
Abstract
The phase changes of gypsum were considered to be sufficiently understood for a long time. However, due to the increasing importance of gypsum recalcination (connected with the planned closure of thermal power plants resulting in a lack of FGD gypsum) and new detailed research associated with it, gaps in the current knowledge have been discovered. This paper clarifies phenomena occurring during the production of gypsum binder, anhydrite, and potentially eco-friendly calcium sulfate-based ceramics at 22–1300 °C by in-situ XRD, thermal analysis, and SEM. It was found that undesirable insoluble anhydrite II arises even in the temperature range from 40 to 90 °C despite the current state-of-the-art. New knowledge about the existence of the individual phases, microstructure (cracking of crystals; fragmentation and sintering of anhydrite AII at temperatures above 700 °C when a ceramic-like material is formed), and their external consequences (changes in moisture and heat transport properties) has been gained.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Although the phase changes of calcium sulfate were considered to be sufficiently understood for a long time, this paper reveals some discrepancies and gaps in current (often old and outdated) knowledge. The importance of this research area is currently increasing because gypsum manufacturers have been worried about the future lack of calcined gypsum (CaSO4·0.5H2O) [1]—one of the most sustainable construction binders [2]. Gypsum (CaSO4·2H2O), the primary raw material for calcined gypsum production, is formed in significant quantities during the desulphurization of thermal power plants (coal plants) as a by-product and is used as a high-quality secondary raw material for the manufacturing of a wide range of new gypsum-based products, as a set regulator in Portland cement. Gypsum is also an ideal material for the incorporation of phase change materials [3]. However, in connection with the Green Deal [4] and the planned shutdown of thermal power plants, a significant decrease in the amount of FGD (flue gas desulfurization) gypsum is expected. Therefore, manufacturers have become intensely interested in using recycled gypsum binders, i.e., plaster created by repeated calcination of already hardened plaster (such as gypsum blocks or plasterboard). The reuse of gypsum (i.e., waste from construction, demolition, renovation, and chemical industry) has been studied by many researchers (described in more detail, i.e., by Li et al. [5]), but it has not been effectively used nowadays. According to many studies that have evaluated the life cycle assessment of gypsum [6, 7], gypsum recycling is effective (almost 100% of the material can be reused) and both economically and environmentally beneficial. In particular, these studies concluded that the production of recycled gypsum showed an essential reduction of the environmental impacts compared to natural gypsum production, as can be seen in Table 1, where changes in Global Warming Potential (GPW) are used to quantify environmental impacts. GPW is a metric indicating the effect of produced greenhouse gases on trapping heat in the atmosphere over time, where CO2 is a reference gas. In Table 1, the GWP of natural gypsum was chosen as the reference (100%), so values lower than 100 mean GWP reduction.
Gypsum was also investigated as a partial substitute for ordinary Portland cement. The resulting material is the eco-friendly gypsum-cement composite [8]. For example, the use of 50% recycled gypsum in cement production could save 6.78 E+07 MJ/year in the non-renewable energy category in Spain alone, and in the global warming category, savings could be 4.52 E+06 kg CO2 eq./year [7].
An important point is that the widespread use of gypsum products results in an enormous amount of waste gypsum every year worldwide (about 80 million tons [7]). The low-cost repetitive reuse of these products is possible [5, 6, 9, 12] and can lead to ecological sustainability. Since FGD gypsum has been the primary source of gypsum binder in recent decades, recycling FGD gypsum is an important step in the future. However, the recycling of gypsum products resulting in the production of calcium sulfate-based binder (gypsum plaster, CaSO4·0.5H2O; or anhydrite, CaSO4) leads to the issue of the behavior of already hardened gypsum at high temperatures. In addition, this issue is important in evaluating the material's fire properties (essential due to gypsum's use as fire protection). Prior to the experiments and discussion, a summary of the material's behavior at high temperatures according to the current state-of-the-art must be provided.
At high temperatures, gypsum dehydrates to other calcium sulfate phases [13]. Their transitions at elevated temperatures are usually described as shown in Fig. 1.

The generally accepted theory of calcium sulfate phases transitions under normal atmospheric conditions [14]
The temperature at which the gypsum binder (calcium sulfate hemihydrate) is industrially produced, between 80 and 180 °C, is often mentioned in the literature [14,15,16,17]. However, dehydration from gypsum to basanite (CaSO4·0.5H2O) starts at a much lower temperature. The dehydration temperature depends on the measurement method, the gypsum origin (natural, FGD), the type of required calcium sulfate hemihydrate (α, β), and the conditions during the experiment (pH, humidity, etc.), and this temperature significantly differs in the literature. For instance, values in Table 2 can be found.
A possible transition from gypsum (CaSO4·2H2O) directly to anhydrite II (between 42 ± 1 °C) is also discussed in the literature [25] (especially in relation to the solubility diagram). The research of this transition has a long history, which is well described by Freyer and Wolfgang in [26]—the conclusions are that this transition is estimated between 42 and 60 °C; the anhydrite solubility curve remained still uncertain, and exact in-situ techniques should be used to detect crystallizing phases in the future.
The transition from bassanite to anhydrite III (soluble anhydrite) takes place in the temperature range from 100 to 200 °C [14, 19, 23]. Anhydrite III is metastable and easily rehydrates to bassanite [27]. The structures of bassanite and soluble anhydrite III are very similar, but it was proved that they can be distinguished by XRD [15]. The difference between anhydrite III (soluble) and anhydrite II (insoluble) is considerable. More specifically, anhydrite II is stable and has a higher density. The densities of calcium sulfate-based phases, according to Wirsching [14], are in Supplementary materials (Fig. 1). Different densities of calcium sulfate phases lead to changes in sample dimensions during heating and cooling.
