
Abstract
Macrophage activation syndrome (MAS) is a potentially fatal complication of systemic inflammation. High mobility group box 1 (HMGB1) is a nuclear protein extensively leaked extracellularly during necrotic cell death or actively secreted by natural killer (NK) cells, macrophages and additional cells during infection or sterile injury. Extracellular HMGB1 orchestrates key events in inflammation as a prototypic alarmin. The redox states of its three cysteines render the molecule mutually exclusive functions: fully reduced “all-thiol HMGB1” exerts chemotactic activity; “disulfide HMGB1” has cytokine-inducing, toll-like receptor 4 (TLR4)-mediated effects—while terminally oxidized “sulfonyl HMGB1” lacks inflammatory activity. This study examines the kinetic pattern of systemic HMGB1 isoform expression during therapy in four children with severe MAS. Three of the four patients with underlying systemic rheumatic diseases were treated with biologics and two suffered from triggering herpes virus infections at the onset of MAS. All patients required intensive care unit therapy due to life-threatening illness. Tandem mass-spectrometric analysis revealed dramatically increased systemic levels of the cytokine-inducing HMGB1 isoform during early MAS. Disease control coincided with supplementary etoposide therapy initiated to boost apoptotic cell death, when systemic HMGB1 levels drastically declined and the molecule emerged mainly in its oxidized, noninflammatory isoform. Systemic interferon (IFN)-γ and ferritin peaked concomitantly with HMGB1, whereas interleukin (IL)-18 and monocyte chemotactic protein (MCP)-1 levels developed differently. In conclusion, this work provides new insights in HMGB1 biology, suggesting that the molecule is not merely a biomarker of inflammation, but most likely also contributes to the pathogenesis of MAS. These observations encourage further studies of disulfide HMGB1 antagonists to improve outcome of MAS.
Similar content being viewed by others

Introduction
Macrophage activation syndrome (MAS) is a severe and potentially life-threatening complication of systemic inflammatory disorders. It may occur in response to an infection (often viral), malignancy or a rheumatic disease (1). MAS typically appears in patients with systemic onset juvenile idiopathic arthritis (SoJIA) and its adult equivalent, adult-onset Still disease (1); it also is reported in other pediatric inflammatory disorders including juvenile systemic lupus erythematosus (SLE) (2) and Kawasaki disease (3). Symptoms and signs of MAS include fever, hepatosplenomegaly, lymphoadenopathy, profound depletion of all cellular blood elements, liver dysfunction, disseminated intravascular coagulation and central nervous system dysfunction (1).
MAS expresses a close clinical resemblance to a group of histiocytic cell disorders collectively known as hemophagocytic lymphohistiocytosis (HLH). MAS is classified among the secondary, or acquired forms of HLH (4,5). Primary HLH is a genetic disorder of immune regulation caused by mutations in genes encoding proteins required for the cytolytic activity exerted by NK cells and cytotoxic T cells (6). Impaired cytolytic capacity also is postulated as a key event in the pathogenesis of MAS, diminishing the ability to induce apoptosis needed for an immunologically silent elimination of target cells (7). Hence, cell death by other mechanisms, including necrosis and pyroptosis, will dominate in MAS and HLH, leading to excessive activation and survival of macrophages, NK cells and T lymphocytes generating an overwhelming inflammatory reaction. A properly functioning cytotoxic defense system is needed to eliminate virally infected cells and transformed cells and to terminate immune reactions by killing autologous activated cells mediating inflammation. MAS, caused by compromised cytolytic capacity, may occur spontaneously as a consequence of uncontrolled systemic inflammation or may be triggered by a viral infection, commonly belonging to the herpes group family. Drug therapy, in particular based on biologics, is another route that may lead to MAS development (8,9).
HMGB1 is a ubiquitous nuclear protein with proinflammatory properties when released to the extracellular space, thus establishing HMGB1 as a prototypic alarmin (10,11). HMGB1 is passively leaked out of necrotic cells. During apoptosis, HMGB1 will be terminally oxidized, strongly bound to the chromatin and retained in apoptotic bodies (12). The assembly of large multiproteins complexed to activated inflammasomes generates caspase-1 formation that controls the release of IL-1β, IL-18 and proinflammatory isoforms of HMGB1, and consequently results in a programmed proinflammatory cell death called pyroptosis (13,14). IL-1 and IL-18 are well established and important mediators in MAS/HLH (1,15), while a functional role of HMGB1 in these conditions remains to be studied.
