
Abstract
Pulmonary hypertension (PH) is a disease with high morbidity and mortality. The prevalence of idiopathic pulmonary arterial hypertension (IPAH) and hereditary pulmonary arterial hypertension (HPAH) is approximately two- to four-fold higher in women than in men. Paradoxically, there is an opposite male bias in typical rodent models of PH (chronic hypoxia or monocrotaline); in these models, administration of estrogenic compounds (for example, estradiol-17β [E2]) is protective. Further complexities are observed in humans ingesting anorexigens (female bias) and in rodent models, such as after hypoxia plus SU5416/Sugen (little sex bias) or involving serotonin transporter overexpression or dexfenfluramine administration (female bias). These complexities in sex bias in PH remain incompletely understood. We recently discovered that conditional deletion of signal transducer and activator of transcription 5a/b (STAT5a/b) in vascular smooth muscle cells abrogated the male bias in PH in hypoxic mice and that late-stage obliterative lesions in patients of both sexes with IPAH and HPAH showed reduced STAT5a/b, reduced Tyr-P-STAT5 and reduced B-cell lymphoma 6 protein (BCL6). In trying to understand the significance of these observations, we realized that there existed a well-characterized E2-sensitive central neuroendocrine mechanism of sex bias, studied over the last 40 years, that, at its peripheral end, culminated in species-specific male (“pulsatile”) versus female (“more continuous”) temporal patterns of circulating growth hormone (GH) levels leading to male versus female patterned activation of STAT5a/b in peripheral tissues and thus sex-biased expression of hundreds of genes. In this report, we consider the contribution of this neuroendocrine mechanism (hypothalamus-GH-STAT5) in the generation of sex bias in different PH situations.
Similar content being viewed by others


Introduction
Pulmonary hypertension (PH) is a disease with high morbidity and mortality, characterized by pulmonary arterial remodeling with the classic onion-skin obliterative and plexiform lesions (1–4). (Some investigators use the phrase “pulmonary arterial hypertension [PAH]” to generically encompass the human disease and rodent models. Others use the phrase “pulmonary hypertension [PH]” as the generic term and reserve PAH for different forms of the human disease [such as idiopathic PAH (IPAH) and hereditary PAH (HPAH)]. In this report, we follow the second approach.) In IPAH, there is a sexual dimorphism with the prevalence approximately two- to four-fold higher in postpubertal women than in men, with an earlier onset in women (median age: third decade in women, fourth decade in men), but a better outcome after diagnosis in women than in men (1–8). In HPAH, autosomal-dominant mutations, mainly in the gene for bone morphogenetic protein receptor 2 (BMPR2), underlie the disease and its female dominance, but with low penetrance (10–15%) and a delayed onset (1–8). A subset of patients, typically women, taking anorexigens develop PH (9,10). In systemic sclerosis (SSc)-associated PH, SSc is itself 4–10 times more prevalent in women, thus the occurrence of PH in this disease is more prevalent in women (11,12). However, after excluding PH diagnosed at baseline, male SSc patients have a higher likelihood of developing PH and a worse outcome (11,12). Schistosomiasisinduced PH shows no sex bias (13), whereas HIV-induced PH shows a small (1.2-fold) male bias (14). The mechanisms that underlie these sexual dimorphisms in humans, especially the female bias in disease prevalence in IPAH and HPAH, are incompletely understood (8,15).
There is an opposite male bias in the typical rodent models of PH induced by chronic hypoxia or monocrotaline (MCT) (8,15,16). In these rodent models, estrogenic compounds [(estradiol-17β (E2) and 2-methoxyestradiol (2-ME)] typically inhibit PH (8,15,16). Strikingly, rats or mice exposed to hypoxia plus the inhibitor SU5416 (Sugen) develop PH without any sex bias, and E2 administration has little protective effect on development of increased right ventricular systolic pressure in this model (8,17,18). (The phrase “sex bias” refers to biological and biochemical differences, and “gender bias” refers to cognitive, behavioral and cultural differences. Thus, in the present essay, the phrase “sex bias” is used throughout. The phrase “sex specific” refers to entities that are observed exclusively in one or the other sex [for example, expression of genes located on the Y chromosome]. In contrast, the phrase “sex biased” refers to entities that show a quantitative phenotypic difference between the two sexes.) In converse examples, female mice over-expressing the serotonin transporter or the S100 calcium-binding protein S100A4/mts1, or administered the anorexigen dexfenfluramine (but not male mice), spontaneously develop modest PH by 5 months (15,19–22). Although only up to one-third of male mice expressing the BMPR2R899X trans gene spontaneously developed right ventricular systolic pressure >30 mmHg, this proportion increased to two-thirds when the mice were fed a high-fat diet (23); in this model, 2-ME was not protective (24). The mechanisms underlying these disparate and contrasting sex-bias observations in different rodent models, and the differences between rodent models and the human disease, are not understood. The juxtaposition of a female bias in prevalence of human IPAH and HPAH with a male bias in disease development in the chronic hypoxia- or MCT-induced rodent models, together with the observation that administration of E2 was protective in the rodent models, led to the concept of an “estrogen paradox” to describe these observations (5,8,15–18). Any hypothesis (Figure 1) that provides a path toward clarifying these puzzling sex-bias observations in PH in humans and rodents would be of great value.

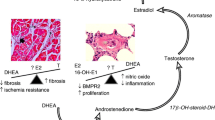
Hypothesis: central neuroendocrine and peripheral mechanisms in generation of sex bias in PH. Male (M: “pulsatile”) versus female (F: “more continuous”) patterned secretion of GH by the pituitary followed by patterned activation of the STAT5a/b-BCL6 axis represents a well-established pathway (27) to functionally connect the hypothalamus/pituitary on the one hand and pulmonary vascular tissues on the other hand as a mechanism for generating sex-biased gene expression in the latter. Upstream of pituitary/GH would be multifactorial central sex-bias mechanisms that impinge on the hypothalamus and effectuate M versus F patterns of secretion of GHRH; these include estradiol-17β (E2), serotonin (5-HT), cytokines and BMPs. Downstream of STAT5-BCL6 in peripheral pulmonary vascular cells would be hundreds of responsive genes for which patterned expression together contributes to the sex-biased disease process involving cell proliferation, cell hypertrophy, resistance to apoptosis and cytokine/growth factor secretion. The hypothesis also includes direct effects of various mediators (for example, cytokines and growth factors) at the level of the peripheral pulmonary vascular cells in a sex- and species-biased manner due to underlying gene expression changes (of on the order of perhaps 500–1,000 genes) already effectuated through the GH-STAT5 axis.
A Gap in Knowledge in the PH Literature Concerning Sex Bias Mechanisms and Effects of Exogenously Administered E2
We begin by identifying a gap in knowledge in the PH literature about a critical aspect of how exogenous E2 injected into an animal “feminizes” gene expression. We note that all previous work on sex bias in rodent-based PH models, including after administration of steroid sex hormones (for example, E2, 2-ME or testosterone) and/or gonadectomy, have focused on direct effects of steroid hormones at the level of peripheral vascular tissues in the lung (5,6,8,15–26). (However, the terminology used can often be confusing and, indeed, misleading. Thus, in the context of a “neuroendocrine hypothesis,” the term “central” refers to the hypothalamus and pituitary [the “neuro-” part], while the terms “distal” or “peripheral” refer to all locations in the body where a hormone can circulate [the “-endocrine” part]). Thus, there have been extensive studies of steroid hormone effects (E2, 2-ME, testosterone) in lung tissues in rodent models, in isolated human and rodent pulmonary vascular cells (smooth muscle cells [SMCs] and endothelial cells [ECs]) and on vascular cell proliferation, transcriptional regulation of BMPR2 gene expression and BMPR2-initiated cell signaling (5,6,8,15–26). This exclusive focus on peripheral tissue effects of steroid hormones is somewhat at odds with insights gleaned over the last 30–40 years in a sister field (sex-biased expression of liver genes) and the mechanism by which exogenously administered E2 influences this sex bias (27,28). The critical insight from the 1970s has been that effects of E2 and testosterone on sex-biased gene expression in distal tissues (liver in this case) depend on the pituitary; hypophysectomy blocks effects of E2 or testosterone on sex-specific gene expression (27) (Figure 1). In terms of vascular biology, it was reported in 1978 (29) and later confirmed in 1992 (30) that hypophysectomy in the male rat markedly impaired arterial remodeling after aortic balloon injury, especially vascular smooth muscle cell proliferation and myointima formation, but the mechanisms remain undefined.