According to the literature, anhydrite II is formed from anhydrite III at a temperature above 181 °C [28], above 200 °C [14, 29], above 280 °C from α-hemihydrate [23], at 340 °C [22], above 225 °C [30]. Anhydrite II is commonly used for self-leveling screeds in civil engineering and is also the dominant phase in the so-called estrich gypsum. This binder is produced at high temperatures (above 700 °C [14]) and contains a small amount of free lime (CaO) that acts as a catalyst when mixed with water. The binder is slow-setting due to slow dissolution. It can be used, for example, for the production of artificial marble (scagliola) or mosaics. Its advantage is great hardness after hardening (compared to standard gypsum plaster), thanks to which it can be used even in more demanding techniques such as pietra dura (= inlay technique of using cut and fitted, highly polished colored pieces of stone to create images) [31].
The presence of anhydrite I is generally mentioned at temperatures above 1180 °C [14]. Due to its instability at temperatures below approximately 1180 °C, its properties have not yet been described.
This system of five calcium sulfate phases seems easy to understand and is often discussed in the literature, but there are still uncertainties. For example, the coexistence of individual phases is not satisfactorily described (amount of these phases during heating and after heat treatment), as well as phase transition temperatures as mentioned above. Furthermore, the reason for the decrease in strength between temperatures of 100 and 150 °C (measured on naturally cooled samples) is not fully understood [32]. Little information is known, especially about the behavior of calcium sulfate-based materials at temperatures above 500 °C and about its properties after cooling to room temperature. Recently, the sintering of CaSO4·nH2O-based materials has also become a new area of research [22, 30, 33,34,35]. The processes occurring during the secondary calcination of gypsum are not completely simple, and the literature that describes the formation of the individual phases of calcium sulfate is outdated and incomplete [15, 18, 20, 24, 25]. The motivation of this research was, therefore, to supplement existing information or to verify old information using modern methods. This research is focused on FGD gypsum, while the generally accepted information is based mainly on natural gypsum research. The processes occurring at temperatures from 22 to 1300 °C are studied, as these are essential in the production of a slow-setting binder (anhydrite) and potentially in the production of novel eco-friendly ceramics. These ceramics have already been tested, and the results are promising [34, 35]. The mineralogical changes in calcium sulfate phases (β-forms) at high temperatures are described using XRD (in situ and after heat treatment). The effect of phase composition on thermal and moisture properties is described as well, and some disagreements in testing phase changes on large samples [33] compared to small samples are explained. This article also clarifies and complements the results obtained by Vimmrová et al. [33] and answers why more anhydrite II is formed in the samples dried at 50 °C than at 100 °C—the formation of anhydrite AII at such low temperatures has not yet been described in the literature. The gained results also clarify various phenomena such as volume changes, strength fluctuations, and changes in the shape of crystals at high temperatures. These findings are essential for effective future gypsum recycling.
2 Materials and methods
The gypsum binder (Rigips, Saint-Gobain Construction Products CZ a.s.) was made of FGD gypsum, and its phase composition determined by XRD analysis was bassanite (calcium sulfate hemihydrate; 74.3% ± 0.19%), gypsum (14.3% ± 0.13%), insoluble anhydrite II (7.3% ± 0.12%), and calcite (CaCO3; 4.1% ± 0.12%). The elemental composition of the gypsum binder analyzed by X-ray fluorescence (XRF) analysis using the device Spectroscan MAKC GVII was Ca (64.2 ± 0.24), S 34.5 ± 0.24, Si (0.39 ± 0.031), Mg (0.34 ± 0.029), Al (0.28 ± 0.026), Fe (0.23 ± 0.024), and others ( < 0.1). This source of gypsum has been chosen for its relatively high content of basanite and absence of admixtures (compared to natural gypsum or other commercial products) and also for the current need for recycling products made of FGD gypsum.
Prismatic samples (40 × 40 × 160 mm) were prepared by mixing water and a gypsum binder (water/gypsum ratio was 0.6), complying with EN 13279-1. The samples were prepared according to EN 13454-2, but the mixing time was reduced to one minute (30 s manually and 30 s in an automatic mixer at a low speed of 140 rpm) due to the short setting time of the gypsum binder. Moulds with the fresh mixture were manually compacted by ten strokes on the table. The samples were unmolded within two hours and stored under laboratory conditions at 22 ± 2 °C and relative humidity of 50 ± 5% for 28 days.
2.1 Thermal analysis (TG, DSC, and TD) and XRD analysis
Thermogravimetry (TG) and differential scanning calorimetry (DSC) were performed on small pieces taken from prismatic samples and crushed into powder. The analysis was carried out using a Labsys Evo device (Setaram, France). The setup consisted of two identical crucibles made of an inert material, alumina (Al2O3), of the size of 100 mm3. A dynamic argon atmosphere of 40 mL/min was applied. Testing was performed using heating rates of 3 °C/min and 5 °C/min. Two heating rates have been chosen because some peaks could merge into one in case of an unsuitable choice of heating rate (and because of this, some phenomena may not be found). The mass of tested samples was from 26 to 46 g. Temperature intervals were up to 1500 °C. Such a high temperature was not repeated because the crucible used had been destroyed during the measurement.
Dilatometry (TD) was performed on a cylindrical sample with a length of 49 mm and a diameter of 5 mm prepared in a special mould (Fig. 10 in Supplementary materials). The NETZSCH dilatometer (DIL 402SE) with an alumina (Al2O3) holder was used. The selected temperature range was from 25 to 800 °C, and the heating rate was 10 °C/min.
The XRD was carried out in three different procedures (their scheme is provided in Figs. 11 and 12 in Supplementary materials).
