
Abstract
Toll-like receptors (TLRs) play an important role in regulating muscle regeneration and angiogenesis in response to ischemia. TLR2 knockout mice exhibit pronounced skeletal muscle necrosis and abnormal vessel architecture after femoral artery ligation, suggesting that TLR2 signaling is protective during ischemia. TLR4, an important receptor in inflammatory signaling, has been shown to regulate TLR2 expression in other systems. We hypothesize that a similar relationship between TLR4 and TLR2 may exist in hindlimb ischemia in which TLR4 upregulates TLR2, a mediator of angiogenesis and perfusion recovery. We examined the expression of TLR2 in unstimulated and in TLR-agonist treated endothelial cells (ECs). TLR2 expression (low in control ECs) was upregulated by lipopolysaccharide, the danger signal high mobility group box-1, and hypoxia in a TLR4-dependent manner. Endothelial tube formation on Matrigel as well as EC permeability was assessed as in vitro measures of angiogenesis. Time-lapse imaging demonstrated that ECs lacking TLR4 formed more tubes, whereas TLR2 knockdown ECs exhibited attenuated tube formation. TLR2 also mediated EC permeability, an initial step during angiogenesis, in response to high-mobility group box-1 (HMGB1) that is released by cells during hypoxic injury. In vivo, ischemia-induced upregulation of TLR2 required intact TLR4 signaling that mediated systemic inflammation, as measured by local and systemic IL-6 levels. Similar to our in vitro findings, vascular density and limb perfusion were both enhanced in the absence of TLR4 signaling, but not if TLR2 was deleted. These findings indicate that TLR2, in the absence of TLR4, improves angiogenesis and perfusion recovery in response to ischemia.
Similar content being viewed by others


Introduction
Peripheral arterial occlusive disease affects 4–5% of the population over age 40 years in the United States and can result in limb loss in its advanced and severe stages (1). Considerable research has focused on the mechanisms by which angiogenesis is induced in the setting of ischemia and how they can be enhanced to promote limb salvage (2–6). Whereas most have focused on the identification and testing of proangiogenic compounds or cellular therapies, there has been much less emphasis on evaluating the role of modulating inflammation to improve tissue recovery from the ischemic insult. We and others have investigated the role of toll-like receptor (TLR) activation in promoting angiogenesis and muscle recovery in a murine model of hindlimb ischemia (7–14). TLRs mediate innate immune responses to bacterial components, including gram-negative bacterial lipopolysaccharide (LPS) (TLR4) and gram-positive bacterial lipoproteins (TLR2) (15–18). TLRs also sense endogenously released damage-associated molecular patterns (DAMPS), such as high-mobility group box-1 (HMGB1) to initiate an immune response (19). We have previously demonstrated that HMGB1, TLR4 and TLR2 modulate inflammation and muscle recovery after ischemia (8–10). TLR2 knockout (TLR2KO) mice develop abnormal vasculature, possibly influencing the regenerative process, whereas TLR4KO mice exhibit a robust regenerative phenotype. These studies independently suggest that TLR4 and TLR2 mediate different responses to skeletal muscle ischemia. However, the way in which these receptors interact with one another in this process is not known.
Cross-talk between TLR4 and TLR2 has been reported in other models and appears to exacerbate inflammation of lung endothelial cells (ECs) during shock (20–22). Whether TLR2 is upregulated in a TLR4-dependent manner during limb ischemia has not been investigated. Our previous studies would suggest that TLR2 is necessary for regenerative responses and may attenuate TLR4-mediated inflammation, both encouraging recovery. In this study, we hypothesize that TLR4 activation induces systemic inflammation and upregulates TLR2 to promote angiogenesis in response to limb ischemia. To test this hypothesis, we examined the effect of TLR4 activation on the expression of TLR2 and TLR4 in ECs. By using siRNA to block TLR2 or TLR4 expression, we evaluated TLR2 levels as a function of TLR4 and assessed the role of each receptor in endothelial tube formation and cell permeability as in vitro measures of angiogenesis. In vivo, we determined the dependence of TLR2 expression on TLR4 function in hindlimb ischemia and the impact of this relationship on local and systemic inflammation and perfusion recovery. We show that TLR2 upregulation in ischemic muscle depends on TLR4 and that TLR2 is required for adequate angiogenesis following muscle ischemia.