Once released into the extracellular milieu, HMGB1 binds and signals via a number of different reciprocal cell-surface receptors dependent on the redox state of the cysteines of the molecule (16,17). HMGB1 contains three conserved redoxsensitive cysteines (C23, C45 and C106), and modification of these cysteines determines the bioactivity of extracellular HMGB1 (Table 1). A systematic nomenclature recently has been derived to classify the structure-activity relationship of the various redox-dependent isoforms (18). The cytokine-stimulating activity of HMGB1 requires a disulfide linkage between C23 and C45, with C106 remaining in its reduced form with a thiol group (HMGB1.C23-C45.C106h). This disulfide HMGB1 enables binding and signaling via the TLR4/MD-2 complex to stimulate cytokine release in macrophages and other cells (17,19). When extracellular HMGB1 appears as a fully reduced molecule with all cysteines expressing thiol groups (HMGB1.C23h.C45h.C106h) it exerts potent chemotactic signals by forming a heterocomplex with CXCL12 that will bind to CXCR4 mediating synergistic chemotaxis (20). However, this fully reduced HMGB1 does not bind to TLR4 and cannot activate the recruited inflammatory cells. The redox states of disulfide and fully reduced-HMGB1 are reversible processes. However, terminal oxidation of any of the three cysteine residues into sulfonyl HMGB1 (HMGB1.C23so.C45so.C106so) eliminates the proinflammatory capacity of HMGB1 in an irreversible fashion (17). Whether this isoform of HMGB1 exerts any other biological activity is presently unknown. Active HMGB1 release requires additional posttranslational modifications including hyperacetylation of multiple lysine residues within the two nuclear location sequences (NLSs) that prevents nuclear reentry of HMGB1, thus resulting in a cytoplasmic accumulation of HMGB1 needed for extracellular export (21). The extracellular presence of hypoacetylated HMGB1 specifies an origin by passive release during primary necrotic or secondary necrotic apoptotic cell death, while hyperacetylated HMGB1 indicates an active release via caspase-1 mediated pyroptosis in NK cells (22–24), macrophages (21,25) and other cells (Table 2).
Recent advances regarding analyses using high-resolution and sensitive liquid chromatography followed by tandem mass spectrometry (LC-MS/MS), offer novel opportunities to determine post-translationally modified HMGB1 in biological samples informative of bioactivity and mode of release. This information provides further understanding of pathogenetic mechanisms operating in various diseases including MAS, which is the focus of the present study.
Materials and Methods
Patients
Four children aged 3 to 15 years, three previously diagnosed with systemic onset juvenile idiopathic arthritis and one with systemic lupus erythematosus, who all presented with MAS, were studied during a one-year period of November 2010 to October 2011 and fulfilled current MAS criteria (2,26). The diagnosis of SoJIA was made on the basis of the criteria of the International League of Associations for Rheumatology (27) and the SLE patient fulfilled the American College of Rheumatology (ACR) revised SLE criteria. MAS was diagnosed on the combination of clinical features, including cytopenia or sudden decrease in white blood cell counts and/or platelet counts, coagulopathy and liver dysfunction, according to the guidelines proposed by Ravelli et al. and Parodi et al. (2,26). All patients also fulfilled diagnostic criteria for inherited HLH, according to HLH-2004 criteria (28). All patients had markedly elevated inflammatory parameters, including CRP and ferritin, and needed intensive care treatment. Three of the patients had involvement of the central nervous system (CNS), which was severe in two. Prior to the development of MAS, all four patients had been administered high doses of corticosteroids, and, in addition, cyclosporin A (CsA), plaquenil, etanercept and/or anakinra. At the time of developing MAS, therapy included tocilizumab and methotrexate; tocilizumab, methotrexate and prednisolone; anakinra, CsA and betamethasone; and plaquenil and prednisolone, respectively. In the two patients with tocilizumab therapy, infections with Epstein-Barr virus and varicella-zoster virus preceded the development of MAS. Owing to the severe clinical MAS presentations including CNS affection in three patients and rapidly progressive pancytopenia in the fourth, and the similarities between MAS and hemophagocytic lymphohistiocytosis (HLH) (5), we chose to provide therapy with etoposide and corticosteroids, which is standard therapy in HLH (28,29). However, since the treatment protocols HLH-94 and HLH-2004 were originally designed for infants with primary HLH (familial hemophagocytic lymphohistiocytosis, FHL), we administered etoposide at lower doses (50–120 mg/m2) and less frequent intervals (scheduled once weekly) than suggested in the HLH protocols (150 mg/m2, initially twice weekly). The duration of the etoposide treatment ranged from 4 to 7 wks. Although the two patients with severe CNS involvement at onset of etoposide therapy still have impaired CNS function at follow-up, all patients responded with dramatic improvement to the addition of etoposide therapy. The clinical characteristics of the patients are summarized in Table 3.