-
(A)
The phase composition of the pastes was determined after heat treatment at defined temperatures. Prismatic samples (160 × 40 × 40 mm) stored for 28 days in laboratory conditions were dried at 50 °C for 24 h. Each sample was exposed to one of the following temperatures: 50, 100, 150, 200, 300, 400, 500, 600, 700, 800, and 1000 °C. Additionally, one prismatic sample was left as a reference without heating and was dried in a vacuum in the form of small pieces (22 ± 2 °C). The Ecocell 22 climatic chamber (BMT Medical Technology, Czech Republic) was used for temperatures from 50 to 200 °C, and the samples were heated for 12 h. The heating element was not in the chamber and heating was carried out using warm air. The furnace (CLASIC CZ, Czech Republic) was used for higher temperatures. The temperature was increased by 1 °C/min, and the desired temperature was maintained for 4 h. The heating was realized by a hot wire in the chamber. Infrared radiation from the wire could have particularly impacted the sample's surface. To eliminate this effect, samples for XRD analysis were taken from the middle parts of the prismatic samples. After natural cooling, samples were tested at a laboratory temperature. Small pieces were taken from the samples and ground into powder. About 2 g of the powder was used for the XRD analysis for each sample.
-
(B)
The phase composition of the pastes was determined in situ (during heating). Prismatic samples were dried at 50 °C for 24 h. Small pieces of the samples were taken, dried again at 50 °C for 24 h, and then immersed in acetone for 24 h for more effective drying. Afterward, acetone was allowed to evaporate from samples in a vacuum for 24 h before observation. During the XRD analysis, the temperature was gradually increased (10 °C/min). At selected values, the desired temperature was maintained for three hours, and the phase composition was detected. The temperature was then increased to the next value.
-
(C)
The phase composition of the pastes was determined in situ, as during procedure B. However, at selected temperatures, the temperature was maintained for a few hours or days to ensure a stable phase composition. Small pieces were taken from the sample and immersed in acetone (as in procedure B). After the acetone evaporated, the pieces were observed. The temperature was gradually increased (10 °C/min), and at the desired values, it was maintained for a few hours. During that time, the phase composition was regularly determined. When the composition stopped changing, the temperature was increased. Procedure C was added to supplement knowledge gained from procedures A and B, particularly regarding the steady-state composition at high temperatures and the gradual in situ formation of AII. The samples were not pre-dried at 50 °C, as this would have resulted in the formation of AII, as verified in procedures A and B.
The phase composition after heat treatment (procedure A) was examined using a PANalytical Aeris X-ray diffractometer equipped with a CoKα tube operating at 40 kV and 7.5 mA. For the determination of phase composition during heating (procedures B and C), the device PANalytical X´Pert Pro-MPD X-ray diffractometer with CuKα source, operating at 40 kV and 35 mA (in some measurements CoKα; 35 kV; 35 mA) was used. Powdered samples were placed on a platinum strip heater (“direct heater”) or in a corundum bowl (“environmental heater”). Direct heating can lead to a temperature deviation between the measured temperature of the sample holder and the surface temperature of the sample. This effect was eliminated in procedure C (by waiting for the stable composition). In contrast, the environmental heater transferred the heat homogeneously from all sides to the sample, but infrared (IR) radiation could additionally influence the phase composition. The atmosphere was air. Data were evaluated using Rietveld refinement with Profex software (ver. 3.12.1) [36]. The PDFs used for analysis in the Profex software are in Supplementary materials (Sect. 3, Table 1). A back calculation of Rietveld quantitative phase analysis was performed to eliminate the effect of chemically bound water release on the results. All the XRD data were normalized to grams of the initial paste.
2.2 Scanning electron microscopy
The microstructural morphology of the materials was studied by scanning electron microscopy (SEM) using a ZEISS Merlin electron microscope with a Gemini II column and a Phenom XL microscope (Thermo Fisher Scientific). Before observing, the samples were dried and immersed in acetone for 24 h for more effective drying. Afterward, acetone was allowed to evaporate from samples in a vacuum for 24 h. Samples have not been covered by any coating.
2.3 Determination of pH
The approximate pH value was determined by applying a universal indicator paper to the moistened piece of the prismatic sample. The color of the indicator paper was compared to a reference scale after 3 min and 30 min of the paper application.
2.4 Thermal conductivity coefficient λ
The thermal conductivity coefficient λ [W/(m·K)] was tested on cubic samples (70 × 70 × 70 mm) by the dynamic impulse method using the Isomet 2114 (Applied Precision). Measurement is based on the analysis of the temperature response of the tested material to heat flow impulses. More details about the measurement can be found in [37].
2.5 Moisture transport properties
The moisture transport properties (of samples dried at 50 °C and samples heated at 1000 °C) were determined on cylindrical samples with a length of 30 mm and a diameter of 120 mm by the dry cup and wet cup methods (ČSN EN ISO 12572 and ČSN EN 1015-19). The tested moisture parameters were water vapor diffusion coefficient D [m2/s], water vapor permeability coefficient δ [s], and the diffusion resistance factor μ [–].
3 Results and discussion
3.1 Thermal analysis (TG, DSC, and TD) and XRD analysis
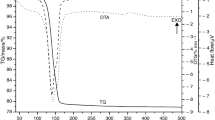
The heat flow curve of the studied GP is shown in Fig. 2. Other thermal analysis results (with additional comments) are provided in Supplementary materials in Sect. 2. The results of the XRD analysis are shown in Figs. 2, 3, 4 and 5, and in Supplementary materials (Figs. 13–18). Impurities (calcite 4.1% in particular) are purposely not included for a better comparison of the amount of the calcium sulfate phases, which are of interest. The phase composition after heat treatment (procedure A) is in Fig. 3 [33]. Figure 3a was additionally supplemented with the reference measurement (22 ± 2 °C) to make the effect of drying at 50 °C visible. The phase composition tested in situ (procedures B and C) is summarized in Fig. 3b and c. In Fig. 3c, the values on the horizontal axis show temperature/time (time measured from the beginning of the experiment).