Materials and Methods
Reagents
LPS and synthetic triacylated lipoprotein Pam3CysSerLys4 (Pam3CSK4) were obtained from Sigma-Aldrich. Recombinant HMGB1 was isolated from yeast as described (23). HMGB1 formulation buffer (25 mmol/L Tris chloride, pH 8, 150 mmol/L KCl, 2 mmol/L dithiothreitol, 10% glycerol) was used as a control for HMGB1 administration. HMGB1-neutralizing antibody (2g7; a gift from Kevin Tracey, The Feinstein Institute for Medical Research, Manhasset, NY) was developed in rabbit and prepared as described (24). The doses used in these experiments have been reported to attenuate murine sepsis (20 µg/mL, final concentration) (25). Rabbit polyclonal IgG served as the negative control (Sigma-Aldrich). Growth factor-reduced Matrigel (BD Biosciences) was stored at 4°C and allowed to solidify for 30 min at 37°C before use in endothelial tube formation experiments. Antibodies were obtained from Abcam (TLR2, TLR4) and Cell Signaling (β-actin). siRNA SMARTpool duplexes were obtained from Thermo Fisher Scientific Dharmacon. Custom polymerase chain reaction (PCR) primers and Lipofectamine RNAiMAX Reagent were purchased from Invitrogen (Life Technologies), and conditions for transfection were optimized following product instructions. The following reagents were used following manufacturer’s instructions: Pierce BCA protein assay (ThermoScientific), RNA STAT-60 reagent (Amsbio), iScript™ Reverse Transcription Supermix (Bio-Rad), GoTaq® Green Master Mix (Promega) and IL-6 enzyme-linked immunosorbent assay (ELISA) (R&D Systems).
Cell Culture
Human dermal microvascular endothelial cells (HDMVECs) (VEC Technologies) were maintained on gelatin-coated dishes in a 1:1 mix of MCDB131 complete media (VEC Technologies) and Dulbecco modified Eagle medium (DMEM) with 5% fetal bovine serum (FBS), penicillin-streptomycin (P/S) and L-glutamine. Cells were used between passages 3–12. When hypoxic conditions were required, cells were placed into a hypoxia chamber (1% O2, 95% nitrogen and 5% CO2; COY Laboratory Products).
TLR2 and TLR4 Suppression Using Short Inhibitory RNA (siRNA)
HDMVECs were transfected for 48 h with SMARTpool siRNA duplexes against TLR2, TLR4 and GAPDH, as well as with nonspecific duplexes in the presence of lipofectamine. Cell lysates were analyzed for the expression of TLR2, TLR4 and GAPDH by PCR to quantify the success of silencing (26).
Western Blot
Cells were lysed in phenylmethanesulfonylfluoride (PMSF), and protein was quantified with BCA assay. Proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, blocked with 1% bovine serum albumin and probed with antibody to TLR2, TLR4 or β-actin (Abcam).
Endothelial Cell Activation and Expression of TLRs
To assess TLR2 expression, HDMVECs were treated with LPS (1 µg/mL) or HMGB1 (1 µg/mL) or were cultured under hypoxic conditions in the absence or presence of 2g7 (20 µg/mL) for 6 h in serum-free DMEM. Isolation of total RNA, reverse transcription and amplification was performed as described above to assess levels of TLR2 and GAPDH mRNA. Western blot for TLR2 was performed on cell lysates.
Tube Formation Assay
HDMVECs were transfected with siRNA to nonspecific target (negative siRNA), TLR2 or TLR4. After transfection, cells were seeded onto 24-well plates coated with Matrigel. Tube formation was captured digitally after 6 h of incubation in normoxic or hypoxic conditions. The number of tubes in five non-overlapping images was counted. In other experiments, HDMVECs were transfected with siRNA and then plated into the bottom well of an angioslide (ibidi GmbH). The slides were mounted in a temperature and CO2-controlled microincubator (Zeiss) on an inverted Olympus IX81 microscope with motorized stage and CCD camera (Q-Imaging, Retiga EXi). Differential interference contrast (DIC) images were collected every 15 min for 360 min using a 10× objective. Images were compiled into movies by using MetaMorph (Molecular Devices). Tube number was calculated from still images obtained at 30-min intervals. Experiments were performed a minimum of three times in transfected and untransfected cells.
Permeability Assays
The vascular permeability assay kit (96-well; Trevigen) was used according to the manufacturer’s instructions to test EC permeability in response to TLR2 and TLR4 agonists. In some experiments, HDMVECs were transfected with siRNA to a nonspecific target, TLR2 or TLR4 24 h before plating. After 48 h of incubation, the cells were stimulated with LPS (1 µg/mL), HMGB1 (1 µg/mL) or PAM3CSK4 (1 µg/mL), and permeability to fluorescein isothiocyanate (FITC) dextran (1:40) at 20 min at room temperature was assayed.
Animals
Male TLR2KO, TLR4KO, TLR2/4 double KO (TLR2/4KO) and control C57B6 mice, fed a standard diet and housed in groups of four, were used at 10–12 wks of age (~30 g). TLR2KO mice were a generous gift from Jay Kolls (Children’s Hospital of Pittsburgh). Control C57B6 mice were obtained from The Jackson Laboratory and were age-and weight-matched. The other strains are bred within the Department of Surgery at the University of Pittsburgh. All procedures conformed to the Guide for the Care and Use of Laboratory Animals adopted by the U.S. National Institutes of Health (8th edition; available at https://doi.org/grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf) and the policies of the Institutional Animal Use and Care Committee of the University of Pittsburgh (approved, protocol #13021224).