Overall, the literature in the PH field today is where the “sex bias in liver gene expression” field was in the early 1970s. At that time, the effects of E2 and/or gonadectomy on sex-biased drug metabolism and thus on the sex-biased expression of cytochrome P450 (P450 CYP) enzymes were thought to be due solely to direct effects of sex steroid hormones on the hepatocyte (27,31). However, it was shown in 1973 by Colby et al. (32), and extensively confirmed by numerous studies since then (27,33–43), that the feminizing effect on sex-biased liver gene expression of an injection of E2 into a rat or mouse was indirect and had an absolute dependence on the pituitary (Figure 1). As explained in detail by Waxman and colleagues over the last 2 decades, this neuroendocrine mechanism of sex bias, working through an axis consisting of the hypothalamic arcuate nucleus-growth hormone releasing hormone (GHRH)-growth hormone (GH)-signal transducer and activator of transcription 5 (STAT5) accounts for sex-biased expression of >1,000 genes in the liver and also of body growth and body weight (27,41–48) (Figures 2, 3). Parenthetically, STAT5 is the major transcription factor activated by the binding of GH to its receptor on all cell types (27,41–48). This activation involves Tyr phosphorylation of receptor-associated Janus kinases, with the latter then mediating Tyr phosphorylation of STAT5. Additional ligand-induced Ser phosphorylation is also observed on STAT5. Thus, much of the biological activity of GH on different cell types is mediated by this activation of STAT5. From among the family of STAT transcription factors, STAT5a and STAT5b are the only transcription factors that mediate sex bias (27,41–48).

Sex bias in circulating GH patterns in rats and humans (note that the y axes in the human panels are logarithmic). In rats (panels A and B on left side), male pattern is “pulsatile” with low interpulse levels; female pattern is “more continuous” with higher frequency of pulses and with GH levels of “non-zero” between pulses. In humans (panels A and B on the right side), males are pulsatile with low interpulse levels, but in human females, the interpulse levels are high. (Rat data shown are adapted from ref. (62) in compliance with the terms of usage of the Proceedings of the National Academy of Sciences, USA; human data shown are adapted from ref. (56) with permission of The Endocrine Society). Additionally, insets in the rat data panels show sex-biased expression of CYP species by Western blotting, especially the male-specific expression of the 3A subfamily (3A2) (62). For comparison, in mice, M and F pulse heights are similar, the frequency is higher in F and interpulse levels can be low in both M and F (ref. (55); not shown).

Temporal relationships between the sexually dimorphic spontaneous GH secretory profiles and hepatic PY-STAT5 activity levels (DNA-binding gel-shift assay) in the normal rat (adapted from ref. (43) with permission of The Association for the Study of Internal Secretions and The Endocrine Society). Black arrows point to when the liver tissue was harvested for PY-STAT5 analyses. In males, PY-STAT5 peak heights are higher and interpulse levels very low (left panels). In females, peak height is lower and interpulse levels higher than in males.
The targets of E2 and testosterone include neuronal cells in the arcuate nucleus and other ventromedial hypothalamic nuclei (49–54) (Figure 4). The downstream elements of this sex-bias mechanism are male versus female patterned secretion of GHRH from the hypothalamus into the hypophyseal portal circulation and then corresponding patterned secretion of GH by the pituitary into the general circulation. This finding was discovered by Edén in 1979 (55) and, since then, has been extensively confirmed in mice, rats and humans (27,45–50,56–62). Circulating GH levels in the male (M) are usually called “pulsatile” (two to four peaks per day) with very low interpulse levels; in the female (F), there is a higher frequency of pulses (seven or more peaks per day) with significant interpulse levels; thus, this is called “more continuous” (Figures 1, 2). This scenario results in M versus F patterned activation of PY-STAT5a/b in the distal tissues and, downstream of that, a major cascade of sex-biased gene expression of >1,000 genes (Figure 3) (27,41–48). Even when assayed at a single time point in humans (in the ambulatory state in the morning after an overnight fast), the median value of serum GH levels was 80- to 120-fold higher in women than in men (61). This is a higher sexual dimorphic ratio than that for E2 (ratio: 2.2 female bias) or testosterone (ratio: 14 male bias) observed in the same sera (61). Thus, while the GH-STAT5 axis of sex bias is well documented in the literature, it seems not to have been a consideration in the PH literature.

Expression of BMPR2 RNA in the mouse brain and includes the arcuate nucleus of the hypothalamus (white arrows). This image was taken from the atlas, available online from The Jackson Laboratory. Figure shows result of a cRNA hybridization (adapted from ref. (78), a PLOS Biology paper).
A Novel Starting Point for Our Stat5 Studies on Sex Bias in PH
For a variety of reasons (28,63,64), we initiated an investigation into the involvement of STAT5a/b species in the pathogenesis of PH produced in mice exposed to chronic hypoxia. Mice with heterozygous or homozygous conditional deletions of the STAT5a/b locus in vascular SMCs were generated in crosses between STAT5a/bfl/fl and transgelin (SM22α)-Cre+/+ parents (28) (Figure 5). Wild-type (wt) males subjected to chronic hypoxia showed significant PH and pulmonary arterial remodeling, with wt females showing minimal changes (a male-dominant phenotype) (28) (Figure 6). However, in conditional STAT5−/− mice, hypoxic females showed the most severe manifestations of PH (a female-dominant phenotype) (28) (Figure 6). The reversal of sex bias in this model of PH did not require the complete loss of STAT5a/b genes. Even heterozygous conditional STAT5a/b+/− mice (thus, with a 50% loss of STAT5a/b) abrogated the maledominant PH phenotype (28).

Immunofluorescence studies using mouse tissues showing selective loss of STAT5a/b from SMA-positive tunica media (dark arrows: green staining) in main pulmonary artery (PA) (A) or PA segment in a lung section (B) of the conditional STAT5a/b−/− mice. Quantitation is shown on the right of the respective panels; *P < 0.05. Scale bars = 10 µm. (C) Western blots of extracts of cultured arterial smooth muscle cells derived from pools of five mice, each of the indicated groups, with SMC-specific conditional homozygous deletion of STAT5a/b verifying the loss of expression of STAT5a (using L-20 pAb from Santa Cruz Biotechnology), STAT5a + b (using C-17 pAb from Santa Cruz Biotechnology) and male-biased expression of BCL6. Adapted from ref. (28). pAb, polyclonal antibody.

Female-dominant PH in homozygous SM22-Cre, STAT5a/b−/− mice after chronic hypoxia. Female homozygous knockout mice developed highest mean right ventricular systolic pressure (RVSP) measured by Millar catheterization (A), greatest right ventricular hypertrophy (B) measured by the Fulton index (right ventricle weight/(left ventricle weight + septum weight)), and greatest PA remodeling (C: wall thickness; D: SMA positive vessels per 10× field; and E: elastin staining using Verhoeff-van Gieson stain); scale bar = 45 µm. Wt and KO mice were matched littermates (n = 5/group). *P < 0.05 (adapted from ref. (28)).
Immunofluorescence studies on human lung sections from patients with late-stage idiopathic or hereditary pulmonary arterial hypertension (IPAH or HPAH) showed that SMCs in obliterative pulmonary arterial lesions, both male and female, had, overall, reduced STAT5a/b, reduced PY-STAT5 and reduced B-cell lymphoma 6 protein (BCL6) (Figure 7), but with interpatient and interlesion variability even in the same patient (28). In concordance with these observations, studies of SMC and EC cell lines derived from vessels isolated from lungs of male and female IPAH patients and controls, revealed instances of coordinate reductions in STAT5a and STAT5b in IPAH-derived cells, including in SMCs and ECs from the same patient (28).

Marked reduction of STAT5, PY-STAT5 and BCL6 in cells in obliterative lesions in IPAH patients. Compiled figure adapted from ref. (28) summarizing data from two studies based on lung sections provided by the Department of Pathology, The Johns Hopkins School of Medicine (JHU) and the Pulmonary Hypertension Breakthrough Initiative (PHBI). The top half of this composite summarizes immunofluorescence data from study 1 showing loss of STAT5a (red) in SMA-positive (green) cells in obliterative lesions in IPAH. Quantitation of images in (A) is shown in (B) (mean ± standard error of the mean (SE)), and various controls are shown in (C) and (D). Scale bar = 45 µm. The bottom half of this composite summarizes study 2 in terms of the quantitation of immunofluorescence data for STAT5a + b, PY-STAT5 and BCL6 (mean ± SE) in obliterative lesions in male and female patients. *P < 0.05.
Taken together, these data (Figures 5–7) identified two aspects of STAT5 biology relevant in the pathogenesis of PH: first, the contribution of differential male versus female PY-STAT5 activation patterns in the early stages of this disease to generating the sex bias, and second, the loss of STAT5 in cells in vascular lesions in many patients of both sexes in the late stages of the disease (28). These observations led us to consider more deeply what was known about the hypothalamus-GH-STAT5 axis in determining sex bias in gene expression and to consider whether those insights might be applicable to sex bias in PH.