Heat flow of the studied GP (crucibles were covered by a lid)

XRD analysis a analysis after heat treatment (procedure A); b in situ XRD analysis (procedure B, platinum strip) in times and related achieved temperatures: 2 h (50 °C), 5 h (100 °C), 8 h (150 °C), 11 h (200 °C), 16 h (300 °C), 19 h (350 °C) 22 h (400 °C) 25 h (500 °C); c in situ XRD analysis (procedure C, corundum bowl)

XRD analysis—diffractograms after heat treatment at defined temperatures (procedure A)

In situ XRD analysis—diffractograms (procedure B, platinum strip)
The existence of individual calcium sulfate phases at elevated temperatures based on the results of thermal analysis, XRD analysis and dilatometry is described below and sorted by the phases (gypsum, bassanite, anhydrite III, anhydrite II, anhydrite I) for a more explicit discussion of the results.
3.1.1 Phase occurrence
3.1.1.1 Gypsum
The dehydration (transition from gypsum through bassanite to anhydrite) was characterized by a mass loss (caused by water vapor release) and started at a temperature of approximately 80 °C according to TG, DTG, and DSC. The exact value depended on the mass of the sample. However, the XRD analysis showed that the partial dehydration started at a much lower temperature. Samples dried at 50 °C were composed of approximately 23.4% of bassanite, 9.5% of anhydrite II, and 62.3% of gypsum after procedure A. In situ XRD analysis showed even lower gypsum content at 50 °C (in the case of pre-dried samples). It is in compliance with some previous research—according to Deutsch et al. [38], dehydration to bassanite can begin even at 40 °C (during long-term heating, especially for samples prepared from synthetic gypsum). The different results between XRD and TG (or DSC) are given by the principle of these methods and their settings—faster temperature growth during TG and DSC did not allow dehydration to occur at lower temperatures.
The dehydration from gypsum further continued up to 90–100 °C (XRD). The measured data show that the temperature range of 90–100 °C is sufficient for the complete dehydration of gypsum (to bassanite or anhydrite) when the thermal exposure is long enough (as seen in Fig. 3a, c, and Section 3 in Supplementary materials). TD showed (Fig. 10 in Supplementary materials) that the heating at temperatures of approximately 40–140 °C is accompanied by thermal expansion, which is reversible and cannot be detected after cooling.
3.1.1.2 Bassanite
As aforementioned, bassanite was already formed at the temperature of 50 °C. Between 50 and 100 °C, the bassanite amount significantly increased. This transition is accompanied by a slight shrinkage, as shown on the TD curve in Supplementary materials (Fig. 10), when the gypsum crystals burst along the single cleavage planes, according to Pritzel et al. [22]. The amount of bassanite at 100 °C in procedure A was higher than this amount in procedures B and C because the soluble anhydrite III (which is formed at 100 °C) transformed back to bassanite after cooling, which is in accordance with the literature [14]. Bassanite was detected up to 600 °C [33] after heat treatment in the case of prismatic samples (procedure A). However, in the case of small samples (procedures B and C) and long time periods, the bassanite amount considerably decreased, and its total dehydration occurred at 300 °C (Fig. 14 in Supplementary materials), which is in agreement with previous studies [14, 36, 39] as well.
In the case of prismatic samples (procedure A, Fig. 3a), bassanite was detected in large quantities even after heating at temperatures over 100 °C for two reasons:
-
(1)
Samples were large—large samples heat up more slowly;
-
(2)
A large amount of bassanite was formed by the rehydration of soluble anhydrite III after cooling.
After heating above 200 °C, the gradual transformation of bassanite residues to soluble anhydrite III and its transformation to insoluble anhydrite II was observed.
3.1.1.3 Anhydrite III
The presence of soluble anhydrite III was very well observable by in situ XRD analysis (procedures B and C). Bassanite partially dehydrated to anhydrite III at 90 °C after 54 h of heating, as can be seen in Supplementary material (Fig. 13). This transition was faster at 94 °C and 100 °C. The transition of bassanite to anhydrite III is not accompanied by length changes [22, 27]. It was due to the similar structure of bassanite and anhydrite III. As can be seen in Supplementary materials (Fig. 10), the length of the sample fluctuated only slightly (± 0.1% of the original length) in the temperature range from approximately 160 to 360 °C. Figure 3c showed that soluble anhydrite III occurred at temperatures between 94 and 400 °C. After 32 h at 100 °C, more than 50% of the sample consisted of soluble anhydrite III (Fig. 14 in Supplementary materials), and only 20% consisted of bassanite. The amount of anhydrite III increased with an increasing temperature up to approximately 200 °C. Between 200 and 400 °C, most of the anhydrite III transformed to insoluble anhydrite II, and it completely converted to insoluble anhydrite II below 500 °C, respectively below 400 °C in procedure C. The transition from anhydrite III to anhydrite II was also detected as a recrystallization (exothermic) peak on the heat flow curve at a temperature of approximately 400 °C (Fig. 2), which is in accordance with [22, 38]. Dilatometry (Fig. 10 in Supplementary materials) showed a significant shrinkage during this transition (between 350 and 500 °C), which is related to the greater density of anhydrite II.
3.1.1.4 Anhydrite II
A small amount of anhydrite II was already present in the gypsum binder used. XRD analysis showed 7.3 wt% insoluble anhydrite II, as mentioned in the “Materials and methods” section. Due to its low solubility, it did not react with water and remained in the samples even after hydration (approximately 2.9 wt% of the paste). More anhydrite II appeared after drying at 50 °C (Fig. 3a). This amount was higher than at 22 °C and 100 °C. The details of what happens in the temperature range between 81 and 100 °C can be seen in Fig. 3c. It is obvious that anhydrite II already formed at temperatures between approx. 50 °C and 90 °C (before the formation of anhydrite III started). In the literature [15, 29], the phase transitions of CaSO4·nH2O phases are usually reported in order gypsum → bassanite → anhydrite III → anhydrite II → anhydrite I. Results of the experiments provided in this study revealed possible dehydration in this order: gypsum → bassanite → anhydrite II (at temperatures between approx. 50 °C and 90 °C, at which anhydrite III does not occur). This is a new discovery that has not been described before.
