. Abendaño, R. A. Juste, and M. Alonso-Hearn, “Anti-inflammatory and antiapoptotic responses to infection: a common denominator of human and bovine macrophages infected with Mycobacterium avium subsp. paratuberculosis,” Biomed. Res. Int., vol. 2013, pp. 908348, January 2013.
D. J. Begg, K. de Silva, N. Carter, K. M. Plain, A. Purdie, et al., “Does a Th1 over Th2 dominancy really exist in the early stages of Mycobacterium avium subspecies paratuberculosis infections?,” Immunobiology, vol. 216, pp. 840–846, July 2011.
A. N. Malone, D. M. Fletcher, M. B. Vogt, S. K. Meyer, A. M. Hess, et al., “Early weight development of goats experimentally infected with Mycobacterium avium subsp. paratuberculosis,” PLoS ONE, vol. 8, pp. e84049, December 2013.
M. E. Hines, 2nd, J. R. Stabel, R. W. Sweeney, F. Griffin, A. M. Talaat, et al., “Experimental challenge models for Johne’s disease: a review and proposed international guidelines.,” Vet. Microbiol., vol. 122, pp. 197–222, June 2007.
D. M. Fletcher, M. B. Vogt, H. E. Pirner, A. M. Hess, R. A. Bowen, et al., “Flow cytometry baseline on peripheral leukocyte cell profiles for dairy goat kids,” Am. J. Anim. Vet. Sci., vol. 8, pp. 177–189, September 2013.
Y. Benjamini and Y. Hochberg, “Controlling the false discovery rate: A practical and powerful approach to multiple testing,” J. Royal Statist. Soc. B, vol. 57, pp. 289–300, January 1995.
M. A. Lanéelle, M. Tropis, and M. Daffé, “Current knowledge on mycolic acids in Corynebacterium glutamicum and their relevance for biotechnological processes,” Appl. Microbiol. Biotechnol., vol. 97, pp. 9923–9930, December 2013.
D. N. McMurray, “Mycobacteria and Nocardia,” In: Baron S, editor. “Medical Microbiology”, 4th edition. Galveston (TX): University of Texas Medical Branch at Galveston; 1996. Chapter 33.
E. J. Manning, H. F. Cushing, S. Hietala, and C. B. Wolf, “Impact of Corynebacterium pseudotuberculosis infection on serologic surveillance for Johne’s disease in goats,” J. Vet. Diagn. Invest., vol. 19, pp. 187–190, March 2007.
D. J. Stewart, J. A. Vaughan, P. L. Stiles, P. J. Noske, M. L. Tizard, et al., “A long-term study in Angora goats experimentally infected with Mycobacterium avium subsp. paratuberculosis: clinical disease, faecal culture and immunological studies,” Vet. Microbiol., vol. 113, pp. 13–24, May 2006.
K. R. Lybeck, A. K. Storset, B. Djønne, M. Valheim, and I. Olsen, “Faecal shedding detected earlier than immune responses in goats naturally infected with Mycobacterium avium subsp. paratuberculosis,” Res. Vet. Sci., vol. 91, pp. 32–39, August 2011.
P. M. Beard, S. M. Rhind, M. C. Sinclair, L. A. Wildblood, K. Stevenson, et al., “Modulation of gammadelta T cells and CD1 in Mycobacterium avium subsp. paratuberculosis infection,” Vet. Immunol. Immunopathol., vol. 77, pp. 311–319, December 2000.
Koo HC, Park YH, Hamilton MJ, Barrington GM, Davies CJ, et al. (2004) Analysis of the immune response to Mycobacterium avium subsp. paratuberculosis in experimentally infected calves. Infect Immun 72: 6870–83.
C. W. Wu, M. Livesey, S. K. Schmoller, E. J. Manning, H. Steinberg, et al., “Invasion and persistence of Mycobacterium avium subsp. paratuberculosis during early stages of Johne’s disease in calves,” Infect. Immun., vol. 75, pp. 2110–2119, December 2004.
F. A. Badi, A. I. Haroon, and A. M. Alluwaimi, “The gammadelta cells as marker of non-seroconverted cattle naturally infected with Mycobacterium avium subspecies paratuberculosis,” Res. Vet. Sci., vol. 88, pp. 72–76, February 2010.
Author information
Authors and Affiliations
Additional information
This research was funded by the Agriculture and Food Research Initiative (AFRI) competitive grant no. 2012-67015-19379 from the USDA National Institute of Food and Agriculture.
Dr. Torsten Eckstein is an assistant professor in microbiology specialized in bacterial lipidomics and glycomics for their use as tools for new diagnostics therapeutics and adjuvants for zoonotic infectious diseases.
Dr. Marcela Henao-Tamayo is an assistant professor in immunology specialized in the cellular immune responses of mycobacterial diseases with a strong emphasis on tuberculosis.
Dr. Richard Bowen is a professor in biomedical sciences and is specialized in animal models for bacterial and viral diseases.
Dr. Ann Hess is an assistant professor in statistics and works closely together with scientists in the research on infectious diseases.
Darcy Fletcher and Megan Vogt are graduate students in the field of immunology, while Stephen Meyer is a graduate student in microbiology.
Allison Genis, Madeline Hayes, and Hannah Pirner are undergraduate students at the time the research was performed.
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fletcher, D., Vogt, M., Genis, A. et al. Silent Phase of Johne’s Disease in Experimentally Infected Goats – A Study on New and Established Diagnostic Approaches Using Specific and Non-Specific Parameters. GSTF J Vet Sci 1, 2 (2015). https://doi.org/10.7603/s40871-015-0002-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.7603/s40871-015-0002-2