The Hypothalamic (GHRH)-Pituitary (GH)-Distal Tissues (Stat5, BCL6) Axis
Several insights in the prior hypothalamic-GH-STAT5 sex-bias literature are specifically relevant to understanding sex bias issues in PH. First, how does administration of exogenous E2 (or similar sex steroid) into an animal produce its effects? As mentioned earlier, it has been shown extensively that effects of exogenously injected E2 depend on generating the “feminine plasma growth hormone pattern” through central hypothalamic/pituitary mechanisms (27,32–40) (Figure 1). Specifically, a single injection of E2 into a rodent affects the function of cells in the arcuate and ventromedial nucleus in the hypothalamus and additional nuclei therein (27,49–54). These are the very cells that secrete GHRH in male and female patterns (27). This result then induces the patterned secretion of GH by the pituitary and then STAT5-driven sex-biased patterns of gene expression in distal tissues (27,41–48). Critically, that the administration of E2 may work on distal tissues in terms of sex-bias effects indirectly through the hypothalamus is an insight missing from the PH literature (Figure 1).
Second, what is the relevant difference between a female human and a female rodent? While, overall, in mice, rats and humans, males have a “pulsatile” GH pattern and females have a “more continuous” GH pattern (27), there are quantitative differences in the three species (Figure 2). In adult male rats, GH is released into the circulation in discrete pulses approximately every 3.5 h with little or no circulating GH detectable during the interpulse interval (62) (Figure 2). In female rats, the pulses are more frequent, the pulse heights are lower and the interpulse levels are higher (62) (Figure 2). In mice, the male circulating GH levels are also pulsatile and with very low levels through the interpulse intervals (27,38,57). However, in the female mouse GH levels pulse more frequently and with the same pulse heights as in the male mouse, but still with low levels during the interpulse intervals (27,38,57). In contrast, men show infrequent GH pulses of high magnitude, with very low interpulse levels (Figure 2); however, women show a more continuous high level of GH (Figure 2) (39,40,56,59,60). Thus, a difference between female humans and female rodents is in the much higher continuous GH levels in women compared with female rodents (Figure 2). Reconstruction experiments in rats show that exogenous administration of GH in 3-min pulses for up to six pulses per day generated expression of male-specific CYP2A2 and 3A2 subfamily genes in the liver (65). Different aspects of the specific GH patterns (pulse frequency, pulse height, interpulse interval and interpulse levels) affected sex-specific expression of different CYP enzymes differently, even in the same species (65–69). In an example specifically relevant to the PH focus of this essay, these differences included that the CYP3A subfamily is male-specific in rats, but female-specific in mice and humans (27,62,65–69). We note that it is the CYP3A subfamily of enzymes that convert the injected inactive MCT to the bioactive monocrotaline pyrrole (MCTP) in male rats that then induces PH (70,71).
Third, the major consequence of the interaction of GH with its receptor at the cell surface is activation (Tyr phosphorylation) of the STAT5a and STAT5b (“JakSTAT signaling”) (27). Indeed, STAT5 was identified 2 decades ago as the key transcription factor that mediated the sex-bias effects of different GH patterns at the level of distal tissues (27,41–48). Thus, respective M or F patterns of circulating GH levels are converted into respective patterned activation of PY-STAT5 in the liver (Figure 3). This patterned PY-STAT5 activation transcriptionally activated or repressed >1,000 different genes through combinations with other preexisting transcription factors (HNF4, C/EBP) as well as by regulating expression of a master transcription repressor such as BCL6 (27,41–48). This generates a cascade of sex-biased gene expression in distal tissues but without the direct effect of any steroid sex hormone.
In the context of the present PH-focused essay (and the MCT comment above), it was shown 2 decades ago that the sex-biased transcriptional expression of the CYP3A subfamily by GH is mediated by STAT5 (27,72). Moreover, it is known that hypoxia also activates STAT5 through Tyr phosphorylation (73) and that STAT5 affects many target genes already shown to be relevant in PH pathogenesis (summarized in Figure 9 in the article by Yang et al. [28]).
Fourth, the sex-biased expression of genes is duplicated in isolated human or rat hepatocytes exposed to the respective high pulsatile (male) or low, continuous (female) levels of GH in cell culture (27,67). Thus, cells isolated from the body replicate sex-biased gene expression in culture in response to patterns of GH in the absence of any exposure to sex hormones.
Fifth, at the molecular level, the patterned activation of PY-STAT5 and downstream cascades of transcription factors lead to sex-specific expression of target genes in several categories (27,41–48). Class I male genes require pulsatile GH/PY-STAT5 activation, whereas class II male genes are primarily regulated by repression by the continuous female pattern of GH. Female class I genes require a continuous GH/PY-STAT5 activation, whereas female class II genes are strongly derepressed (thus upregulated) after hypophysectomy. It is critical to understand that GH-STAT5-initiated transcriptional mechanisms that generate sex-biased changes in expression of hundreds of genes differently is not a simple on/off switch in a digital sense. Waxman and colleagues, and others, have shown in detailed studies (27,41–48) that sex-biased expression of genes by GH-STAT5 activation in the hepatocyte depends on a “dynamical” signaling process that involves multiple activation and inactivation cycles (frequency as in “pulsatile” or “continuous”), differences in magnitude of signal strength (“level” of GH) and the rates of these changes (different slopes), resulting in different rates of association of different transcription regulatory proteins (including PY-STAT5a/b) at the level of the chromatin encompassing different genes. It is these differences in signal strength, frequency, slopes of the activation or inactivation reactions, and in coassociated proteins that lead to different chromatin conformations (active or inactive for RNA transcription) in the DNA context of different genes (27,41–48). The net result is a cellular phenotype driven by the GH-STAT5 axis consisting of a large array (500–1,000) of genes expressed in a sex-biased manner differently in different species (Figure 1).
This patterned activation of cells includes longer-lived chromatin remodeling at the respective male- and female-specific genes in that respective male-derived hepatocytes were more responsive to the male pulsatile pattern of GH in culture, and female-derived were less so (27,67). From this, we anticipate that this remodeled chromatin will retain sex-biased phenotype when primary cells are continued to be maintained in culture.
Sixth, upstream of the pituitary is the arcuate nucleus in the hypothalamus, which secretes GHRH (36,74,75). BMPR1, BMPR2 and caveolin-1 (cav-1) and various BMPs (-2, -4, -6, -7) are also expressed and function in arcuate neurons of the hypothalamus (74–78) (Figures 1, 4). Moreover, these are the very neurons that are the target of exogenously administered E2 converting the GHRH secretion pattern into a feminine continuous pattern (49–54). Thus, it is necessary to consider the possibility that pathogenesis of hereditary PAH might involve contributions of BMPR2 and cav-1 mutations to changes in signaling and altered intracellular trafficking at the level of neuronal cell bodies in the arcuate nucleus of the hypothalamus (Figure 4). This result would affect patterns of GHRH secretion and, thus, the neuroendocrine pathway culminating in STAT5-generated sex-bias in pulmonary vascular tissues. This neurogenic component has received little attention in the PH literature, even though it is known that chronic hypoxia as well as intermittent hypoxia decrease plasma GH levels by effects that include the hypothalamus/pituitary (79,80).
Seventh, STAT5 is a critical transcription factor in hypothalamic neuronal cells (74) (Figure 1). Instillation of granulocyte-macrophage colony-stimulating factor (GM-CSF) into the arcuate nucleus of the hypothalamus activates PY-STAT5 and affects appetite and thus body fat (74). Nestin-Cre-driven deletion of STAT5a/b generates mice that show insulin resistance, put on weight and become obese, but in a sex-biased manner; female mice accumulate more body fat but less lean mass (74). These Nestin-Cre-STAT5−/− mutant mice no longer change their food-intake behavior upon GM-CSF instillation into the hypothalamus (74) (Figure 1). Moreover, STAT5 and STAT3 also mediate the effects of leptin and the gp130 receptor in hypothalamic neuronal cells (74,75).
Eighth, the cytokine BMP9 has drawn recent attention as a “quiescence” promoting factor in vascular biology and in PH (79,80). Curiously, BMP9 is expressed largely by the liver, and its promoter region has binding sites for STAT5a/b, HNF4α and C/EBPα (81,82; Qiagen CHiP database). BMP9, which is also expressed in the basal forebrain (83), stimulates GH expression (84). Moreover, there is synergism between BMP9 and GH intracellular signaling pathways in murine multilineage cells, especially at the level of the Jak-STAT3 and -STAT5 pathways (84). Inhibitors of Jak1, STAT3 or STAT5 each inhibited this synergism (84).