The amount of insoluble anhydrite II did not change at 100 °C (its amount remained between 23 and 27%), while the amount of bassanite increased. This means that soluble anhydrite III arises from the dehydration of bassanite. The insoluble anhydrite II formed again at a temperature of 200 °C and became the dominant phase at temperatures above 300 °C. Similar results were achieved by Christensen et al. [23]. The phase transformation of anhydrite III to anhydrite II, occurring between 350 and 500 °C, caused significant shrinkage, as showed by TD, because anhydrite II has a greater density.
At 500 °C, the samples measured during heating (procedures B and C) contained only insoluble anhydrite II. In the case of prismatic samples (Fig. 3a—procedure A), the transformation was not completed at 500 °C due to the sample dimensions and insufficient time for which the temperature was held. Between 500 and 1000 °C, the phase composition remained the same (samples contained anhydrite II). The positions of peaks in the diffractogram between 500 and 1000 °C did not change (Fig. 4). The only difference observed was the increase in intensities after heat treatment at 1000 °C, indicating a change in the crystallite shape and size. The shrinkage at temperatures above 500 °C observed by TD (Fig. 10 in Supplementary materials) was probably caused by the formation of cracks on the crystals—their partial disintegration and degradation [30]. Pritzel et al. [22] stated that this shrinkage is caused by sintering. However, based on the performed experiments, sintering does not occur below 700 °C.
3.1.2 Coexistence of anhydrite III, basanite, and anhydrite II
XRD analysis results showed that soluble anhydrite III, basanite, and insoluble anhydrite II coexist within a wide temperature range. Additional experiments were focused on the phase composition after rapid changes (Supplementary materials, Figs. 15 and 16)—heating from 24 to 125 °C, natural cooling (from 125 to 39 °C and from 250 to 30 °C), and reheating (to 188 °C and to 250 °C). It can be noted that only soluble anhydrite III and bassanite appeared at 125 °C. These observations are in agreement with the results described above (anhydrite II did not form in the temperature range from 90 to 200 °C). Anhydrite III is not stable, and after cooling to 39 °C, it hydrated to bassanite. Its thermal instability has already been mentioned in the literature [27, 40]. Anhydrite III should be stable during cooling until 36 °C (at 50% relative humidity), according to Ohno et al. [41]. However, based on the results reported in this study, anhydrite III is not stable either at 39 °C or at 30 °C. After reheating (188 °C; 250 °C), anhydrite III formed again. At 250 °C, anhydrite III, anhydrite II, and bassanite coexisted. Anhydrite III hydrated to bassanite after cooling to 30 °C. After reheating to 250 °C, the three phases (bassanite, anhydrite III, and anhydrite II) coexisted again.
3.1.2.1 Lime
Free lime (CaO), a solid product of the decomposition of anhydrite (CaSO4), appeared in percent units at temperatures from 700 to 900 °C and reached approximately 9.5% at 1000 °C (based on XRD results normalized to the initial paste). A small amount of free lime arising between 700 and 800 °C was a result of the thermal decomposition of calcite (CaCO3)—an impurity of nonhydrated gypsum binder, as shown in Fig. 2. In the case of the sample tested up to 1500 °C, the TG and DTG curves (Supplementary materials, Figs. 3, 5) showed a significant mass decrease caused by the decomposition of anhydrite at temperatures between 1100 and 1500 °C, when calcium oxide and sulfur oxide are products of the reaction. The occurence of free lime was also proved by the XRD analysis, as shown on diffractograms in Supplementary materials (Figs. 17, 18).
3.1.2.2 Anhydrite I
No suitable high-temperature anhydrite I model was found in the databases; therefore, the Profex software could not evaluate the presence of this anhydrite. For this reason, the phase composition was also analyzed using diffractograms provided in Fig. 5 (procedure B; copper radiation was used instead of cobalt radiation in Fig. 4). Intensities changed during in situ XRD between 800 and 1100 °C. This refers to the change of crystallites. Moreover, a peak at a diffraction angle of approximately 46 [2θ] appeared. This peak belongs to a platinum substrate, which could be detected through the sample deformed at high temperatures. Other peaks belong to anhydrite II. Supplementary materials (Figs. 17, 18) also include diffractograms gained after heat treatment at 1200 °C and 1300 °C. The diffractograms did not prove the existence of high-temperature anhydrite I.
A summary Table 3 and Fig. 6 have been added was created for a better overview of the obtained results. It shows the heating of gypsum and the formation of CaSO4·nH2O phases differently than has been published so far. The existence of calcium sulfate phases in Fig. 6 represents a stable state with samples dried in a vacuum (without pre-drying at high temperature) at atmospheric pressure. The temperature intervals on the horizontal axis are not regular because some sections would be less clear. The composition strongly depends on the heating procedure, with deviations estimated to be about 10%.

Existence of calcium sulfate phases at high temperatures
3.2 Microstructural observations (SEM)
The SEM pictures of GP samples heated at 100 °C show that dehydration causes cracks and crystal disruptions, as can be seen in Fig. 7a and b.

Crystals of GP after heating at 700 °C
Interestingly, the microstructure of GP samples does not visually change much up to a temperature of 700 °C. At 700 °C (Fig. 7c), the crystals do not have flat surfaces as observed at lower temperatures [33]—fragmentation starts (as Payraudeau et al. [30] called this phenomenon).