This study was approved by the Stockholm Ethical Committee, Stockholm, Sweden. Parents and patients gave informed consent before inclusion.
Blood Samples
Sera were obtained from blood samples collected in tubes without additive and plasma was collected in EDTA tubes. Blood samples were centrifuged at 1440g for 10 min and cells were removed and stored at −80°C until assayed.
We serially determined the serum levels of inflammasome-associated HMGB1, IL-1α, IL-1β and IL-18 and additionally IFN-γ, MCP-1 and ferritin in the patients before, during and after therapeutic intervention until inflammation had resolved.
Serum samples from patients with oligoarticular, polyarticular, enthesitis-related or other type of JIA according to the ILAR criteria, as well as serum samples from 10 healthy pediatric controls (range 2–14 years), were collected at Astrid Lindgren Children’s Hospital and analyzed for comparison of total HMGB1 levels.
ELISA Assays for HMGB1 Detection
HMGB1 levels were measured in undiluted plasma using HMGB1 enzyme-linked immunosorbent assay (ELISA) kit II, according to the instructions of the manufacturer (IBL International, Hamburg, Germany). The lower limit for detection was 0.3 ng/mL.
Cytometric Bead Array (CBA) and Bioplex for Detection of IFN-γ, IL-1α, IL-1β, IL-18 and MCP-1
All sera were diluted 1:2–1:8 before analysis. IL-1α, IL-1β, IL-18 and IFN-γ were measured by Bioplex (Bio-Rad Laboratories, Hercules, CA, USA) with a lower detection limit of 4–9 pg/mL. Levels of MCP-1 were measured by CBA using Human Soluble Protein Flex Sets and Human Soluble Protein Master Buffer Kits (BD Biosciences, San Jose, CA, USA) with a lower detection limit of 20 pg/mL. All analyses were performed according to the instructions of the manufacturers.
Serum levels of ferritin were analyzed at the clinical laboratory at Karolinska University Hospital in Stockholm, Sweden.
Characterization of HMGB1 Isoforms by Electrospray Ionization-Liquid Chromatography-Tandem Mass Spectrometry (ESI-LC-MS/MS)
All chemicals and solvents used were of the highest available grade (Sigma-Aldrich, St. Louis, MO, USA). Samples were precleared with 50-µL Protein G-Sepharose beads for 1 h at 4°C. HMGB1 present in 100 µL serum was immunoprecipitated with 5 µg of rabbit anti-HMGB1 (ab18256; Abcam, Cambridge, UK) for 16 h at 4°C as described previously (30). For the analysis of HMGB1 redox posttranslational modifications, free thiol groups within HMGB1 were alkylated with 90 mmol/L N-ethylmaleimide (NEM) at 4°C for 10 min. Cysteine residues in disulfide bonds were then reduced with 30 mmol/L dithiothreitol (DTT) at 4°C for 1 h. Newly DTT reduced cysteines then were alkylated with a heavy labeled d5NEM (NEM + 5× deuterium) which yields a mass shift of 130 atomic mass unit (amu) compared with the NEM-unexposed protein and 5 amu compared with the NEM-alkylated protein. Terminal oxidation of HMGB1 cysteines was characterized as described previously (16,17,31). Samples were subjected to trypsin (Promega, Madison, WI, USA) or GluC (New England Biolabs, Ipswich, MA, USA) digestion according to manufacturer’s instructions and desalted using C18 zip-tips (Millipore, Billerica, MA, USA). Assessment of HMGB1 acetyl modifications was determined as previously described (32). Peptide analysis was determined as described previously using an AB Sciex QTRAP 5500 equipped with a NanoSpray II source (AB Sciex, Framingham, MA, USA) by inline liquid chromatography using a U3000 HPLC System (Dionex [Thermo Scientific], Sunnyvale, CA, USA), connected to a 180 µm × 20 mm nanoAcquity UPLC C18 trap column and a 75 µm × 15 cm nanoAcquity UPLC BEH130 C18 column (Waters, Milford, MA, USA) via reducing unions. A gradient from 0.05% trifluoroacetic acid (TFA) (v/v) to 50% acetonitrile (ACN)/0.08% TFA (v/v) in 40 min was applied at a flow rate of 200 nL/min. The ion spray potential was set to 2,200–3,500 V, the nebulizer gas to 19 and the interface heater to 150°C.