Synthesis of the Literature on Sex Bias Mediated by the Neuroendocrine-GH-Stat5 Axis with That on Sex Bias in PH
The above-enumerated insights involving the neuroendocrine-GH-STAT5 axis allowed us to compile the following synthesis, potentially explaining many of the puzzling sex bias-related observations in the PH literature.
First, why is the prevalence of IPAH higher in women than in men? On average, women have 80- to 120-fold higher levels of circulating GH than men (61). Moreover, this is in a more continuous pattern than in men (Figure 2) (27,56). It is known that GH promotes vascular smooth muscle cell proliferation and migration and is required for normal vascular reactivity and modeling (85–88). Indeed, the prevalence of systemic hypertension is 20–50% in patients with acromegaly (in whom the plasma GH levels are high) because of “stiffer arteries” (85,87); no information is available on PH in acromegaly. We propose that these 80-to 120-fold higher levels and a more continuous pattern of GH, and thus activation of different sets of cell cycle, cell proliferation and cell migration regulatory genes (27), is why IPAH is more prevalent in women than men. The identification of molecular targets activated, directly or indirectly, in vascular cells by differences in male versus female patterns of circulating GH (and thus PY-STAT5 activation) is unexplored. It is already known that these gene targets in liver include BMP2, BMP5, BMP10, Id1, Id2, Id3, BMPER, Smad5 and Smad7 as female-biased and BMP4 and BMP6 as male-biased (45–48). We anticipate occurrence of these and also interactions between BMP9 and the GH-STAT5 axis in pulmonary vascular cells (see comments on BMP9 in previous section and refs. 79–84).
Second, what is the relevant difference between a female human and a female rodent underlying differences in sex bias in PH? There are quantitative differences in patterns of circulating GH in males and females in mice, rats and humans resulting in the same gene being regulated differently in terms of sex specificity in the three species (Figure 2) (discussed in detail in refs. 27,56–62,65–69). The major relevant difference between female humans and female rodents is in the much higher continuous GH levels in women compared with female rodents (Figure 2). Thus, expression of P450 CYP 3A subfamily genes is male-biased in the rat (62,65), but female-biased in humans (68). We anticipate that such differences will underlie sex bias in the development of PH in the different species.
Third, is there really an estrogen paradox? We suggest that the basis for why women get more IPAH than men may be unrelated to E2 levels in women. Importantly, women have an 80- to 120-fold higher level of GH than men and in a “more continuous” pattern. This difference far exceeds the 2.2-fold difference in E2 between women and men or the 14fold difference in testosterone (61). Also, it has not been appreciated before that E2 may have little to do directly with IPAH prevalence in women. In animal models, typically in male rodents, E2 would be protective by targeting the hypothalamus directly (49–54). Thus, as has been shown in the liver literature, E2 would “feminize” GH patterns, changing the male pattern of expression in rodents to a female pattern (Figure 1). Therefore, the apparent paradox can be explained by involving GH in human IPAH and by remembering that E2 directly targets the hypothalamus, not so much the peripheral vascular tissue.
Fourth, what is the basis for the ghrelin paradox? Ghrelin is a peptide produced by, for example, the stomach, which stimulates hypothalamic neurons to secrete GHRH and thus increase GH secretion (89). Ghrelin levels are three-fold higher in women than in men (90). Increased levels of circulating ghrelin are observed in IPAH (91). However, paradoxically, in MCT or hypoxic rat models the administration of ghrelin reduces PH (92,93). Thus, ghrelin is protective in rodent models but elevated in IPAH. In this instance, ghrelin has a central effect (stimulates GHRH and thus GH secretion, as in humans) and a different peripheral effect (antagonizes endothelin-1) (91).
Fifth, why is the MCT model male-biased in the rat (8,15,16)? Downstream of the GH/STAT5 axis is the sex-specific expression P450 CYP3A subfamily members (27,62,65–69). STAT5 is the transcription factor that upregulates CYP3A (27,72), and it is the CYP3A subfamily enzymes that metabolize MCT to its active MCTP (70,71). In the rat, this is male-specific (27,62,66) but is female-specific in mouse and humans (27,62,65–69). It should not then be a surprise that a single injection of MCT efficiently induces PH in the male rat.
Sixth, why is there no sex bias in the hypoxia-SU5416 model (8,17,18)? SU5416 (Sugen) has been described in the PH literature as an inhibitor of receptor 2 for vascular endothelial growth factor (VEGF R2) (8). However, it has been shown already that SU5416 inhibits multiple receptor tyrosine kinases (VEGFR, platelet-derived growth factor receptor [PDGFR], colony stimulating factor receptor 1 [CSF-1R], Fms-related tyrosine kinase 3 [Flt3], receptor tyrosine kinase protein [Kit], proto-oncogene “rearranged during transfection” [Ret], fibroblast growth factor receptor 1 [FGF-R1]) (88). Thus, SU5416 inhibits Flt3-dependent activation of PY-STAT5 (95). It is also an agonist of the aryl hydrocarbon receptor (96) and even inhibits expression and activity of neuronal nitric oxide synthase, thus protecting neurons from apoptosis (97). The plethora of these effects of SU5416, especially inhibition of PY-STAT5 activation, suggests why this model does not show a sex bias. In ovariectomized mice, SU5416 by itself in normoxia produces PH, and administration of E2 does not affect this (17). This lack of effect of E2 administration would be the consequence of SU5416 already inhibiting PY-STAT5 activation. Indeed, in humans, the PY-STAT5 inhibitor dasatinib (98) is known to be accompanied by the occasional development of PH mainly in female patients (99). As for the differences between rat and mouse in their response to E2 in the hypoxia-Sugen model (8,17,18), we note that it has already been shown that members of the same GH-STAT5-responsive gene subfamily (for example, the CYP 3A subfamily) can be male-specific in the rat and female-specific in the mouse (27,62,65–69). Thus, we expect that effects of E2 will indeed be different in the mouse and the rat given how the GH-STAT5 pathway is already known to show different sex bias in the regulation of similar target genes in the two species.
Seventh, why do female mice over-expressing the serotonin transporter or the S100 calcium-binding protein S100A4/mts1 or the dexfenfluramine-administered mice, but not male mice, develop modest PH by 5 months (15,19–22)? Although increased serotonin (5-HT) has been implicated in the pathogenesis of PH (10,15,26,100), especially in the female-specific mouse models developed by MacLean et al. (19–21), and after administration of an anorectic drug dexfenfluramine (22,100), the mechanistic focus has largely been on effects of 5-HT on distal vascular tissues (10,15,19–22,26,100). We note that it is already known that the PH-causing anorexigens fenfluramine, aminorex, phentermine and fluoxetine increase 5-HT in the hypothalamus (101–105) and that fenfluramine blunted GH responsiveness to GHRH (105). Moreover, estrogens are themselves anorexigenic through effects at the level of the hypothalamus (106,107) showing that the central effects of anorexigens can be female-biased. Additionally, it is already known that PY-STAT5 signaling in the hypothalamus is involved in regulating appetite and sex-biased changes in body weight (74–77) (Figure 1). Remarkably, it has been shown already that 5-HT suppresses STAT5 expression and PY-STAT5 activation (108), and 5-HT receptor and dopaminergic D1 and D2 receptor antagonists also inhibit PY-STAT5 activation (for example, pimozide) (109,110). Interestingly, the neuroleptic pimozide is now sold commercially as a low-molecular-weight inhibitor of PY-STAT5 activation.
Although Launay et al. (100) reported that deletion of the serotonin 2B receptor gene in mice reduced development of chronic hypoxia-induced increase in right ventricular systolic pressure and also blocked its further enhancement by dexfenfluramine, these investigators used mice that had a whole-body knockout of the 5-HTR2B gene (111). These mice had cardiac defects, and this genetic deletion would also involve the serotonin system in the hypothalamus (111). We suggest that consideration of mechanisms in such models also include the central effects of 5-HT at the level of the hypothalamus and sex-specific changes in the patterns of GH secretion and STAT5 activation.
Eighth, a small subset (3 of 11) of male mice expressing BMPR2 R899X in smooth muscle cells developed increased right ventricular systolic pressure >30 mmHg (23). However, this was reported as increased to 7 of 11 when these mice were fed a high-fat diet (23). This result was not affected by the estrogen 2-ME (24). The investigators focused on insulin resistance at the level of peripheral tissues in this model, but did not consider the participation of the hypothalamic targets of a high-fat diet in this pathogenesis. The hypothalamic mechanisms would have been apparent from the data of Lee et al. (74), who showed that Nestin-Cre-driven deletion of STAT5a/b in the central nervous system (including in the hypothalamus) led to increased body fat and increased insulin resistance in mice in a sexually dimorphic manner. Thus, central neuroendocrine mechanisms, no longer responsive to estrogens because of high-fat diet feeding, should be considered in this model.