Fragmentation causes a change in the shape of the crystals. Clearly visible fragments locally occur at 800 °C (Fig. 8a). These fragments are not needle-shaped as the crystals of gypsum. Fragments are smooth and packed very close to each other. At temperatures above 700 °C, also sintering starts (sintering is also mentioned by Payraudeau et al. [30]). With increasing temperature, larger and thicker crystals (fragments) appear (Fig. 8b) until the material becomes compact (Fig. 8c). Sintered calcium sulfate-based material is dense, compact, and less porous. The continual change of the crystals (needle-shaped → cracked needle-shaped → fragments → fused fragments → sintered material) leads to ceramic-like material with relatively high strength, as described in [33]. The evolution of strengths has already been described, but the links between phase composition (tested in situ and after heat treatment), morphology, pH, and solubility have never been described (pH and solubility are described below). The whole GP heating process is illustrated in Fig. 8d.

Crystals of GP after heating at a 800 °C, b 1000 °C, and c 1260 °C; d dehydration, fragmentation, and sintering of gypsum paste
3.3 Relation between thermal load and pH
Results (in Fig. 9 and in Supplementary materials, section 4) showed that the pH value of the samples heated up to 400 °C was close to 6, while the pH value of the samples heated at temperatures from 500 to 1000 °C exceeded 10. The increase in pH value is affected by the formation of small quantities of CaO, which occurs during the decomposition of calcite and anhydrite.

Samples 3 min after the contact with the pH indicator
This experiment also showed different solubilities, respective reactivity of the samples. The pH indicator placed on the sample heated at 700 °C changed color first—this sample was at least partially soluble and had higher reactivity compared to other samples heated at temperatures from 500 to 1000 °C. The sample (heated at 700 °C) had a high specific surface area of crystals without smooth surfaces, according to the images taken by the scanning electron microscope. The sample heated at 800 °C reacted as the second—this sample was partially fragmented. Then solubility decreased with increasing temperatures (900 °C and 1000 °C). This was caused by the denser structure of sintered samples and by larger crystals with a smoother surface.
3.4 Thermal conductivity coefficient λ
The coefficient of thermal conductivity λ [W/(m K)] of GP samples after exposure to elevated temperatures is summarized in Fig. 10. This coefficient depends on the density, porosity, and pore size distribution of samples.

Thermal conductivity coefficient
After drying at 50 °C, the coefficient of thermal conductivity of the gypsum paste was 0.37 W/(m·K). During the dehydration to bassanite (100 °C), the total porosity [33] increased, and therefore, the coefficient of thermal conductivity decreased. The dehydration to anhydrite III (150 °C) was accompanied by a very slight increase in λ. (The structures of bassanite and anhydrite III are very similar, but anhydrite III has, for example, higher strength [33, 41]. As a result, some researchers point to an increase in strength between temperatures of 100 °C and 200 °C [42].) The transition from anhydrite III to anhydrite II (especially between 300 and 400 °C) caused an increase in λ due to the higher density of anhydrite II. Further growth in thermal conductivity can be attributed to the shrinkage of the tested material and to sintering. Sintering was a significant factor and started at temperatures above 800 °C. A comparison of sample dried at 50 °C and sintered sample after heating at 1000 °C is shown in Fig. 11. An increase in thermal conductivity (in plasterboard) at a temperature of 1150 °C was also described, for example, by Ghazi Wakili et al. [43], who mentioned an increase in contact surfaces between crystals during sintering. The gained trend of the values in Fig. 10 is similar to the results obtained by plasterboard testing and reported by other authors [44].

Comparison of 70 × 70 × 70 mm sample dried at 50 °C and heated at 1000 °C
3.5 Moisture transport properties
The values of the water vapor diffusion coefficient D [m2/s], the water vapor permeability coefficient δ [s], and the diffusion resistance factor μ [–] of GP dried at 50 °C and heated at 1000 °C are summarized in Table 4. Samples with a higher μ [–] showed a smaller D [m2/s] and δ [s]. Samples heated at 1000 °C had a lower porosity [32, 33] and a higher specific gravity due to the sintering and, thus, larger contact surfaces between crystals. Therefore, the diffusion resistance factor μ [–] of the sample heated at 1000 °C was higher, and its coefficients D [m2/s] and δ [–] were lower. As for the wet cup method, the diffusion resistance factors µ [–] were lower than in the case of the dry cup method, which is common for all materials.
4 Conclusions
The results obtained in this research point to some phenomena (especially regarding the formation of anhydrite II at temperatures up to 100 °C and the description of fragmentation and sintering and their effect on material properties after heat treatment) that were not previously investigated or were not emphasized in previous studies. The obtained results brought the following new insights:
-
Partial dehydration to anhydrite II (insoluble) takes place already at temperatures below 90 °C before the formation of soluble anhydrite III. Dehydration below 90 °C occurs in the following order: calcium sulfate dihydrate → calcium sulfate hemihydrate → insoluble anhydrite II.
-
The amount of anhydrite II does not significantly change in the temperature range from 90 to 200 °C, and the amount of anhydrite III increases. Between 200 and 400 °C, the anhydrite II amount increases again, and both these anhydrites coexist in various proportions.
-
Morphological changes at temperatures between 700 and 1100 °C (fragmentation, sintering, and changes of crystallites) are not connected with the phase changes—only anhydrite II is present in this temperature range.
The results of this research can be used by gypsum plaster manufacturers and they are essential for effective gypsum recycling in the future. The gained data are useful even for temperatures higher than 200 °C for these reasons:
-
For “pure” gypsum board recycling to bassanite, temperatures higher than 200 °C are not required. However, our data showed that heating or drying at the temperature range from 50 to 90 °C should be avoided. Otherwise, anhydrite II arises. This impurity (almost) does not react with water without an activator.