Results
Four children with exceptionally severe MAS (clinical data are outlined in Table 3) were studied retrospectively regarding systemic overall HMGB1 levels and the kinetic expression of systemic HMGB1 isoforms generated by posttranslational modifications in particular. First-line therapy based on high intravenous doses of corticosteroids and cyclosporine A (CsA) to suppress the cytokine storm did not control the excessive inflammation in any of the MAS patients. Systemic etoposide treatment subsequently was introduced to improve the deficient ability to mediate apoptosis in critical target cells driving the uncontrolled inflammation. Particular focus was directed on HMGB1 isoform analysis in blood samples obtained closely before and after etoposide infusions. Information about the acetylation state of the extracellular HMGB1 discriminates between active and passive modes of release (Table 2), while HMGB1 redox isoform analysis relates to reciprocal receptor usage and thus the functional role of HMGB1 in the inflammatory process (Table 1). Studies of well-known proinflammatory mediators and biomarkers known to reflect the clinical course of MAS were conducted in parallel. These serum analyses included assessments of ferritin, IFN-γ, IL-1α, IL-1β, IL-18 and monocyte chemotactic protein (MCP-1).
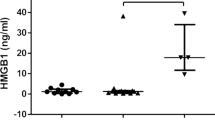
Total serum HMGB1 levels in MAS patients before initiation of etoposide were significantly higher (p < 0.05) compared with those observed in JIA patients without MAS and healthy children (Figure 1). Peak systemic HMGB1 levels were recorded at the same time as the MAS patients expressed maximal symptoms and signs. The predominant part of the total HMGB1 pool was at this stage hyperacetylated (Figures 2A, 3A, 4A, 5A) indicating that the systemic HMGB1 in these patients was actively secreted rather than passively released from dying cells (14,21).

High systemic HMGB1 levels in MAS patients. Serum concentrations of total HMGB1 levels were markedly increased in plasma during severe MAS as compared with children with uncomplicated JIA and healthy pediatric controls. HMGB1 levels were measured by ELISA and correlation was calculated using the Spearman rank correlation test. The HMGB1 levels in the same cohort of JIA patients and healthy control children have been published previously in (38).

Longitudinal serum analyses before and after introduction of etoposide treatment in patient 1. Serum concentration of total HMGB1 and hyperacetylated HMGB1 during the course of MAS are demonstrated in (A) with the different HMGB1 redox-forms illustrated in (B). High levels of HMGB1 in both fully reduced and disulfide isoforms were documented during severe disease, which rapidly declined after initiation of etoposide treatment concomitantly with serum concentrations of (C) the clinical biomarker ferritin as well as (D) IFN-γ. In the clinical resolution phase, serum HMGB1 appeared predominantly in the terminally oxidized sulfonyl isoform. Serum concentrations of (E) IL-18 and (F) MCP-1 peaked weeks later when the patient was recovering. MP-pulses: methylprednisolone pulses.

Longitudinal serum analyses before and after introduction of etoposide treatment in patient 2. Serum concentration of total HMGB1 and hyperacetylated HMGB1 are illustrated in (A) with the different HMGB1 redox-forms illustrated in (B). The first two serum samples were collected at onset of SoJIA without MAS manifestations when HMGB1 levels appeared mainly in its cytokine-inducing, hyperacetylated isoform. These HMGB1 levels increased at onset of MAS and declined promptly after treatment with etoposide infusions. Increased serum concentrations of (C) ferritin and (D) IFN-γ were documented during MAS followed by a normalization post etoposide treatment. (E) Serum IL-18 levels were distinctly increased during the entire observation period with peak values during MAS. (F) MCP-1 did not mimic the clinical course. MP-pulses: methylprednisolone pulses.

Longitudinal serum analyses before and after introduction of etoposide treatment in patient 3. Serum concentration of total HMGB1 and hyperacetylated HMGB1 are demonstrated in (A) with the different HMGB1 redox-forms illustrated in (B). The expression of serum levels of HMGB1 as well as (C) ferritin and (D) IFN-γ corresponded very well to the clinical course of MAS with a rapid decline and clinical improvement after etoposide treatment. IL-18 and MCP-1 levels were increased during critical disease with a prolonged release period (E,F). CsA: cyclosporine A; MP-pulses: methylprednisolone pulses.