Hindlimb Ischemia Model
Mice were anesthetized with pentobarbital (0.1 mL/g intraperitoneally). After hair removal and betadine preparation, transverse incisions were made in each groin, and the femoral structures were isolated. On the right, the external iliac and femoral veins and arteries and all visible branches were ligated with 6-0 silk suture (27,28). Care was taken to preserve the femoral nerve. On the left, the femoral vessels were exposed but not ligated. Animals were kept warm with a heating lamp. Mice were euthanized by an overdose of inhaled isoflurane followed by cervical dislocation. PCR and immunohistochemical analysis were performed on tibialis anterior muscle samples from the right (ischemic) and left (nonischemic) hindlimbs. The time points of harvest were 1 h, 6 h, 1 d, 3 d and 14 d after femoral artery ligation (FAL).
Laser Doppler Perfusion Imaging
Animals were anesthetized with inhaled isoflurane for the duration of the procedure. After preparing each hindlimb with depilatory cream, the blood flow to both hindlimbs was measured using a Laser Doppler blood-flow meter (PERIMED III, Stockholm, Sweden). Three sequential images of the entire hindlimb were obtained and averaged. Perfusion was expressed as a ratio of the ischemic to nonischemic limb at each time point. Laser Doppler perfusion imaging was performed before and 1, 7 and 14 d after FAL.
RT-PCR Analysis
Total RNA was isolated by using RNA STAT-60 reagent following the manufacturer’s instructions. Total RNA was then reverse-transcribed to cDNA by using iScript™ Reverse Transcription Supermix. Amplification reactions for target genes used GoTaq® Green Master Mix (Promega). Primers for amplification were as follows: mouse IL-6, forward, 5′-TCCAGTTGCCTTCTTGGGACTGAT-3′, reverse, 5′-TTGGATGGTCTTGGTCCTTAGCCA-3′; mouse TLR2, forward, 5′-GTTCTCCCAGCATTTAAAATCATT-3′, reverse, 5′-GTCTCCAGTTTGGGAAAAAGAACC-3′; mouse TLR4 forward, 5′-AGTGGGTCAAGGAACAGAAGCA-3′, reverse, 5′-CTTTACCAGCTCATTTCTCACC-3′; GAPDH, forward, 5′-AACCTGCCAAGTATGATGAC-3′, reverse, 5′-ATACCAGGAAATGAGCTTGA-3′; human TLR2, forward, 5′-CCAGCACACGAATACACAGT-3′, reverse, 5′-CAAATGAAGTTATTGCCACC-3′; and human TLR4 forward, 5′-TCCCTCCAGGTTCTTGATTACAGTC-3′, reverse, 5′-TGCTCAGAAACTGCCAGGTCTG-3′. PCR products were separated on 1% agarose gel and identified by ethidium bromide staining. Relative reversetranscriptase (RT)-PCR quantification was performed by color inversion of the exposed gel followed by band analysis by using ImageJ software. The target gene signal was expressed relative to GAPDH signal.
Immunohistochemistry
At 1 h, 6 h, 1 d, 3 d and 14 d after is-chemic injury, tibialis anterior muscle from both ischemic and nonischemic limbs were harvested for immunohisto-chemical analysis. Tissues were fixed, embedded in paraffin and serially sectioned in 8-µm sections, and three sections were obtained per animal. Paraffin embedding has assisted in the maintenance of muscle architecture during sectioning. After deparaffinization, sections were stained with FITC isolectin to detect endothelium and DAPI to elucidate nuclei. TLR2 and TLR4 were identified by using anti-TLR2 and -TLR4 antibodies (Biolegend) and Cy-3-conjugated secondary antibody. Images were obtained with the 60× objective of an Olympus microscope equipped with a Nikon camera at the Centers of Biologic Imaging at the University of Pittsburgh. Four images per section were digitally acquired for a total of 12 images per animal. Tibialis anterior muscle harvested 3 d after FAL was also evaluated by using the standard hematoxylin and eosin (H&E) stain to assess overall architecture as well as polymorphonuclear (PMN) leukocyte recruitment on the basis of nuclear morphology. Muscles harvested 14 d after FAL were additionally stained for CD31 by using horseradish peroxidase-conjugated secondary antibody to assess capillary density.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM) of at least three experiments. Analysis of variance was used to compare multiple means, whereas t test was used to evaluate differences between two means.
Results
TLR4 Activation Upregulates TLR2 in ECs
TLR4 has been reported to regulate TLR2 expression in pulmonary ECs. Thus, we tested the role of TLR4 in mediating TLR2 expression in cultured HD-MVECs. TLR4 activation was achieved with LPS and HMGB1. At baseline, TLR2 mRNA and protein levels were low in HDMVECs. However, both increased after the cells were treated with LPS, HMGB1 and hypoxia (Figure 1A). The upregulation of TLR2 by hypoxia was attenuated by treatment with TLR4 siRNA and suggests that TLR4 signaling mediated this effect of hypoxia (Figure 1A). TLR2 and TLR4 knockdown was successfully achieved in the ECs by using siRNA to each receptor (Figure 1B). LPS, HMGB1 and hypoxia all failed to increase TLR2 expression in the TLR4KD cells (Figures 1C, D).