Ninth, in primary human pulmonary artery smooth muscle cells (PASMCs) in culture (from disease-free individuals), the expression of BMPR2, Id1 and Id3 but not Smad1 are higher in male-derived cells compared with female-derived cells (25). Thus, these genes show retention of sex bias in their expression (25). Correspondingly, female-derived PASMCs proliferate modestly faster in response to 5-HT, PDGF or BMP4 (25). These observations are similar to the findings of Thangavel et al. (67), who observed that GH-mediated inducibility of male-biased isoforms of cytochrome P450 was better when cultures were derived from male rats but not female rats. Taken together, these data show the retention of sex bias in cells in culture in the absence of any direct steroid hormone additions. Waxman and Holloway (27) suggested that this results from lasting effects of sex-biased chromatin remodeling in female versus male cells.
Tenth, we emphasize that in the hypothesis outlined in Figure 1, we specifically combine both central neuroendocrine mechanisms with peripheral tissue-level mechanisms in the pathogenesis of PH. For example, we clearly visualize that biologically activated MCTP would directly affect pulmonary vascular cells in the process of generating a PH phenotype in male rats (64). The male dominance in this model would derive from how the inactive MCT is converted into the active MCTP through CYP3A enzymes, which are themselves upregulated by the GH-STAT5 axis in a sex-biased manner (27,70,71).
Eleventh, the male bias in disease worsening in SSc-associated PH (11,12), the lack of sex bias in schistosomiasis-associated PH (13) and the small male bias in development of HIV-associated PH (14) are likely due to localized pulmonary vascular-level activation of immune processes that include STAT5 activation (112–117).
Stat5a/b and BCL6 in Vascular Cells
STAT5a/b transcription factors are activated by Tyr phosphorylation by a range of cytokines and growth factors including interleukin (IL)-2, GM-CSF, IL-6, PDGF and erythropoietin and participate in mediating Th2 responses (112). Waxman and colleagues have identified BCL6, a master transcriptional repressor affecting hundreds of genes, as a male-biased downstream effector of PY-STAT5 activation (27,46–48). Our attention to BCL6 resulted from (a) the knowledge that this master regulator is involved in regulating B-cell development and function and follicular T helper cell function, and its genetic deletion results in a hypercytokine production state, which includes pulmonary vasculitis (118–120); (b) that STAT5a/b and BCL6 are expressed ubiquitously in different tissues and function, respectively, as master transcription activator and master transcription repressor (27); and (c) that several investigators have proposed that PH pathogenesis involves a component of localized pulmonary vascular inflammation (121–123). Thus, a reduction in BCL6 in cells in obliterative lesions of PH (Figure 7) would enhance localized proinflammatory cytokine production (plus changes in expression of hundreds of additional genes) (27,118,119). We would expect the conditional localized deletion of BCL6 in vascular smooth muscle cells in mice to also lead to an abrogation of sex bias in the chronic hypoxia model of PH (Figure 1).
It has been established over the last 2 decades that the expression levels of STAT5a and STAT5b in different adult tissues are approximately equal in males and females in rodents and humans (27,28,112). Thus, the sex-specific differences between males and females in STAT5 biology in adult tissues is in terms of patterns of activation of the transcription factor (PY-STAT5) and not regulation of expression per se. In contrast, BCL6 expression, mediated by PY-STAT5 itself, is male-biased (27,46,47).
STAT5 has also been identified as a tumor suppressor (118,119). In terms of clinical outcome, a reduction in STAT5a expression is associated with worse prognosis in breast and prostate cancer (124,125). Thus, a reduction in STAT5 levels corresponds to increased cell proliferation and tumorigenesis. Reduced STAT5 content (nonphosphorylated and phosphorylated) in cells in obliterative lesions in PH (28) (Figure 7) is consistent with this view of a loss of a tumor suppressor in a proliferative lesion.
We posit that sex bias in PH pathogenesis would be generated early in the disease process by the patterned activation of intact levels of STAT5a/b and BCL6 (Figure 1). This result would transition in late-stage disease to an overt reduction in STAT5a/b, PY-STAT5 and BCL6 levels in lesions in both female and male patients. This loss of STAT5a/b, PY-STAT5 and BCL6 in late-stage disease would set the stage for enhanced cell proliferation, cell hypertrophy and increased localized cytokine production and localized inflammation, and thus arterial remodeling.
What regulates the level of expression of STAT5 in vascular tissues? There is an increase in STAT5a and STAT5b expression in mammary epithelium in the female at puberty (126). However, changes in expression of STAT5a/b in vascular tissues at puberty and its sex dependence have not been investigated. Some mechanisms that regulate STAT5a/b expression levels and function include BCL6 itself (as repressor), microRNA 222 (miR222), transforming growth factor (TGF)-β and BMP/Smad signaling and the HIV negative regulatory factor (nef) protein (reviewed in 28). At this time, what downregulates STAT5a/b levels in SMCs in obliterative IPAH lesions in late-stage disease in both men and women is not known.
Sex-Bias Effects Depend on the Sum of a Plethora of Changes in Gene Expression
It is clear from the work of Waxman and colleagues that sex bias in hepatocyte biology due to the operation of the GH-STAT5-BCL6 axis is the net result of changes that include the altered expression of at least 500–1,000 genes (Figure 1) (27). We anticipate that a similar large pool of gene expression changes will underlie the sex-bias phenotypes observed in PH. We anticipate that a different spectrum of changes will be observed in different vascular cell types (smooth muscle, endothelial cells) under different PH situations. We emphasize that the GH-STAT5-BCL6 axis regulates target genes through multiple dynamical parameters (GH pulse height, pulse frequency, interpulse intervals, interpulse levels, speed of onset of PY-STAT5 activation, termination of PY-STAT5 activation, rate of decrease in nuclear PY-STAT5 levels, activation and repression of a cascade of additional master transcription factors and repressors, sex-biased chromatin remodeling of the hundreds of different target genes differently in different cell types) (27), such that at the present time, it would be premature to single out one or more mediators. We project that these downstream events will include altered expression of multiple mediators of intracellular regulatory pathways (transcription, translation and intracellular trafficking) as well as extracellular communication (cytokines and growth factors) acting in concert to generate the overall sex-biased phenotype. As to the question to what extent does PY-STAT5 activation protect against PH or cause PH, the answers would be “both” and “neither,” depending upon the species, sex and PH circumstance. Despite these complexities, the purpose of this essay is to draw attention to two specific entities (GH and STAT5) in establishing a functional connection between the hypothalamus on the one hand and pulmonary vascular tissues on the other hand as a mechanism for generating sex bias in the latter. Upstream of GH would be the multifactorial sex-bias mechanisms that impinge on the hypothalamus, and downstream of STAT5 in pulmonary vascular cells would be hundreds of “pattern” responsive genes that together comprise the sex-biased disease process.
Conclusion
Fifty years ago, Frantz and Rabkin (127) pointed out that the fasting ambulatory levels of plasma GH in women were markedly higher than levels in men. Physical activity enhanced GH levels, particularly in women. These authors also reported that men administered estrogen (diethylstilbestrol) showed the “female pattern” of ambulatory GH (that is, marked elevation of GH levels) and postulated that this was due to the effect of estrogen to enhance “pituitary sensitivity, or that of higher centers” to “physical activity and possibly other stimuli” (127). Today, the neuroendocrine-GH-STAT5 axis represents a novel way to think about sex bias in the pathogenesis of a vascular disease—pulmonary hypertension. This hypothesis connects the hypothalamus/pituitary through well-established regulatory mechanisms to sex-biased changes in gene expression in pulmonary vascular tissues. It also provides a path toward explaining many of the puzzlements observed in sex bias in human PH and in rodent models of this disease. The detailed elucidation of these GH-STAT5-based molecular mechanisms may lead to identification of novel targets for therapy and, perhaps more importantly, prophylaxis of this disease.