Serum analyses in patient 4 before and after treatment with etoposide. Serum concentration of total HMGB1 and hyperacetylated HMGB1 are presented in (A) with the different HMGB1 redox-forms illustrated in (B). Two serum samples were analyzed where normalized levels of (A) HMGB1, (C) ferritin and (D) IFN-γ were documented after intervention with etoposide and subsequent clinical improvement. IL-18 declined but was still elevated (E) while MCP-1 increased (F). MP-pulses: methylprednisolone pulses.
Control of inflammation with a clinical stabilization in the patients coincided with the initiation of supplementary etoposide treatment, when a prompt decrease of the systemic HMGB1 levels occurred. Redox state analysis of the expressed HMGB1 isoforms revealed additional useful clinical information. Serum samples taken before initiation of etoposide treatment mainly expressed the fully reduced and disulfide HMGB1 isoforms. These isoforms mediate inflammation via promotion of chemotactic signals, accumulating professional inflammatory cells or activating these via the TLR4 receptor complex, respectively. Disulfide HMGB1 binds to the extracellular adaptor protein MD-2 of the TLR4 receptor complex, in the same fashion as lipopolysaccharide (LPS), generating potent proinflammatory cytokine production, which is one of the hallmarks of MAS. By contrast, serum samples obtained after etoposide treatment predominantly expressed HMGB1 that was terminally oxidized expressing sulfonic acid (SO3H) groups on the cysteine residues (Figures 2B, 3B, 4B, 5B). This is an HMGB1 isoform without known proinflammatory activities. The clinical signs of disease were considerably milder at this stage. Taken together, these findings suggest that etoposide treatment contributed to a reduction of total HMGB1 levels and a conversion of the remaining HMGB1 to a noninflammatory isoform.
Serum ferritin is a protein that stores iron in a soluble form and its synthesis is regulated by intracellular iron, inflammatory cytokines and oxidative stress. Serum ferritin levels are especially increased in HLH and MAS, since hemophagocytosis results in enhanced uptake of haptoglobin-hemoglobin complexes by macrophages triggering a production of ferritin to sequester the excessive amount of free iron. Serum ferritin levels are used as the golden standard parameter to monitor the clinical course of MAS/HLH (33,34). All our studied MAS patients expressed serum ferritin levels that followed a parallel course to that of hyperacetylated serum HMGB1 levels and all these parameters mirrored the clinical course with decreased levels in response to etoposide treatment combined with clinical improvement (Figures 2C, 3C, 4C, 5C). Patient 1 in particular demonstrated a dramatic decline in serum ferritin levels from 121,937 to 13,416 µg/L (upper normal range should be below 100 µg/L) within a few days after etoposide infusions (see Figure 2C).
IFN-γ is the main macrophage-activating cytokine and is released from activated NK cells and T lymphocytes. IFN-γ primes the capacity of macrophages for phagocytosis and for proinflammatory cytokine production and is thus a central cytokine in the pathogenesis of MAS (1,35). Serum levels of IFN-γ were increased markedly during periods with severe disease in all studied MAS patients and these levels promptly decreased during clinical resolution after etoposide treatment (Figures 2D, 3D, 4D, 5C).
IL-1 and IL-18 also have been demonstrated to be central pathogenic mediators in MAS (36,37) and both molecules are released during pyroptosis, which likewise is an important pathway for HMGB1 release (13,14). Increased serum levels of IL-18 were documented in all our patients, with peak values appearing later than serum HMGB1, IFN-γ and ferritin levels (Figures 2E, 3E, 4E, 5E). Serum IL-1α and IL-1β were not detected at any time point in any of the studied MAS patients (data not shown). IL-1 exerts potent biological effects at the low pg/mL range, and we believe that our detection methods were not sensitive enough to discover these low levels. The chemokine MCP-1 is increased in JIA patients (38) and has been implicated in the pathogenesis of HLH where serum concentrations correlate well to disease activity (39). In the present study, serum MCP-1 levels did not change in any consistent manner in response to treatment and the levels peaked around 3–4 wks after the initiation of therapy (Figures 2F, 3F, 4F, 5F).
To summarize, the results were uniform in all four MAS patients with high levels of inflammatory HMGB1 isoforms promptly declining during clinical resolution induced by therapeutic intervention with etoposide, corticosteroids and cyclosporin A. Systemic levels of ferritin and IFN-γ appeared concomitantly with HMGB1, whereas levels of IL-18 and MCP-1 did not correlate as closely with the clinical course.