Activation of TLR4 upregulates TLR2 on microvascular endothelial cells. HDMVECs were activated for 6 h with LPS (1 µg/mL), HMGB1 (1 µg/mL) and hypoxia (1% O2) with or without 20 µg/mL anti-HMGB1 antibody. (A) RT-PCR and Western blot were used to test the level of TLR2. Data are shown as a ratio to GAPDH (PCR) and β-actin (Western blot) and are expressed as a fold-increase when compared with unstimulated cells. *p < 0.001, hypoxia versus no stimulation, LPS versus no stimulation, LPS versus anti-HMGB1 antibody, hypoxia alone versus hypoxia plus anti-HMGB1 antibody; **p < 0.01, LPS versus hypoxia plus anti-HMGB1 antibody, hypoxia versus hypoxia plus anti-HMGB1 antibody, HMGB1 versus no stimulation, HMGB1 versus anti-HMGB1 antibody, HMGB1 versus hypoxia plus anti-HMGB1 antibody; N = 3 experiments, ANOVA, SEM expressed. (B) Western blots for TLR2 and TLR4 demonstrate knockdown efficiency 48 h after transfection of HDMVECs with siRNA to a nonspecific target (negative siRNA), TLR2 and TLR4. (C, D) HDMVECs were stimulated with LPS, HMGB1 or hypoxia (1% O2) for 6 h with and without treatment with siRNA to TLR4 to abolish TLR4-mediated signaling. RT-PCR (C) or Western blot (D) evaluated TLR2 expression as a fraction of GAPDH or β-actin, respectively. (C) *p < 0.03, hypoxia to no stimulation, wild-type (WT) to TLR4KD cells in hypoxia; **p < 0.01, WT to TLR4KD cells treated with HMGB1; ***p< 0.06, LPS to no stimulation in WT cells, HMGB1 to no stimulation in WT cells, WT to TLR4KD cells treated with LPS. (D) *p < 0.01, LPS versus no stimulation, HMGB1 versus no stimulation; **p < 0.01, hypoxia versus no stimulation, WT versus TLR4KD cells treated with LPS, WT versus TLR4KD cells treated with HMGB1; ***p < 0.08, N = 3 experiments.
TLR2 Is Required for EC Angiogenic Activity In Vitro
ECs manifest angiogenic activity in vitro by forming tube-like structures when cultured in Matrigel. ECs treated with negative siRNA or siRNA targeted against TLR2 or TLR4 were seeded on Matrigel, and endothelial tube formation was assessed after 6 h. In parallel experiments, time lapse imaging was used to capture tube formation at 15-min intervals for 360 min and quantified in 30-min segments. Knockdown of TLR2 reduced EC tube formation compared with control cells and cells treated with negative siRNA or TLR4 siRNA. Similar effects were observed under normoxic and hypoxic conditions (Figures 2A, B). In other experiments, HDMVECs were treated with HMGB1 under normoxic and hypoxic conditions. ECs treated with negative siRNA or TLR2 siRNA were also exposed to HMGB1. HMGB1 significantly increased tube formation in all treatment groups except the TLR2KD cells (Figure 2C), suggesting that TLR2 signaling is required for HMGB1-induced angiogenesis. Under normoxic conditions, time-lapse imaging revealed that TLR2KD cells formed significantly fewer tubes than TLR4KD cells at 300–360 min, whereas cells treated with a nonspecific siRNA formed intermediate numbers of tubes (Table 1).

Endothelial tube formation is attenuated in TLR2KD endothelial cells. (A) HDMVECs were transfected with siRNA to a nonspecific target, TLR2 or TLR4. Nontransfected cells were also evaluated. After transfection, cells were seeded onto growth factor-reduced Matrigel. Tube formation experiments were performed in normoxia (A) or hypoxia (B) (1% O2). Six hours after incubation, tubes were imaged by microscopy and counted at 20×. *p < 0.05, TLR2KD cells versus other groups; **p < 0.03, TLR2KD cells versus other groups, N = 3; ANOVA. Scale bar = 100 µm. (C) Tube formation experiments were also performed in the presence and absence of HMGB1, in both hypoxic and normoxic conditions, and in the presence of competent or incompetent TLR2. TLR2KD was achieved by using siRNA as described. HMGB1 increased tube formation in normoxia, hypoxia and the presence of negative siRNA. Similar in previous experiments, knockdown of TLR2 attenuated tube formation, which was not increased by addition of HMGB1. *p < 0.001, normoxic cells treated with and without HMGB1; **p < 0.05, hypoxic cells treated with and without HMGB1; ***p < 0.001, cells pretreated with nonspecific, negative siRNA with and without HMGB1.
Another measure of EC angiogenic activity is the development of vascular permeability that allows ECs to separate from one another, divide and migrate to form new vessels (29). To assess the role of TLR2 and TLR4 on this function, we measured EC permeability in vitro in response to specific TLR agonists with and without TLR knockdown. HDMVECs treated with LPS, HMGB1 or Pam3CSK4, a TLR2 agonist, all exhibited increased permeability as measured by the diffusion of FITC dextran through EC monolayers (Figure 3). Cells treated with TLR2 or TLR4 siRNA developed less permeability in response to Pam3CSK4 and LPS, respectively. HMGB1 mediated permeability was diminished in TLR2KD but not TLR4KD EC monolayers, suggesting that TLR2 is still present and functional in the setting of TLR4KO and mediates EC permeability to HMGB1.