Note Added in Proof
Recently, Savai et al. (128) have drawn attention to the reduction in expression of the forkhead transcription factors FoxO1, FoxO3 and BCL6 (and additional “FoxO1 targets”) in SMCs in pulmonary vascular lesions and in microdissected vessels from patients with IPAH (both sexes). FoxO1, FoxO3 and BCL6 were also reduced in microdissected vessels from male rats with PH in the hypoxia-Sugen model, but only FoxO1 and BCL6 were reduced in the MCT male rat model. These investigators did not investigate STAT5a/b expression. SMC-specific deletion of FoxO1 using the SM22-Cre approach generated mice that showed PH under normoxic conditions and a more severe PH in response to hypoxia compared with wt mice (males and females were pooled). Enhancing FoxO1 expression by administering FoxO1 adenovirus or paclitaxel reduced the severity of PH in the MCT rat and the hypoxia-Sugen/rat models. Savai et al. (128) suggested that a pro-proliferative and inflammatory state, generated by the loss of FoxO1, BCL6, growth arrest and DNA damage (GADD45) and other molecules, contributes to the development of PH. However, these investigators did not discuss the relationships between FoxO transcription factors, BCL6 and the GH-STAT5 axis. Our data showing the loss of BCL6 in cells in obliterative lesions in both male and female late-stage IPAH patients (Figure 7) confirm the loss of BCL6 observed by Savai et al. in IPAH (128). However, there is an already known interplay between STAT5a/b, BCL6, FoxO1 and FoxO3. Both the FoxO1 and FoxO3 promoters have STAT5-binding sites (128) (Qiagen CHiP database). FoxO1 is repressed by STAT5 binding (129). The GH-STAT5 axis is known to functionally repress a large cluster of genes targeted for activation by FoxO1 (130). Activated STAT5 upregulates expression of miR-182, which targets FoxO1 mRNA for degradation (131). We note that Waxman and colleagues previously showed that expression in murine hepatocytes of BCL6 is male-biased and GH-STAT5-dependent and that expression of various forkhead transcription factors is also sex-biased in a STAT5-dependent manner (27,41–48).
In recent years, several investigators suggested that insulin resistance phenotypes, obesity and metabolic dysfunction contribute to pathogenesis of PH in humans and in animal models (132–137). As examples, male mice with ApoE−/− deletion but not female mice developed PH (134), and there was an increased prevalence of markers of insulin resistance phenotype in female patients with PH (135,136). Remarkably, it has been known for >5 decades that insulin potently affects the hypothalamic-pituitary-GH axis, including in the context of diet and obesity, and that this axis is dysfunctional in insulin resistance (74,127,138–142). Indeed, today, the GH secretion response in patients to administered insulin is taken to be a gold standard to evaluate the integrity of these neuroendocrine mechanisms (138–142), and the relationships (typically inverse) between insulin resistance and GH levels have been studied extensively (74,140,142). Nevertheless, even though the relationships between the GH axis, estrogen administration, insulin resistance and sex-biased obesity phenotypes are well established (74,127,138–142), to date, the literature in the area of insulin resistance, obesity and PH pathogenesis (132–135) lacks consideration of any of these GH-anchored neuroendocrine mechanisms.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Tuder RM, Marecki JC, Richter A, Fijlkowska I, Flores S. (2007) Pathology of pulmonary hypertension. Clin. Chest. Med. 28:23–42.
Rabinovitch M. (2008) Molecular pathogenesis of pulmonary hypertension. J. Clin. Invest. 118:2372–9.
Morrell NW (2010). Role of bone morphogenetic protein receptors in the development of pulmonary hypertension. Adv. Exp. Med. Biol. 661:251–64.
Stacher E, et al. (2012) Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186:261–72.
Austin ED, et al. (2009) Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 34:1093–9.
Austin ED, et al. (2012) BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol. Sex Differ. 3:6.
Fessel JP, Loyd JE, Austin ED. (2011) The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm. Circ. 1:305–19.
Lahm T, Tuder R, Petrache I. (2014) Progress in solving the sex hormone paradox in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 307:L7–26.
Fishman AP. (1999) Minorex to Fen/Phen: an epidemic foretold. Circulation. 99:156–161.
MacLean MR. (1999) Pulmonary hypertension, anorerexigens and 5-HT: pharmacological synergism in action? TiPS. 20:490–5.
Launay D, et al. (2013) Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann. Rheum. Dis. 72:1940–6.
Elhai M, et al. (2014) A gender gap in primary and secondary heart dysfunctions in systemic sclerosis: a EUSTAR prospective study. Ann. Rheum. Dis. 2014, Oct 23. [Epub ahead of print].
Armstrong AC, et al. (2013) Pulmonary artery pressure, gender, menopause, and pregnancy in schistosomiasis-associated pulmonary hypertension. Arq. Bras. Cardiol. 101:154–9.
Almodovar S, Cicalini S, Petrosillo N, Flores SC. (2010) Pulmonary hypertension associated with HIV infection-pulmonary vascular disease: the global perspective. Chest. 137:6S–12S.
Mair KM, Johansen AKZ, Wright AF, Wallace E, MacLean MR. (2014) Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Brit. J. Pharmacol. 171:567–79.
Umar S, Rabinovitch M, Eghbali M. (2012) Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am. J. Respir. Crit. Care Med. 186:125–31.
Liu A, et al. (2014) Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 307:H273–83.
Frump AL, et al. (2015) Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell. Mol. Physiol. 308:L873–90.
MacLean MR, et al. (2004) Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation.109:2150–5.
White K, et al. (2011) The serotonin transporter, gender, and 17β estradiol in the development of pulmonary arterial hypertension. Cardiovasc. Res. 90:373–82.
Dempsie Y, et al. (20111) Development of pulmonary arterial hypertension in mice over-expresssing S100A4/Mts1 is specific to females. Respir. Res. 12:159.
Dempsie Y, et al. (2008) Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation. 117:2928–37.
West J, et al. (2013) A potential role for insulin resistance in experimental pulmonary hypertension. Eur. Respir. J. 41:861–71.
Fessel JP, et al. (2013) Interaction between bone morphogenetic protein receptor type 2 and estrogeneic compounds in pulmonary arterial hypertension. Pulm. Circ. 3:564–77.
Mair KM, et al. (2015) Sex affects BMPR-II signaling in pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 191:693–703.
Wright AF, et al. (2015) Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc. Res. 106:206–16.
Waxman DJ, Holloway MG. (2009) Sex differences in the expression of hepatic drug metabolizing enzymes. Mol. Pharmacol. 76:215–28.
Yang YM, et al. (2014) Deletion of STAT5a/b in vascular smooth muscle abrogates the male bias in hypoxic pulmonary hypertension in mice: implications in the human disease. Mol. Med. 20:625–38.
Tiell ML, Stemerman MB, Spaet TH. (1978) The influence of the pituitary on arterial intimal proliferation in the rat. Circ. Res. 42:644–9.
Khorsandi M, Fagin JA, Fishbein MC, Forrester JS, Cercek B. (1992) Effects of hypophysectomy on vascular insulin-like growth factor-I gene expression after balloon denudation in rats. Atherosclerosis. 93:115–22.
Kato R, Onoda K. (1970) Studies on the regulation of the activity of drug oxidation in rat liver microsomes by androgen and estrogen. Biochem. Pharmacol. 19:1649–60.
Colby HD, Gaskin JH, Kitay JI. (1973) Requirement of the pituitary gland for gonadal hormone effects on hepatic corticosteroid metabolism in rats and hamsters. Endocrinology. 92:769–74.
Kramer RE, Greiner JW, Rumbaugh RC, Sweeney TD, Colby HD. (1979) Requirement of the pituitary gland for gonadal hormone effects on hepatic drug metabolism in rats. J. Pharmacol. Exp. Ther. 208:19–23.
Rumbaugh RC, Colby HD. (1980) Is growth hormone the pituitary feminizing factor mediating the actions of estradiol on hepatic drug and steroid metabolism? Endocrinology. 107:719–24.
Sakuma T, et al. (2002) Regulation of the expression of two female-predominant CYP3A mRNAs (CYP3A1 and CYP3A44) in mouse liver by sex and growth hormone. Arch. Biochem. Biophys. 404:234–42.
Muller EE, Locatelli V, Cocchi D. (1999) Neuroendocrine control of growth hormone secretion. Physiol. Rev. 79:511–607.
Nishida Y, Yoshioka M, St. Amand J. (2005) Sexually dimorphic gene expression in the hypothalamus, pituitary gland, and cortex. Genomics. 85:679–87.
MacLeod JN, Pampori NA, Shapiro BH. (1991) Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J. Endocrinol. 131:395–9.
Low MJ, et al. (2001) Somatostatin is required for masculinization of growth hormone-regulated hepatic gene expression but not of somatic growth. J. Clin. Invest. 107:1571–80.
Coutant R, Lahlou N, Bouvattier C, Bougneres P. (1998) Circulating leptin level and growth hormone response to stimulation in obese and normal children. Eur. J. Endocrinol. 139:591–7.
Waxman DJ, Ram PA, Park SH, Choi HK. (1995) Intermittent plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein: proposed role as an intracellular regulator of male-specific liver gene transcription. J. Biol. Chem. 27:13262–70.