Discussion
In this report, we present four children with severe MAS, where serum levels of total HMGB1 and its redox isoforms were monitored longitudinally at clinically important stages. We revealed markedly elevated serum HMGB1 levels in active MAS in all patients. Moreover, the systemic HMGB1 was expressed in immunological active isoforms promoting either cytokine induction, phagocytosis or chemotaxis during critical illness. Control of clinical inflammation was achieved in all patients when etoposide treatment was added to the conventional antiinflammatory treatment with a marked decline of the extracellular presence of HMGB1 that then mainly appeared irreversibly oxidized to the inflammatory inactive isoform, sulfonyl HMGB1. This in line with several publications that have demonstrated that the oxidative stress during apoptotic cell death induces potent intracellular oxidation events resulting in terminal oxidation of HMGB1 (40–42). Sulfonyl HMGB1 is generally the end product of apoptotic cell death and shows caspase-3 dependence (40). Etoposide is a chemotherapeutic drug that inhibits topoisomerase II, resulting in errors in DNA synthesis leading to apoptotic cell deaths in rapidly dividing or activated cells. It was recently demonstrated in an animal HLH model based on a lymphocytic choriomeningitis virus infection in perforin-deficient mice that etoposide selectively ablated activated pathogenic T cells via induction of apoptotic cell death. Resting T cells, including memory T cells, were not influenced by the treatment (43). Etoposide is the mainstay of treatment for HLH patients (44), but has not yet undergone controlled trials for MAS patients. The life-threatening courses of MAS in our patients, who were refractory to high-dose corticosteroids, required additional treatment options. Since the clinical expressions of HLH and MAS are indistinguishable, we chose etoposide in a somewhat reduced dose than commonly used for HLH treatment. A considerable risk regarding the use of etoposide is an elimination of too many immunocompetent cells necessary for the defense against pathogens, as in patient #2 who developed neutropenia and bacterial sepsis after the etoposide infusion, and was successfully treated. All patients had ongoing immunomodulatory therapy at onset of MAS when additional treatment with intravenous corticosteroid pulses only or corticosteroid pulses plus CsA was initiated. Inadequate clinical responses in the treated patients led to supplementary etoposide treatment that was followed by drastically reduced HMGB1 levels, terminal HMGB1 oxidation and clinical improvement. Whether the observed outcome parameters were due to effect generated by etoposide alone or etoposide acting synergistically with other therapeutic agents cannot be resolved in our small study.
Significant amounts of extracellular HMGB1 may be discharged either by active secretion from activated immune cells or by passive release from necrotic, pyroptotic or damaged cells, but not from apoptotic cells (10). Acetylation of multiple lysine residues within the two NLS sites of HMGB1 is a key regulatory mechanism promoting the active release of HMGB1 from secreting cells. The high levels of hyperacetylated HMGB1 during severe MAS imply that the largest contribution to the systemic HMGB1 pool came via secretion from stimulated professional immune cells. The systemic levels of hyperacetylated HMGB1 demonstrated in our MAS patients were exceptionally high and even exceeded levels recorded when identical LC-MS/MS methods were employed to assess acetylated HMGB1 serum levels in acetaminophen (APAP)-intoxicated patients, who either died or needed a liver transplant (45). It is well established that acetylated HMGB1 is a major pathogenetic factor causing liver failure during APAP intoxication (30,42,45). Serum levels of acetylated HMGB1 have been demonstrated to be a highly sensitive and specific biomarker to predict the clinical outcome in patients with APAP-induced hepatotoxicity (45). Future prospective studies are warranted to estimate the value of assessing acetylated HMGB1 levels in MAS/HLH as a prognostic tool to predict outcome in these conditions.
Conclusion
How could then extracellular HMGB1 contribute to the systemic inflammation in MAS? There are several HMGB1-dependent pathways that may be involved if we postulate that the initiating event in MAS in a given sterile or infectious insult is an impaired capacity to provide key cytolytic molecules needed for induction of apoptosis in critical target cells. Other modes of cell deaths including necrosis or pyroptosis will then take place instead. These cellular events will generate extracellular fully reduced HMGB1 as well as disulfide HMGB1 pools that will drive inflammation via chemotactic signals via CXCR4 (20) and activation of the recruited inflammatory cells via TLR4/MD2 (19,46) to produce proinflammatory cytokines and to induce powerful phagocytic responses. A deficient cytolytic capacity to eliminate autologous activated NK cells and cytotoxic T cells, which are key producer cells of both IFN-γ and HMGB1, will further contribute to the macrophage activation in the patients. The almost identical temporal changes observed in our patients regarding systemic levels of IFN-γ and acetylated HMGB1 in response to therapy are in line with this assumption (Figures 2–5). It is important to consider that it is most likely only the cytolytic pathway that is selectively compromised in cytotoxic NK cells and CD8-positive T cells in MAS/HLH patients. When these cells get activated they will produce much IFN-γ and HMGB1 and other mediators in their normal repertoire. Furthermore, the poor cytolytic activity seen in HLH/MAS patients may lead to a failure to remove autologous virus-infected cells and thus the source for antigen stimulation will persist leading to long-lasting antigen-driven activation of the immune system escalating the inflammatory response.
