
Abstract
Raman spectroscopy combined with confocal imaging, i.e. confocal Raman microscopy (CRM) is a relatively new technique with huge potential for high-resolution chemical mapping of phase composition and spatial distribution in cement-based materials. However, the effects of sample preparation and various operating parameters on mapping quality has not been systematically studied. This paper optimises CRM for spectral mapping of carbonated and non-carbonated cement-based materials. The effects of sample preparation and scanning parameters on the detection of four main phases (calcite, portlandite, ettringite and unreacted cement) were investigated. Results show that although freshly cut cementitious samples can be analysed as-is, the Raman signals improve with short gentle drying and surface grinding/polishing prior to analysis. Increasing laser power, exposure time and scan accumulation, and short laser wavelength yields higher signal-to-noise (SNR) ratio in the obtained spectrum. The use of a 4.15 mW laser power, 2 s exposure time and scan accumulation of 2 with 532 nm laser represents a good operating condition for Raman analysis of cement-based materials. This produces SNR > 10 for all investigated phases at short testing time and low risk of laser-induced damage. Microcracking caused by localised heating during closely-spaced mapping can be limited by impregnating the sample with epoxy to protect the microstructure. We show for the first time that CRM can be used to quantify the volume fraction of calcium carbonate and portlandite at high resolution when combined with SEM. The advantages and limitations of CRM for mapping cement-based materials are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The phase assemblage of hardened cement-based materials is highly complex, consisting of capillary pores, unreacted cement and various hydration products including amorphous calcium-silicate-hydrate (C-S-H) and minerals such as portlandite (CH), ettringite (AFt), gypsum, etc. Depending on the curing and exposure conditions, the phase assemblage evolves over time, influencing the mechanical properties and durability of concrete. When exposed to carbonation, calcium-bearing phases (particularly portlandite and C-S-H) react with atmospheric CO2 in the pore solution to form various calcium carbonate (CaCO3) polymorphs including calcite, vaterite and aragonite [1]. This leads to a pH drop in concrete, causing depassivation and corrosion initiation of steel reinforcement [2]. Carbonation is also relevant to research on developing carbon capture, utilisation and storage strategies such as accelerated CO2 curing of concrete and carbonation of waste concrete fines. Therefore, the ability to map the phase assemblage composition and characterise its spatio-temporal distribution is highly valuable for many fundamental research applications.
X-ray diffraction (XRD), thermogravimetric analysis (TGA) and infrared spectroscopy (IR) are techniques commonly used to characterise phase composition of hardened cement pastes [3,4,5,6]. However, these techniques are mainly for bulk analysis of powdered samples. They require destructive sample preparation and hence are unable to provide spatial information on the microstructural level, which is important for understanding, monitoring and predicting concrete deterioration and carbon capture. For example, knowledge on the spatial distribution of portlandite and CaCO3 enables precise profiling of carbonation in concrete. However, these techniques are impractical for analysing mortar or concrete due to the presence of aggregate particles, in particular limestone aggregates that are difficult to separate out [6].
Petrographic examination with optical microscopy has long been employed for direct characterisation of the physical and mineralogical properties of concrete [7,8,9]. Different phases can be distinguished based on their colour, morphology and optical properties under normal, fluorescent or polarised light. For example, portlandite and CaCO3 can be readily distinguished from C-S-H under polarised light based on their high birefringence [10,11,12]. This technique however requires laborious sample preparation to produce thin sections for analysis, and extensive knowledge and experience to interpret the images obtained. Achieving quantitative analysis is difficult.
Backscattered electron (BSE) microscopy is another technique widely used for examining the microstructure and constituent phases of cement-based materials. Different phases are distinguishable based on their grey levels, which increase with the mean atomic number. However, phases such as CaCO3 and hydration products share similar grey levels and hence remain difficult to detect and isolate [13]. Although the use of energy or wavelength-dispersive X-ray spectroscopy can facilitate the segmentation process to some extent, the exact type of polymorphs present cannot be determined [14, 15]. BSE also requires substantial sample preparation including drying, epoxy-impregnation, grinding and polishing.