Gebert CA, Park SH, Waxman DJ. (1997) Regulation of signal transducer and activator of transcription (STAT) 5b activation by the temporal pattern of growth hormone stimulation. Mol. Endocrinol. 11:400–14.
Tannenbaum GS, Choi HK, Gurd W, Waxman DJ. (2001) Temporal relationship between the sexually dimorphic spontaneous GH secretory profiles and hepatic STAT5 activity. Endocrinology. 142:4599–606.
Udy GB, et al. (1997) Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. U. S. A. 94:7239–44.
Holloway MG, et al. (2007) Loss of sexually dimorphic liver gene expression upon hepatocytespecific deletion of Stat5a-Stat5b locus. Endocrinology. 148:1977–86.
Zhang Y, Laz EV, Waxman DJ. (2012) Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol. Cell. Biol. 32:880–96.
Meyer RD, Laz EV, Su T, Waxman DJ. (2009) Male-specific hepatic Bcl6: Growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol. Endocrinol. 23:1914–26.
Sugathan A, Waxman DJ. (2013) Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol. Cell. Biol. 33:3594–610.
Garcia-Segura LM, Baetens D, Naftolin F. (1986) Synaptic remodeling in arcuate nucleus after injection of estradiol valerate in adult female rats. Brain Res. 366:131–6.
Shirasu K, Stumpf WE, Sar M. (1990) Evidence for direct action of estradiol on growth hormone-releasing factor (GRF) in rat hypothalamus: localization of [3H]estradiol in GRF neurons. Endocrinology. 127:344–9.
Senaris RM, et al. (1992) Differential effects of in vivo estrogen administration on hypothalamic growth hormone releasing hormone and somatostatin gene expression. Neurosci. Lett. 141:123–6.
Desjardins GC, Brawer JR, Beaudet A. (1993) Estradiol is selectively neurotoxic to hypothalamic beta-endorphin neurons. Endocrinology. 132:86–93.
Brawer JR, Beaudet A, Desjardins, Schiffer HM. (1993) Pathologic effect of estradiol on the hypothalamus. Biol. Reprod. 49:647–52.
Kelly MJ, Ronnekleiv OK. (2015) Neural signaling of estradiol in the hypothalamus. Mol. Endocrinol. doi:10.1210/me.2014–1397.
Edén S. (1979) Age- and sex-related differences in episodic growth hormone secretion in the rat. Endocrinology. 105:555–60.
Winer LM, Shaw MA, Baumann G. (1990) Basal plasma growth hormone levels in man: new evidence for rhythmicity of growth hormone secretion. J. Clin. Endocrinol. Metab 70:1678–86.
MacLeod JN, Pampori NA, Shapiro BH. (1991) Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J. Endocrinol. 131:395–9.
Painson JC, Tannenbaum GS. (1991) Sexual dimorphism of stomatostatin and growth hormone-releasing factor signaling in the control of pulsatile growth hormone secretion in the rat. Endocrinology.128:2858–66.
van den Berg G, Veldhuis JD, Frolich M, Roelfsema F. (1996) An amplitude-specific divergence in the pulsatile mode of growth hormone (GH) secretion underlies the gender differences in mean GH concentrations in men and premenopausal women. J. Clin. Endocrinol. Metab 81:2460–7.
Pincus SM, et al. (1996) Females secrete growth hormone with more process irregularity than males in both humans and rats. Am. J. Physiol. 270:E107–15.
Engström BE, Karlsson FA, Wide L. (1998) Marked gender differences in ambulatory morning growth hormone values in young adults. Clin. Chem. 44:1289–95.
Dhir RN, Shapiro BH. (2003) Interpulse growth hormone secretion in the episodic plasma profile causes the sex reversal of cytochrome P450s in senescent mate rats. Proc. Natl. Acad. Sci. U. S. A. 100:15224–8.
Lee JE, et al. (2012) Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 302:C804–20.
Yang YM, Lane KB, Sehgal PB. (2013) Subcellular mechanisms in pulmonary arterial hypertension: combinatorial modalities that inhibit anterograde trafficking and cause BMPR2 mislocalization. Pulm. Circ. 3:533–50.
Waxman DJ, Ram PA, Pampori NA, Shapiro BH. (1995) Growth hormone regulation of male-specific rat liver P450s 2A2 and 3A2: induction by intermittent growth hormone pulses in male but not in female rats rendered growth hormone deficient by neonatal monosodium glutamate. Mol. Pharmacol. 48:790–7.
Thangavel C, Garcia MC, Shapiro BH. (2004) Intrinsic sex differences determine expression of growth hormone-regulated female cytochrome P450s. Mol. Cell. Endocrinol. 220:31–9.
Thangavel C, Dworakowski W, Shapiro BH. (2006) Inducibility of male-specific isoforms of cytochrome P450 by sex-dependent growth hormone profiles in hepatocyte cultures from male but not female rats. Drug Metab. Dispos. 34:410–9.
Martignoni M, Groothuis GMM, Kanter R. (2006) Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2:875–94.
Cheung C, et al. (2006) Growth hormone determines sexual dimorphism of hepatic cytochrome P450 3A4 expression in transgenic mice. J. Pharmacol. Exp. Ther. 316:1328–34.
Kasahara Y, et al. (1997) Bioactivation of monocrotaline by P-450 3A in rat liver. J. Cardiovasc. Pharmacol. 30:124–9.
Reid MJ, Lame MW, Morin D, Wilson DW, Segall HJ. (1998) Involvement of cytochrome P450 3A in the metabolism and covalent binding of 14C-monocrotaline in rat liver microsomes. J. Biochem. Mol. Toxicol. 12:157–66.
Subramanian A, Teixeira J, Wang J, Gil G. (1995) A STAT factor mediates the sexually dimorphic regulation of hepatic cytochrome P450 3A10/lithocholic acid 6 beta-hydroxylase gene expression by growth hormone. Mol. Cell. Biol. 15:4672–82.
Joung YH, et al. (2003) Hypoxia activates signal transducers and activators of transcription 5 (STAT5) and increases its binding activity to the GAS element in mammary epithelial cells. Exp. Mol. Med. 35:350–7.
Lee JY, et al. (2008) Loss of cytokine-STAT5 signaling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS One. 3:e1639.
Wada N, et al. (2014) Leptin and its receptors. J. Chem. Neuroanat. 61–62:191–9.
Peng CY, Mukhopadhyay A, Jarrett J, Yoshikawa K, Kessler JA. (2012) BMP receptor 1A regulates development of hypothalamic circuits critical for feeding behavior. J. Neurosci. 32:17211–24.
Townsend KL, et al. (2012) Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. 26:2187–96.
Magdaleno S, et al. (2006) BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 4:e86.
David L, et al. (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 102:914–22.
Long L, et al. (2015) Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 21:777–85.
Bidart M, et al. (2012) BMP9 is produced by hepatocytes and circulates mainly in an inactive mature form complexes to its protodomain. Cell. Mol. Life Sci. 69:313–24.
Herrera B, Dooley S, Brietfopf-Neinleion K. (2014) Potential roles of bone morphigenetic protein (BMP)-9 in human liver diseases. Int. J. Mol. Sci. 15:5199–220.
Lopez-Coviella I, et al. (2006) Developmental pattern of expression of BMP receptors and Smads and activation of Smad1 and Smad5 by BMP9 in mouse basal forebrain. Brain Res. 1088:49–56.
Huang E, et al. (2012) Growth hormone synergizes with BMP9 in osteogenic differentiation by activating the JAK/STAT/IGF1 pathway in murine multilineage cells. J. Bone Min. Res. 27:1566–75.
Isgaard J, Arcopinto M, Karason K, Cittadini A. (2015) GH and the cardiovascular system: an update on a topic at heart. Endocrine. 48:25–35.
Jin MH, et al. (2011) DNA microarray profiling identified a new role of growth hormone in vascular remodeling of rat ductus arteriosus. J. Physiol. Sci. doi:10.1007/s12576-011-0133–3.
Capaldo B, et al. (2001) Abnormal vascular reactivity in growth hormone deficiency. Circulation. 103:520–4.
Borson-Chazot F, et al. (1999) Decrease of carotid intima-media thickness after one year growth hormone (GH) treatment in adults with GH deficiency. J. Clin. Endocrinol. Metab. 84:1329–33.
Dimaraki EV, Jaffe CA. (2006) Role of endogenous ghrelin in growth hormone secretion, appetite regulation and metabolism. Rev. Endocr. Metab. Disord. 7:237–49.
Barkan AL, et al. (2003) Ghrelin secretion in humans is sexually dimorphic, suppressed by somatostatin, and not affected by the ambient growth hormone levels. J. Clin. Endocrinol. Metab. 88:2180–4.