“HMGB1 with a disulfide linkage between cysteine 23 and 45 binds to MD-2 of the TLR4 receptor complex with almost the same avidity as LPS” (2015 letter from Huan Yang to Ulf Andersson; unreferenced; see Acknowledgments) (46). Consequently, high systemic disulfide HMGB1 levels may elicit a cytokine storm analogous to that occurring in gram negative sepsis (46,47). Furthermore, HMGB1 has an inherent ability to form immunostimulatory complexes with other molecules including nucleic acids, nucleosomes, IL-1α, IL-1β, or CXCL12 as well as exogenous factors including LPS and additional TLR-ligands, thereby enhancing proinflammatory responses in a synergistic way via reciprocal receptors for the HMGB1-partner molecules (48,49).
Potent inflammasome activation during exaggerated pyroptotic cell death is another mechanism that may contribute with IL-1β, IL-18 and proinflammatory HMGB1 isoforms (13,14,31,50). Furthermore, it was reported recently that extracellular HMGB1 by itself may induce macrophage pyroptosis and caspase-1 activation through RAGE-mediated endocytosis of HMGB1 (51). No information is yet available regarding HMGB1 isoform requirements for this positive feedback loop that may play an important functional role in the uncontrolled inflammation.
Administration of disulfide HMGB1 to normal animals produces systemic inflammatory responses with sickness behaviors including fever, anorexia, cognitive dysfunction, anemia, acute lung injury, endothelial cell activation, epithelial barrier dysfunction, arthritis and death (52). Therapeutic targeting of HMGB1 with neutralizing antibodies or other specific antagonists has been successful in numerous animal models of systemic inflammation (52–54).
The finding of our limited, retrospective study cannot provide definitive information about any critical step in the pathogenesis of life-threatening MAS, but rather represents a first attempt to explore dynamic changes of HMGB1 isoforms in relationship to clinical presentation and given therapy. Multicenter studies are required to recruit larger patient cohorts for prospective, randomized, controlled studies to provide definitive conclusions on the role of HMGB1 isoforms in MAS/HLH as biomarkers to predict outcome, treatment stratification and as possible target molecules for future therapy.
Disclosures
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Change history
30 December 2020
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1186/s10020-020-00263-2
References
Ravelli A, Grom AA, Behrens EM, Cron RQ. (2012) Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 13:289–98.
Parodi A, et al. (2009) Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 60:3388–99.
Simonini G, et al. (2010) Macrophage activation syndrome/hemophagocytic lymphohistiocytosis and Kawasaki disease. Ped. Blood Canc. 55:592.
Athreya BH. (2002) Is macrophage activation syndrome a new entity? Clin. Exp. Rheumatol. 20:121–3.
Ramanan AV, Schneider R. (2003) Macrophage activation syndrome—what’s in a name! J. Rheumatol. 30:2513–6.
Stepp SE, et al. (1999) Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 286:1957–9.
Villanueva J, et al. (2005) Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res. Ther. 7: R30–7.
Aouba A, De Bandt M, Aslangul E, Atkhen N, Patri B. (2003) Haemophagocytic syndrome in a rheumatoid arthritis patient treated with infliximab. Rheumatology (Oxford). 42:800–2.
Ramanan AV, Schneider R. (2003) Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J. Rheumatol. 30:401–3.
Scaffidi P, Misteli T, Bianchi ME. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–5.
Harris HE, Raucci A. (2006) Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 7:774–8.
Bianchi ME, Manfredi A. (2004) Chromatin and cell death. Biochim. Biophys. Acta. 1677:181–6.
Lamkanfi M, et al. (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185:4385–92.
Lu B, et al. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 488:670–4.
Takada H, et al. (1999) Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br. J. Haematol. 106:182–9.
Venereau E, et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209:1519–28.
Yang H, et al. (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18:250–9.
Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. (2014) A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol. Med. 20:135–7.