TLR2 mediates endothelial cell permeability to HMGB1. HDMVECs were transfected with siRNA to a nonspecific target, TLR2 or TLR4. Twenty-four hours after transfection, cells were seeded into trans-wells at 105 cells per well to form a monolayer. Cells were stimulated with LPS, HMGB1 or Pam3CSK4 (1 µg/mL final concentration). Six hours after stimulation, FITC dextran was added to the trans-well to assess its transport through the cell monolayer. Medium was collected from the bottom well, and the level of fluorescence was measured. (A) LPS, Pam3CSK4 and HMGB1 all resulted in increased permeability through the transwells (*p < 0.003; **p < 0.03). Increased permeability to LPS (B) (*p < 0.005 compared with control) and Pam3CSK4 (C) (*p < 0.001 compared with control) was attenuated with treatment of cells with siRNA to TLR4 (B) and TLR2 (C), respectively. Increased permeability to HMGB1 was attenuated by treatment of cells with siRNA to TLR2 (D), but not with siRNA to TLR4 (**p < 0.05 HMGB1 versus control buffer) or nonspecific target (*p < 0.01 HMGB1 versus control buffer). N = 3 for each condition.
TLR4 Mediates IL-6 and TLR2 Expression after FAL
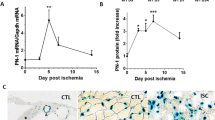
We characterized the temporal expression of TLR2 and TLR4 in ischemic and nonischemic skeletal muscle after FAL. ECs were stained with isolectin to assist in antigen localization within the blood vessel wall. In addition to using the contralateral nonischemic limb as a control, a group of C57B6 mice that did not undergo any surgery was also used for comparison. TLR4 was upregulated by 6 h after FAL and persisted to at least 3 d. TLR2 expression was also upregulated in the ischemic limb over the same time period (Figures 4A, B). By immunostaining, TLR4 expression was increased in ECs within the ischemic muscle by 6 h and looked to be returning toward baseline by d 3 (Figure 4C). TLR2 expression also localized to the ECs as well as in circulating cells within blood vessel lumens in a similar temporal pattern (Figure 4D).

TLR2 and TLR4 are upregulated in ischemic skeletal muscle. (A, B) RT-PCR was performed to evaluate expression of TLR2 and TLR4 mRNA in both hindlimbs at multiple time points after FAL. Results are expressed as a ratio to GAPDH mRNA expression. Representative images are shown after color inversion of the original gel. (B) N = 4–6 animal muscle samples/time point, error bars represent SEM, *p < 0.01, **p < 0.03. Representative images of TLR4 (C) and TLR2 (D) staining in anterior tibialis muscles at the designated time points after FAL. Images were obtained with a 60× objective attached to an Olympus Provis I microscope and Nikon camera. TLR2 and TLR4 were stained using immunohistochemical techniques with a Cy-3-conjugated secondary antibody (red). FITC isolectin was used to identify vascular structures (green), whereas DAPI staining identified nuclei. Scale bar = 100 µm. Magnified view represents 60× magnification of original image.
In our in vitro studies, TLR4 appeared to regulate TLR2 expression. To determine if this relationship also exists in vivo, TLR2 mRNA expression in ischemic muscle was examined by RT-PCR in control and TLR4KO mice. In control mice, ischemia significantly upregulated TLR2 mRNA 24 h after FAL. In TLR4KO, however, TLR2 expression did not change in response to ischemia (Figure 5A). To evaluate whether an inflammatory state had been induced in the ischemic muscle after ligation, IL-6 levels were examined. IL-6 mRNA was undetectable in nonsurgical C57B6 and TLR4KO mice and in the non-ischemic contralateral limbs from mice undergoing FAL. IL-6 was significantly elevated in ischemic skeletal muscle 24 h after FAL in C57B6 mice but remained low in TLR4KO mice (Figure 5A).

Competent TLR4 mediates upregulation of TLR2 and expression of IL-6 in ischemic skeletal muscle. (A) Anterior tibialis (AT) muscle from uninjured control C57B6 and TLR4KO mice, as well as those that underwent FAL were subjected to PCR for TLR2, IL-6 and GAPDH. Representative blots are shown. *p < 0.01, N = 3–6/group, ANOVA. (B) Serum was obtained 6 h after the initiation of limb ischemia with FAL in control C57B6, TLR2/4KO, TLR2KO TLR4KO mice and C57B6 mice that did not undergo surgery. ELISA for IL-6 was performed on all samples. *p < 0.03, TLR4KO to TLR2KO mice; 3–5 animals/group, ANOVA. (C) PMNs were identified on H and E sections by using characteristic nuclear morphology as described (9). Percent PMN nuclei relative to total nuclei is shown from tibialis anterior muscle obtained 3 d after FAL in control, TLR2/4 double KO, TLR2KO and TLR4KO mice (p value nonsignificant among the groups).