Raman spectroscopy is a powerful technique capable of providing chemical and structural information of organic and mineral phases. It is based on the excitation of chemical bonds with a laser to produce Raman scattering. The frequency difference between the incident and scattered light, registered in the form of a spectrum, provides fingerprint information on chemical structure. Raman spectroscopy can detect oxides, carbonates, sulphates and silicates, and hence is particularly suitable for analysing cement-based materials. Since its first application on white cements by Bensted [16], Raman spectroscopy has seen increasing use in the characterisation of various cements [17], supplementary cementitious materials [18, 19], alkali-activated materials [20] and corrosion products [21,22,23,24]. Combining Raman spectroscopy with confocal microscopy enables high-resolution chemical imaging of phase distribution. The use of laser, confocal pinhole and high-power objective enables a very small sampling volume (~ 1 µm3), thus achieving high spatial resolution analysis of local features. Confocal Raman micro-spectroscopy or microscopy (CRM) has been used to image clinker remnants and aggregates in historic cement stone [25], hydration of cement pastes [26,27,28], weathering of anhydrous cements [29], alkali-silica reaction [30], and carbonation of hardened cement pastes [31, 32] and lime paste [33].
Despite the huge potential applications, there has been no systematic study published on understanding the effects of sample preparation and various operating parameters on data quality, and on optimising CRM for spectral mapping of cement-based materials. Previous studies have used different sample preparation protocols, drying and sample types including cast, cut, fractured and polished surfaces for analysis. Scan settings also varied significantly between studies, with laser wavelength ranging from 488 to 1064 nm, laser power from 0.025 to 400 mW, exposure time from 0.1 to 60 s and mapping step size from 0.2 to 150 µm. These parameters are expected to influence results, but the magnitude of the effects on different phases in cement-based materials is unclear.
The aim of this study is to understand these effects and to optimise confocal Raman microscopy for spectral mapping of cement-based materials. We focused on five main phases: unreacted cement (alite, belite), portlandite, calcite and ettringite, in naturally carbonated and non-carbonated cement paste samples. The effects of drying, sample preparation, surface roughness, and operating parameters such as laser power, exposure time, scan accumulation and mapping step size on data quality and signal–noise-ratio were examined. Microstructural changes and damage induced by prolonged laser irradiation were also investigated. Finally, some recommendations were made based on the obtained results.
2 Experimental
2.1 Materials and sample preparation
Cement pastes with a water/cement ratio (w/c) of 0.45 were prepared. Portland cement CEM I 52.5 N was used. The mineral composition of the cement was 62.4% C3S, 10.3% C2S, 10.6% C3A, 2.2% C4AF and 4.9% calcite as limestone filler, and the specific gravity and loss on ignition were 3.06 and 2.1 respectively. The cement was mixed with tap water in a Hobart mixer for 3 min and then compacted in two equal layers into a steel mould of 50 mm cube on a vibration table. The compacted samples were covered with plastic sheet and wet hessian for the first 24 h, and then demoulded and cured in a chamber at > 95% relative humidity (RH) and 21 °C for 3 days.
2.2 Conditioning and carbonation
After curing, the samples were transferred to a separate chamber for drying at 65% RH, 21 °C until mass equilibrium. The chamber contained saturated sodium nitrite to maintain the RH, soda lime to keep the CO2 level at < 10 ppm and a motorised fan to circulate air. The samples were then transferred to a CO2 incubator for natural carbonation at the same humidity and temperature (65% RH, 21 °C) for 3 months. All except one cast surface were sealed with two layers of waterproof tape to ensure unidirectional carbonation. The environment inside the incubator was continuously refreshed using an air pump to maintain the CO2 level at ~ 440 ppm. Phenolphthalein spray test confirmed that an average carbonation depth of ~ 2 mm was achieved. After carbonation, each sample was diamond-sectioned using tap water as a coolant to produce four blocks of 50 × 20 × 6 mm3. One of the blocks was surface-dried with paper towel and analysed immediately (Sect. 3.3) whereas the remaining blocks were pre-conditioned at 33% RH, 21 °C in a conditioning chamber containing saturated magnesium chloride, soda lime and motorised fan for 1 month to achieve mass equilibrium for other analyses.