Yang H, et al. (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U. S. A. 107:11942–7.
Schiraldi M, et al. (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 209:551–63.
Bonaldi T, et al. (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo. J. 22:5551–60.
Semino C, Angelini G, Poggi A, Rubartelli A. (2005) NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 106:609–16.
Semino C, et al. (2007) The maturation potential of NK cell clones toward autologous dendritic cells correlates with HMGB1 secretion. J. Leukoc. Biol. 81:92–9.
Li G, Liang X, Lotze MT. (2013) HMGB1: The central cytokine for all lymphoid cells. Front. Immunol. 4:68.
Lu B, et al. (2014) JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc. Natl. Acad. Sci. U. S. A. 111:3068–73.
Ravelli A, et al. (2005) Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J. Ped. 146:598–604.
Petty RE, et al. (1998) Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J. Rheumatol. 25:1991–4.
Henter JI, et al. (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Ped. Blood Cancer. 48:124–31.
Trottestam H, et al. (2011) Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 118:4577–84.
Antoine DJ, et al. (2009) High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol. Sci. 112:521–31.
Nyström S, et al. (2013) TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 32:86–99.
Ge X, et al. (2014) High-mobility group box-1 (HMGB1) Participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 289:22672–91.
Allen CE, Yu X, Kozinetz CA, McClain KL. (2008) Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Ped. Blood Cancer. 50:1227–35.
Grom AA, Mellins ED. (2010) Macrophage activation syndrome: advances towards understanding pathogenesis. Curr. Opin. Rheumatol. 22:561–6.
Zoller EE, et al. (2011) Hemophagocytosis causes a consumptive anemia of inflammation. J. Exp. Med. 208:1203–14.
Shigemura T, et al. (2011) Monitoring serum IL-18 levels is useful for treatment of a patient with systemic juvenile idiopathic arthritis complicated by macrophage activation syndrome. Pediatr. Rheumatol Online J. 9:15.
Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. (2011) Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford). 50:417–9.
Schierbeck H, et al. (2013) HMGB1 levels are increased in patients with juvenile idiopathic arthritis, correlate with early onset of disease, and are independent of disease duration. J. Rheumatol. 40:1604–13.
Tamura K, et al. (2008) Increased serum monocyte chemoattractant protein-1, macrophage inflammatory protein-1beta, and interleukin-8 concentrations in hemophagocytic lymphohistiocytosis. Ped. Blood Cancer. 51:662–8.
Kazama H, et al. (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box1 protein. Immunity. 29:21–32.
Urbonaviciute V, et al. (2009) Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity. 42:305–7.
Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. (2010) Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol. Med. 16:479–90.
Johnson TS, et al. (2014) Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J. Immunol. 192:84–91.
Henter JI, et al. (2002) Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 100:2367–73.
Antoine DJ, et al. (2012) Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatology. 56:1070–9.
Kim S, et al. (2013) Signaling of high mobility group box 1 (HMGB1) through toll-like receptor 4 in macrophages requires CD14. Mol. Med. 19:88–98.
Andersson U, et al. (2000) HMG-1 stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192:565–70.
Sha Y, Zmijewski J, Xu Z, Abraham E. (2008) HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 180:2531–7.
Hreggvidsdottir HS, et al. (2009) The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol. 88:655–62.
Yang H, Antoine DJ, Andersson U, Tracey KJ. (2013) The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 93:865–73.
Xu J, et al. (2014) Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 21:1229–39.
Andersson U, Tracey KJ. (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–62.
Yang H, Wang H, Czura CJ, Tracey KJ. (2005) The cytokine activity of HMGB1. J. Leukoc. Biol. 78:1–8.
Harris HE, Andersson U, Pisetsky DS. (2012) HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatology. 8:195–202.
Acknowledgments
This study was supported financially by grants through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institute, the Swedish Association against Rheumatism, the Swedish Medical Research Council, Berth von Kantzow’s Foundation, the Swedish Society of Medicine, Stiftelsen Allmänna Barnhuset and the Freemason Lodge Barnhuset in Stockholm.
Huan Yang wrote Ulf Andersson in 2015 to give permission for use of the personal communication.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1186/s10020-020-00263-2
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Palmblad, K., Schierbeck, H., Sundberg, E. et al. RETRACTED ARTICLE: High Systemic Levels of the Cytokine-Inducing HMGB1 Isoform Secreted in Severe Macrophage Activation Syndrome. Mol Med 20, 538–547 (2014). https://doi.org/10.2119/molmed.2014.00183
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00183