Because both TLR4 and TLR2 expression increased in ischemia and both can mediate inflammatory responses, we evaluated their individual and combined roles in promoting systemic inflammation after limb ischemia. We previously reported that serum IL-6 levels peak 6 h after FAL and correlate inversely with myofiber size (30). In current experiments, IL-6 was undetectable in nonsurgical mice. Serum IL-6 levels increased at 6 h in all mice undergoing FAL, with the highest levels detected in the TLR2KO mice and lowest in TLR4KO mice (Figure 5B). These results suggest that TLR4 activation in response to muscle ischemia mediates inflammatory cytokine expression as well as TLR2 upregulation. Three days after arterial ligation, neutrophil infiltration was equal among all treatment groups (Figure 5C), suggesting that TLR function did not affect inflammatory cell recruitment at that time point.
TLR4KO Mice Demonstrate Accelerated Recovery of Perfusion and Increased Vascular Density after FAL
To determine the effect of TLRs on the recovery of limb perfusion after ischemia, laser Doppler perfusion imaging measurements were obtained before and 1, 7 and 14 d after FAL. At d 1 after FAL, all mice developed significant ischemia in the right hindlimb. Only TLR4KO mice exhibited significant recovery of perfusion by d 7 (Figures 6A, C). Histologically, ischemic hindlimb muscle from TLR4KO mice had significantly higher vascular density, as indicated by CD31-positive structures, than WT, TLR2KO or TLR2/4KO mice 2 wks after FAL (Figures 6B, D).

Loss of TLR4 signaling accelerates perfusion recovery after hindlimb ischemia, but not in the absence of TLR2. (A, C) Laser Doppler perfusion imaging was performed before the initiation of limb ischemia with FAL and 1, 7 and 14 d after arterial ligation in control C57B6, TLR2/4 double KO, TLR2KO and TLR4KO mice. No reperfusion was performed after the arterial ligation. Perfusion in the ischemic limb was compared with the nonischemic limb at each time point and analyzed using a t test. Comparisons among the group were evaluated with ANOVA. *p < 0.003, before ischemia versus 1 d after ischemia, t test; **p = 0.05, 7 d after ischemia versus 1 d after ischemia in TLR4KO mice t test; ***p = 0.02, 14 d after ischemia in TLR4KO versus TLR2KO and TLR2/4KO mice; ANOVA, 4–8 animals per group. (B, D) The number of CD31-positive vascular structures per high-power field was quantified in anterior tibialis muscle harvested 14 d after femoral arterial ligation. TLR4KO mice demonstrated the highest vascular density among the groups, *p < 0.05, ANOVA.
Discussion
We previously demonstrated that TLR2 and TLR4 mediate key tissue responses to skeletal muscle ischemia. These receptors are known to be expressed within skeletal muscle, where they mediate inflammation in response to LPS (31), play a role in insulin resistance (32) and mediate fatty acid signaling during exercise (33). We have reported that the absence of TLR2 signaling in the setting of ischemia leads to massive muscle necrosis and impaired angiogenesis, whereas the absence of TLR4 promotes regenerative responses. Mice competent in both TLR2 and TLR4 pathways have an intermediate response. Recent studies in models of lung injury show that TLR4 can regulate TLR2 expression (20). On the basis of these prior reports, we examined the interplay between TLR4 and TLR2 in the setting of skeletal muscle ischemia. We show here that TLR4 favors inflammation with increased IL6 production while upregulating TLR2 expression. The ability of TLR4 agonists to stimulate angiogenesis depends on TLR2, suggesting that angiogenesis is independent of TLR4-mediated inflammation. This result is supported by pronounced angiogenesis and recovery observed in TLR4KO mice. Thus, the balance between these TLR receptors determines the final response to ischemia. Sources of TLR signaling in this model include tissue as well as circulating cells such as leukocytes. Pedregosa et al. (34) demonstrated upregulation of TLR2 and TLR4 in both inflammatory cells and resident cells following ischemia-reperfusion injury in kidney, which may have analogy to this model.
In the current study, we identified a relationship between TLR4 and TLR2 in regulating the behavior of ECs. TLR4 agonists upregulated TLR2 expression in HDMVECs, and it was this TLR2 that mediated endothelial tubing and monolayer permeability. HMGB1, an agonist for both TLR4 and TLR2, had a similar proangiogenic effect in vitro. However, TLR2KO cells were unresponsive to HMGB1, indicating that HMGB1 was signaling directly through TLR2 or that HMGB1-TLR4 signaling functioned through downstream effects on TLR2. Interestingly, angiogenesis was observed in TLR4KO cells cultured on Matrigel, although TLR2 expression in these cells was found to be low when cultured on gelatin. This result suggests that baseline levels of TLR2 signaling may be sufficient to support angiogenesis. Alternatively, Matrigel is a laminin-rich substrate and may promote TLR2 expression independent of TLR4. Preliminary experiments suggest that integrin-mediated interactions on specific extracellular matrices may dictate TLR2 expression on EC.