-
For gypsum boards contaminated with organic additives or admixtures, higher temperatures could help obtain a pure binder. Theoretically, the resulting product could be an anhydrite binder, which is usable for self-leveling underlayments, etc. Therefore, understanding also the behavior at higher temperatures is important.
-
The production of novel eco-friendly ceramics is another way to use recycled gypsum boards. Calcium sulfate-based ceramics have already been tested, and the results are promising [34, 35].
References
Clean energy wire, Germany’s gypsum supply threatened by coal exit—report. https://www.cleanenergywire.org/news/germanys-gypsum-supply-threatened-coal-exit-report. Accessed 9 Aug 2023
Lushnikova N, Dvorkin L (2016) 25-Sustainability of gypsum products as a construction material. In: Woodhead publishing series in civil and structural engineering, sustainability of construction materials, 2 edn. Woodhead Publishing, pp 643–681. https://doi.org/10.1016/B978-0-08-100370-1.00025-1
Li CE, Wang TY, Zhang J, Wang Y, Wang XX (2022) Energy performance of buildings with composite phase-change material wallboards in different climatic zones of China. Energy Build 273:112398. https://doi.org/10.1016/j.enbuild.2022.112398
News European Parliament, Green Deal: key to a climate-neutral and sustainable EU. https://www.europarl.europa.eu/news/en/headlines/society/20200618STO81513/green-deal-key-to-a-climate-neutral-and-sustainable-eu?&at_campaign=20234-Green&at_medium=Google_Ads&at_platform=Search&at_creation=RSA&at_goal=TR_G&at_audience=green%20deal&at_topic=Green_Deal&at_location=CZ&gclid=EAIaIQobChMIwO7Zo_vJgAMVh9_tCh1VfQ_nEAAYASAAEgI0d_D_BwE. Accessed 9 Aug 2023
Li ZX, Xu KD, Peng JH, Wang JN, Zhang JW, Li QX (2021) Study on mechanical strength and water resistance of organosilicon waterproofing agent blended recycled gypsum plaster. Case Stud Constr Mater. https://doi.org/10.1016/j.cscm.2021.e00546
Pedreno-Rojas MA, Fořt J, Černý R, Rubio-de-Hita P (2020) Life cycle assessment of natural and recycled gypsum production in the Spanish context. J Clean Prod 253:120056. https://doi.org/10.1016/j.jclepro.2020.120056
Suarez S, Roca X, Gasso S (2016) Product-specific life cycle assessment of recycled gypsum as a replacement for natural gypsum in ordinary Portland cement: application to the Spanish context. J Clean Prod 117:150–159. https://doi.org/10.1016/j.jclepro.2016.01.044
Chernyshova N, Lesovik V, Fediuk R, Timokhin R (2020) Enhancement of fresh properties and performances of the eco-friendly gypsum-cement composite (EGCC). Constr Build Mater. https://doi.org/10.1016/j.conbuildmat.2020.120462
Erbs A, Nagalli A, de Carvalho KQ, Mymrin V, Passig FH, Mazer W (2018) Properties of recycled gypsum from gypsum plasterboards and commercial gypsum throughout recycling cycles. J Clean Prod 183:1314–1322. https://doi.org/10.1016/j.jclepro.2018.02.189
Fořt H, Černý R (2018) Carbon footprint analysis of calcined gypsum production in the Czech Republic. J Clean Prod 177:795–802. https://doi.org/10.1016/j.jclepro.2018.01.002
Baran E, Czernik S, Hynowski M, Michalowski B, Piasecki M, Tomaszewska J, Michalak J (2021) Quantifying environmental burdens of plasters based on natural vs. flue gas desulfurization (FGD) gypsum. Sustainability. https://doi.org/10.3390/su13084298
Papailiopoulou N, Grigoropoulou H, Founti M (2019) Techno-economic impact assessment of recycled gypsum usage in plasterboard manufacturing. J Remanuf 9:141–167. https://doi.org/10.1007/s13243-018-0062-x
Cao WX, Yi W, Peng JH, Li J, Yin SH (2022) Recycling of phosphogypsum to prepare gypsum plaster: effect of calcination temperature. J Build Eng. https://doi.org/10.1016/j.jobe.2021.103511
Wirsching F (2000) Calcium sulfate. In: Ullmann's encyclopedia of industrial chemistry. Wiley-VCH, pp 519–550. https://doi.org/10.1002/14356007.a04_555
Schulze W (1990) Necementové malty a betony, 1st edn. SNTL, Prague
Lancia A, Prisciandaro M, Musmarra D (2012) Calcium sulphate: Kirk-Othmer encyclopedia of chemical technology, 5th edn. Wiley, New York
Dvořák K, Hájková I, Havlíčková K (2014) Monitoring the influence of dehydrating solutions for the production of alpha gypsum. Adv Mater Res. https://doi.org/10.4028/www.scientific.net/AMR.1000.51
Strydom CA, HudsonLamb DL, Potgieter JH, Dagg E (1995) The thermal dehydration of synthetic gypsum. Thermochim Acta 269:631–638. https://doi.org/10.1016/0040-6031(95)02521-9
Černý R, Rovnaníková P, Keršner AZ (2009) Vlastnosti modifikované sádry, 1st edn. Vysoké učení technické v Brně, Brno
Wirsching F (1984) Drying and agglomeration of flue gas gypsum. In: Kuntze R (ed) The chemistry and technology of gypsum. ASTM International, West Conshohocken, pp 160–174
Féjean J, Lanos C, Mélinge Y, Baux AC (2003) Behaviour of fire-proofing materials containing gypsum, modifications induced by incorporation of inert filler. Chem Eng Res Des 81:1230–1236. https://doi.org/10.1205/026387603770866434