Yang D, Liu Z, Yang Z. (2013) Ghrelin and its relation with N-terminal brain natriuretic peptide, endothelin-1 and nitric oxide in patients with idiopathic pulmonary hypertension. Cardiology. 124:241–5.
Henriques-Coelho T, et al. (2004) Endogenous production of ghrelin and beneficial effects of its exogenous administration in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 287:H2885–90.
Xu YP, et al. (2011) Ghrelin ameliorates hypoxia-induced pulmonary hypertension via phosphor-GSK3β/β-catenin signaling in neonatal rats. J. Mol. Endocrinol. 47:33–43.
Roskovski R Jr. (2007) Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 356:323–8.
Yee KW, et al. (2002) SU5416 and SU5614 inhibit kinase activity of wild-type and mutant FLT3 receptor tyrosine kinase. Blood. 100:2941–9.
Mezrich JD, et al. (2012) SU5416, a VEGF receptor inhibitor and ligand of the AHR represents a new alternative for immunomodulation. PLoS One. 7:e44547.
Cui W, et al. (2012) Unexpected neuronal protection of SY5416 against 1-methyl-4-phenylpyridinium ion-induced toxicity via inhibiting neuronal nitric oxide synthase. PLoS One. 7:e46253.
Nam S, et al. (2007) Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol. Cancer Ther. 6:1400–5.
Montani D, et al. (2012) Pulmonary arterial hypertension in patients treated by dasatanib. Circulation. 125:2128–37.
Launay J-M, et al. (2002) Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat. Med. 8:1129–35.
Tao R, et al. (2002) Effects on serotonin in rat hypothalamus of D-fenfluramine, aminorex, phentermine and fluoxetine. Eur. J. Pharmacol. 445:69–81.
Prow MR, Lancashire B, Aspley S, Heal DJ, Kilpatrick IC. (2001) Additive effects on rat brain 5HT release of combining phentermine with dexfenfluramine. Int. J. Obes. Relat. Metab. Disord. 25:1450–3.
Jia Y, El-Haddad M, Gendy A, Nguyen T, Ross MG. (2010) Serotonin-induced region-specific responses of the arcuate nucleus and ventromedial hypothalamic nuclei. Int. J. Neurosci. 120:386–95.
Voigt JP, Fink H. (2015) Serotonin controlling feeding and satiety. Behave Brain Res. 277:14–31.
Argenio GF, et al. (1991) Blunted growth hormone (GH) responsiveness to GH-releasing hormone in obese patients: influence of prolonged administration of serotoninergic drug fenfluramine. Metabolism. 40:724–7.
Litwak SA, et al. (2014) Estradiol prevents fat accumulation and overcomes leptin resistance in female high-fat diet mice. Endocrinology. 155:4447–60.
Wilson ME, Moore CJ, Ethun KF, Johnson ZP. (2014) Understanding the control of ingestive behavior in primates. Horm. Behav. 66:86–94.
Chiba T, et al. (2014) Serotonin suppresses β-casein expression via inhibition of the signal transducer and activator of transcription 5 (STAT5) protein phosphorylation in human mammary epithelial cells MCF-12A. Biol. Pharm. Bull. 37:1336–40.
Lieberman LA, Higgins DE. (2009) A small-molecule screen identifies the antipsychotic drug pimozide as an inhibitor of Listeria monocytogenes infection. Antimicrob. Agents Chemother. 53:756–64.
Nelson EA, et al. (2011) The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 117:3421–9.
Nebigil CG, et al. (2000) Serotonin 2B receptor is required for heart development. Proc. Natl. Acad. Sci. U. S. A. 97:9508–9513.
Sehgal PB, Levy DE, Hirano T, eds. (2003) Signal transducers and activators of transcription (STATs): activation and biology. Dordrecht: Kluwer Academic. 746 pp.
Crosby A, et al. (2010) Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am. J. Respir. Crit. Care Med. 181:279–288.
Mauad T, et al. (2014) Immunopathological aspects of schsitosomiasis-associated pulmonary arterial hypertension. J Infect. 68:90–98.
Pericle F, et al. (1998) HIV-1 infection induces a selective reduction in STAT5 protein expression. J. Immunol. 160:28–31.
Prost S, et al. (2008) Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARγ/STAT5 signaling pathway in macaques. J. Clin. Invest. 118:1765–1775.
Sehgal PB, et al. (2009) Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am. J. Physiol. Lung Cell. Mol. Physiol. 297:L729–37.
Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. (1997) Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 276:589–92.
Toney LM, et al. (2000) BCL-6 regulates chemokine gene transcription in macrophages. Nat. Immunol. 1:214–20.
Liao W, et al. (2014) Opposing actions of IL-2 and IL-21 on Th9 differentiation correlates with their differential regulation of BCL6 expression. Proc. Natl. Acad. Sci. U. S. A. 111:3508–13.
Burke DL, et al. (2009) Sustained hypoxia promotes the development of a pulmonary artery-specific inflammatory microenvironment. Am. J. Physiol. Lung Cell. Mol. Physiol. 297:L238–50.
Soon E, et al. (2010) Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 122:920–7.
Daley E, et al. (2008) Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 205:361–72.
Hu X, et al. (2013) Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. U. S. A. 110:10213–8.
Peck AR, et al. (2012) Low levels of Stat5a protein in breast cancer are associated with tumor progression and unfavorable clinical outcomes. Breast Cancer Res. 14:R130.
Santos SJ, Haslam SZ, Conrad SE. (2008) Estrogen and progesterone are critical regulators of Stat5a expression in the mouse mammary gland. Endocrinology. 149:329–38.
Frantz AG, Rabkin MT. (1965) Effects of estrogen and sex difference on secretion of human growth hormone. J. Clin. Endocr. 25:1470–80.
Savai R, et al. (2014) Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat. Med. 20:1289–300.
Arumugam R, et al. (2008) The interplay of prolactin and the glucocorticoids in the regulation of β-cell gene expression, fatty acid oxidation, and glucose-stimulated insulin secretion: implications for carbohydrate metabolism in pregnancy. Endocrinology. 149:5401–14.
Ono M, et al. (2007) Signal transducer and activator of transcription (Stat) 5b-mediated inhibition of insulin-like growth factor binding protein-1 gene transcription: a mechanism for repression of gene expression by growth hormone. Mol. Endocrinol. 21:1433–57.
Stittrich AB, et al. (2010) The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat. Immunol. 11:1057–62.
Hansmann G, et al. (2007) Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-γ activation. Circulation. 115:1275–84.
Zamanian RT, et al. (2009) Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 33:318–24.
Pugh ME, et al. (2011) Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J. Heart Lung Transplant. 30:904–11.
West J, et al. (2013) A potential role for insulin resistance in experimental pulmonary hypertension. Eur. Respir. J. 41:861–71.
Naderi N, et al. (2014) Insulin resistance in pulmonary arterial hypertension: is it a novel disease modifier? Res. Cardiovasc. Med. 3:e19710.
Chen X, et al. (2015) The estrogen metabolite 16αOHE exacerbates BMPR2-associated PAH through miR-29-mediated modulation of cellular metabolism. Circulation. 2015, Oct 20 [Epub ahead of print].
Roth J, Glick SM, Yalow RS, Berson SA. (1963) Hypoglycemia: a potent stimulus to secretion of growth hormone. Science. 140:987–8.
Hoffman DM, O’Sullivan AJ, Ho KKY, Baxter RC. (1994) Diagnosis of growth hormone deficiency in adults. Lancet. 343:1064–8.
Jorgensen JO, et al. (2004) Growth hormone and glucose homeostasis. Horm. Res. 62 Suppl 3:51–5.
Ho KKY, et al. (2007) Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur. J. Endocrinol. 157:695–700.
Popovic V. (2013) Approach to testing growth hormone (GH) secretion in obese subjects. J. Clin. Endocrinol. Metab. 98:1789–96.
Acknowledgments
We thank Jana Velíšková for insightful neuroscience discussions. This work was supported, in part, by National Heart, Lung, and Blood Institute Grants HL-087176 (to PB Sehgal), HL-114509 (to PB Sehgal) and HL-111469 (to EJ Miller). We also thank the Pulmonary Hypertension Breakthrough Initiative (PHBI) for providing sections of human lung tissue studied in ref. (28) (adapted data illustrated, in part, in the present Figure 7); funding for PHBI was provided by the Cardiovascular Medical Research and Education Fund (CMREF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Sehgal, P.B., Yang, YM. & Miller, E.J. Hypothesis: Neuroendocrine Mechanisms (Hypothalamus-Growth Hormone-STAT5 Axis) Contribute to Sex Bias in Pulmonary Hypertension. Mol Med 21, 688–701 (2015). https://doi.org/10.2119/molmed.2015.00122
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2015.00122