Our previous findings of enhanced muscle injury in response to FAL in TLR2KO mice suggest that TLR2 functions in a protective role in the setting of ischemia. They also indicate that unopposed TLR4 activation results in excessive inflammatory injury. Indeed, we detected increased local and systemic IL-6 production as early as 6 h after FAL. This inflammatory response was blunted in TLR4KO mice, whereas TLR2KO mice had the highest levels of IL-6 in response to ischemia. This finding is consistent with the ability of TLR2 to attenuate TLR4-mediated inflammation. When we examined the expression of TLR4 and TLR2 after ischemia, we observed that TLR2 is present at resting states and is upregulated by 24 h after the ischemic injury. Similar to our in vitro findings, the induction of TLR2 is not observed in the absence of TLR4. Baseline levels of TLR2 expression appear to be sufficient to support the reparative and angiogenic processes observed in the TLR4KO mice. Thus, TLR2 may represent a mechanism to limit TLR4-mediated inflammation and stimulate repair. This result is in contrast to the setting of lung injury after hemorrhagic shock, where upregulation of TLR2 by TLR4 yielded a pronounced inflammatory response to pathogens administered sequentially (20,21). Such divergent roles for TLR2 likely depend on the molecular signals initiated by the injured tissues that dictate the downstream response necessary for recovery and healing. Further studies are needed to delineate how such pathways are determined.
Despite the development of profound ischemia immediately after FAL, mice typically recover perfusion efficiently, achieving near-baseline levels of perfusion by 2 wks. Histologic examination of the hindlimb musculature of WT mice at 2 wks showed regenerated myocytes and increased capillary structures (8). In TLR2KO mice, we have shown that this recovery was not observed with persistence of necrotic myocytes and grossly abnormal vascular structures supporting the role of TLR2 in angiogenesis in vivo (9). This result was confirmed in TLR4KO mice, where perfusion and vascularity in the ischemic hindlimb were increased compared with WT mice. This finding is in keeping with our prior studies that showed mature myocytes and minimal evidence of ischemic injury in these mice (9). In mice lacking TLR2, including TLR2/4 double KO mice, this perfusion recovery was blunted. These findings indicate that the inflammation may not play a key role in the regenerative and angiogenic responses to ischemia. Instead, TLR2 appears to be essential to these processes. This conclusion is supported by West et al. (35), who found that end products of lipid oxidation promote angiogenesis through TLR2 in a vascular endothelial growth factor (VEGF)- and TLR4-independent manner. TLR2 was also shown to be myoprotective. When the TLR2 agonist peptidoglycan (PGN) or Pam3CSK4 was administered before myocardial ischemia/reperfusion, infarct areas were reduced. Additionally, nuclear translocation of nuclear factor (NF)-κB was attenuated with PGN (36). While these results substantiate our claim that TLR2 and TLR4 have opposing roles in muscle injury, both reports studied ischemia/reperfusion injury in myocardium. Our study is unique in its focus on TLR function in skeletal muscle ischemia without reperfusion.
Inflammation is critical to removing damaged myofibers, allowing for proliferation and fusion of satellite cells to the remaining muscle fibers (37). The attenuated inflammation seen in CCR2 knockout mice is associated with fatty infiltration and poor regeneration (38). Others have reported that IL-6 is beneficial for muscle satellite cell proliferation and sustenance (39). Both of these reports suggest that some degree of inflammation is required for skeletal muscle recovery after ischemia. A role for TLRs in myocyte inflammation was reported by Frost et al. (31), who showed that TLR activation can promote IL-6 release from cultured C2C12 myocytes. Our prior findings, in contrast, revealed that the absence of TLR4-mediated inflammation improved muscle regeneration in hindlimb ischemia. Our current studies add to these findings, showing that the suppression of inflammation was associated with a more rapid angiogenesis and perfusion recovery. Similar findings were reported in mice lacking CD180, a molecule that attenuates TLR4-induced inflammation, where profound inflammation and compromised arteriogenesis were observed (40,41). In another report, TLR4KO mice undergoing myocardial ischemia/reperfusion developed smaller infarct areas and less myocardial-activated caspase-3 staining compared with wild-type mice (42).
Conclusion
TLR4 and TLR2 are important regulators of inflammation and revascularization in skeletal muscle ischemia, respectively. This report adds to a growing literature supporting a role for TLR4 in promoting the inflammatory process that, when unopposed, slows regeneration and angiogenesis. However, TLR4 simultaneously upregulates TLR2, which counters the injurious influences of TLR4 by mediating angiogenesis and muscle repair. While previously perceived to be essential to tissue repair, TLR4-mediated inflammation may not be required. Instead, the ability of TLR4 to enhance TLR2 signaling may be the most important influence. Thus, selective modulation of TLR signaling may be an important therapeutic maneuver to attenuate ischemic injury and promote regeneration.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Selvin E, Erlinger TP. (2004) Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation. 110:738–43.
Baumgartner I, et al. (1998) Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation. 97:1114–23.
Lawall H, Bramlage P, Amann B. (2011) Treatment of peripheral arterial disease using stem and progenitor cell therapy. J. Vasc. Surg. 53:445–453.
Lawall H, Bramlage P, Amann B. (2010) Stem cell and progenitor cell therapy in peripheral artery disease: a critical appraisal. Thromb. Haemost. 103:696–709.
Lederman RJ, et al. (2002) Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. Lancet. 359:2053–8.
Powell RJ, Dormandy J, Simons M, Morishita R, Annex BH. (2004) Therapeutic angiogenesis for critical limb ischemia: design of the hepatocyte growth factor therapeutic angiogenesis clinical trial. Vasc. Med. 9:193–8.