Pritzel C, Kowald T, Sakalli Y, Trettin R, Pöllmann H (2017) Binding materials based on calcium sulphates. In: Cementitious materials: composition, properties, application. De Gruyter, Berlin, pp 285–310. https://doi.org/10.1515/9783110473728-010
Christensen AN, Olesen M, Cerenius Y, Jensen TR (2008) Formation and transformation of five different phases in the CaSO4–H2O system: crystal structure of the subhydrate β-CaSO4· 0.5H2O and soluble anhydrite CaSO4. Chem Mater 6:2124–2132. https://doi.org/10.1021/cm7027542
Šatava V (1974) Equilibrium in the system CaSO4· 2H20-CaSO4·1/2 H20–H20. J Am Ceram Soc. https://doi.org/10.1111/j.1151-2916.1974.tb11418.x
Posnjak E (1938) The system CaSO4-H2O. Am J Sci
Freyer D, Wolfgang V (2003) Crystallization and phase stability of CaSO4 and CaSO4-based salts. Monatsh Chem 134:693–719. https://doi.org/10.1007/s00706-003-0590-3
Seufert S, Hesse C, Goetz-Neunhoeffer F, Neibauer J (2009) Quantitative determination of anhydrite III from dehydrated gypsum by XRD. Cem Concr Res 39:936–941. https://doi.org/10.1016/j.cemconres.2009.06.018
Ballirano P, Mells E (2009) The thermal behaviour of γ-CaSO4. Phys Chem Miner 36:319–327. https://doi.org/10.1007/s00269-008-0280-0
Kuthandi SK (2014) Laboratory scale study of calcium sulfate hydration forms. Masters Theses & Specialist Projects. https://digitalcommons.wku.edu/cgi/viewcontent.cgi?article=2471&context=theses
Payraudeau-Le Roux N, Meille S, Chevalier J, Maire E, Adrien J (2016) In situ observation of plaster microstructure evolution during thermal loading. Fire Mater 7:973–984. https://doi.org/10.1002/fam.2357
Massinelli AM (1997) Scagliola L’arte della pietra di luna. Editalia, Rome
Krejsová J, Doleželová M, Vimmrová A (2018) Microstructure of gypsum paste at elevated temperatures. SGEM. https://doi.org/10.5593/sgem2018V/6.4/S09.059
Vimmrová A, Krejsová J, Scheinherrová L, Doleželová M, Keppert M (2020) Changes in structure and composition of gypsum paste at elevated temperatures. JTAC. https://doi.org/10.1007/s10973-020-09528-8
Mirian NM, Perotti M, Indelicato C, Magrini C, Giorgetti G, Capitani G, Viti C (2023) From high-volume industrial waste to new ceramic material: the case of red gypsum muds in the TiO2 industry. Ceram Int 10:15034–15043. https://doi.org/10.1016/j.ceramint.2023.01.086
Li L, Yan H, Hong WB, Wu SY, Che XM (2020) Dense gypsum ceramics prepared by room-temperature cold sintering with greatly improved mechanical properties. J Eur Ceram Soc 13:4689–4693. https://doi.org/10.1016/j.jeurceramsoc.2020.06.003
Döbelin N, Kleeberg R (2015) Profex: a graphical user interface for the Rietveld refinement program BGMN. J Appl Cryst. https://doi.org/10.1107/S1600576715014685
Pavlík Z (2014) Advanced computational methods and experiments in heat transfer XIII: the thermal and mechanical performance of cement-based composites with enhanced thermal insulation properties. Heat Transf 83:251–260
Deutsch Y, Nathan Y, Sarig S (1994) Thermogravimetric evaluation of the kinetics of the gypsum-hemihydrate-soluble anhydrite transitions. J Therm Anal 42:159–174. https://doi.org/10.1007/BF02546998
Sebbahi S, Chameikh MLO, Sahban F, Aride J, Benarafa L, Belkbir L (1997) Thermal behaviour of Moroccan phosphogypsum. Thermochim Acta 1–2:69–75. https://doi.org/10.1016/S0040-6031(97)00159-7
Lundgren CJ, Hesse N (2007) Phase transformation temperature in calcium sulfate hemihydrate measured by vapor sorption analysis. https://www.tainstruments.com/pdf/literature/TA341a%20%20Phase%20Trans%20Temp%20Cal%20Sulf%20Hemihydrate%20by%20VS.pdf. Accessed 15 Jul 2023
Ohno H, Nakano S, Miyakawa O, Watanabe K, Shiokawa N (1982) Effects of phase transformations of silicas and calcium sulfates on the compressive strength of gypsum-bonded investments at high temperatures. J Dent Res 9:1077–1082. https://doi.org/10.1177/00220345820610091001
Doleželová M, Scheinherrová L, Krejsová J, Vimmrová A (2018) Effect of high temperatures on gypsum-based composites. Constr Build Mater 168:82–90. https://doi.org/10.1016/j.conbuildmat.2018.02.101
Ghazi Wakili K, Koebel M, Glaettli T, Hofer M (2015) Thermal conductivity of gypsum boards beyond dehydration temperature. Fire Mater 1:85–94. https://doi.org/10.1002/fam.2234
Thong NV, Hung T (2019) Research on the parameters influencing the thermal conductivity of gypsum plasterboard under fire conditions. Cogent Eng. https://doi.org/10.1080/23311916.2019.1569796
Funding
Open access publishing supported by the National Technical Library in Prague. This work was supported by the project CTU in Prague [ SGS24/116/OHK1/3T/11] and by ERDF "NanoCent" [project No. CZ.02.1.01/0.0/0.0/15_003/0000485].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krejsová, J., Kužel, R., Keppert, M. et al. New insight into the phase changes of gypsum. Mater Struct 57, 128 (2024). https://doi.org/10.1617/s11527-024-02404-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-024-02404-z