Biscetti F, et al. (2010) High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes. 59:1496–505.
Sachdev U, et al. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J. Vasc. Surg. 55:180–91.
Sachdev U, et al. TLR2 and TLR4 mediate differential responses to limb ischemia through MyD88-dependent and independent pathways. PLoS One. 7:e50654.
Sachdev U, Cui X, Tzeng E. (2013) HMGB1 and TLR4 mediate skeletal muscle recovery in a murine model of hindlimb ischemia. J. Vasc. Surg. 58:460–9.
Biscetti F, et al. (2010) High-mobility group box 1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes. 59:1496–505.
De Mori R, et al. (2007) Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler. Thromb. Vasc. Biol. 27:2377–83.
Mitola S, et al. (2006) Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J. Immunol. 176:12–5.
Palumbo R, et al. (2004) Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J. Cell Biol. 164:441–9.
Lien E, et al. (1999) Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274:33419–25.
Yoshimura A, et al. (1999) Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via toll-like receptor 2. J. Immunol. 163:1–5.
Poltorak A, et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–8.
Medzhitov R, Preston-Hurlburt P, Janeway CA. (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–7.
Lotze MT, Tracey KJ. (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5:331–42.
Li Y, et al. (2009) Hemorrhagic shock augments lung endothelial cell activation: role of temporal alterations of TLR4 and TLR2. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297:R1670–80.
Fan J, Frey RS, Malik AB. (2003) TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J. Clin. Invest. 112:1234–43.
Wolfs TGAM, et al. (2002) In vivo expression of toll-like receptor 2 and 4 by renal epithelial cells: IFN-γ and TNF-α mediated upregulation during inflammation. J. Immunol. 168:1286–93.
Ngamkitidechakul C, Twining SS. (2002) Buffered non-fermenter system for lab-scale production of secreted recombinant His-tagged proteins in Saccharomyces cerevisiae. Biotechniques. 33:1296–300.
Wang H, et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–51.
Yang H, et al. (2004) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. U. S. A. 101:296–301.
Kibbe MR, et al. (2002) Potentiation of nitric oxide-induced apoptosis in p53-/- vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 282:C625–34.
Messina LM, Brevetti LS, Chang DS, Paek R, Sarkar R. (2002) Therapeutic angiogenesis for critical limb ischemia: invited commentary. J. Control Release. 78:285–94.
Couffinhal T, et al. (1998) Mouse model of angiogenesis. Am. J. Pathol. 152:1667–79.
Carmeliet P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6:389–95.
Sachdev U, Cui X, Xu J, Xu J, Tzeng E. (2014) MyD88 and TRIF mediate divergent inflammatory and regenerative responses to skeletal muscle ischemia. Physiol. Rep. 2:e12006.
Frost RA, Nystrom GJ, Lang CH. (2006) Multiple 615 Toll-like receptor ligands induce an IL-6 transcriptional response in skeletal myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290:R773–84.
Caricilli AM, et al. (2008) Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J. Endocrinol. 199:399–406.
Zbinden-Foncea H, Deldicque L, Pierre N, Francaux M, Raymackers JM. (2012) TLR2 and TLR4 activation induces p38 MAPK-dependent phosphorylation of S6 kinase 1 in C2C12 myotubes. Cell Biol. Int. 36:1107–13.
Pedregosa JF, et al. (2011) TLR2 and TLR4 expression after kidney ischemia and reperfusion injury in mice treated with FTY720. Int. Immunopharmacol. 11:1311–8.
West XZ, et al. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 467:972–6.
Ha T, et al. (2010) TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc. Res. 87:694–703.
Charge SB, Rudnicki MA. (2004) Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84:209–38.
Contreras-Shannon V, et al. (2007) Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2-/- mice following ischemic injury. Am. J. Physiol. Cell Physiol. 292:C953–67.
McKay BR, et al. (2009) Association of interleukin-6 signalling with the muscle stem cell response following muscle-lengthening contractions in humans. PLoS One. 4:e6027.
Divanovic S, et al. (2005) Negative regulation of toll-like receptor 4 signaling by the toll-like receptor homolog RP105. Nat. Immunol. 6:571–8.
Bastiaansen AJ, et al. (2014) TLR4 accessory molecule RP105 (CD180) regulates monocyte-driven arteriogenesis in a murine hind limb ischemia model. PLoS One. 9:e99882.
Hua F, et al. (2007) Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J. Immunol. 178:7317–24.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K08HL103899 and a Society for Vascular Surgery Foundation/American College of Surgeons Mentored Clinical Scientist Award to U Sachdev. This material is the result of work supported in part with resources and the use of facilities at the VA Pittsburgh Healthcare System (ET). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, Department of Veterans Affairs or the U.S. government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Xu, J., Benabou, K., Cui, X. et al. TLR4 Deters Perfusion Recovery and Upregulates Toll-like Receptor 2 (TLR2) in Ischemic Skeletal Muscle and Endothelial Cells. Mol Med 21, 605–615 (2015). https://doi.org/10.2119/molmed.2014.00260
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00260