2.3 Confocal Raman microscopy
2.3.1 Overview and general considerations
A Renishaw inVia Qontor confocal Raman microscope equipped with 50 mW 532 nm and 300 mW 785 nm diode lasers, and a charged-coupled device (CCD) array detector was used. For the 785 nm laser, a pinhole was inserted into the beam path to produce a circular excitation spot comparable to that of the 532 nm laser. The laser power at the sample surface was adjusted to range from 0.55 to 4.15 mW as part of optimisation (Sect. 3.2). The laser beam was focused onto the sample surface through a long working distance objective of 50 × and 0.5 NA, providing a spatial resolution of 1.3 µm for the 532 nm laser and 1.9 µm for the 785 nm laser. Prior to each scan, spectral calibration was verified using an internal silicon standard with a Raman shift at 520.2 ± 0.5 cm−1. The spectral range was set to 143 to 1385 cm−1 to ensure that the most intense peaks of the main phases of interest were captured, i.e. 1085 cm−1 for calcite, 356 cm−1 for portlandite, 988 cm−1 for ettringite, 864 cm−1 for belite and 835 cm−1 for alite [29, 34]. These phases were selected for analysis because they are the Raman-active phases involved in carbonation. All data acquisition was performed using Renishaw WiRE v.5.2 software. Spectral artefacts including cosmic rays and background were removed using the nearest neighbour method and least squares polynomial fit respectively.
2.3.2 Optimisation of scanning parameters
Spot analysis was performed on the cut surface of one of the pre-conditioned sample blocks to optimise the laser line (532, 785 nm), laser power (0.55, 0.72, 0.89, 2.48, 4.15 mW), exposure time (0.1, 0.5, 1, 2, 5, 10 s) and scan accumulation (1, 2, 3, 5, 10). It should be noted that the laser power here refers to the laser power at the sample surface (measured with Thorlabs Optical Power Meter) and not that of the laser beam. The exposure time represents the duration of each scan whereas scan accumulation represents the number of scans performed on the same spot repeatedly. These parameters influence the intensity of Raman scattering and in turn the overall spectral quality. The parameter ranges covered the values typically used in literature. While optimising the main parameter, the other parameters were kept constant at 532 nm laser line, 4.15 mW laser power, 1 s exposure time and 1 scan accumulation.
Both carbonated and non-carbonated regions were analysed. The phases of interest were located by visual examination combined with Raman spot analysis. Unreacted cement particles were located based on their defined morphology and relatively dark appearance under optical microscopy. Calcite and portlandite can also be distinguished visually as they were present in abundance in the carbonated and non-carbonated zones respectively. Five different locations were analysed for each phase and the results averaged. Signal-to-noise ratio (SNR) of the main peaks was quantified by dividing the signal level by the root mean square (RMS) value of the noise on the background signal according to ASTM E579-04 (2015) [35]. Unless otherwise stated, all other analyses were carried out using the 532 nm laser line, with 4.15 mW laser power, 2 s exposure time and scan accumulation set to 2.
2.3.3 Sample moisture condition and surface roughness
The freshly cut surface-dried sample block was analysed as-is to investigate the effect of moisture on Raman signals and to determine whether sample drying was necessary for Raman analysis. Spot analysis was performed on the phases of interest at several time intervals to monitor any spectral changes as the sample was left to air-dry naturally in the microscope enclosure at ~ 45% RH, 21 °C for up to 9 min. Following that, the sample was dried at 33% RH, 21 °C for 1 month until mass equilibrium and then reanalysed at the exact same locations to observe any differences in the collected spectra. Three different locations were analysed, and the results averaged.
The effect of surface roughness was investigated by progressively grinding and polishing the pre-conditioned sample blocks (33% RH, 21 °C) using silicon carbide (SiC) paper at 1000 grit size (grinding) and non-woven cloth containing diamond abrasives of 6, 3 and 1 µm (polishing) to produce successively finer surfaces. Grinding and polishing were performed at low pressure and slow rotational speed (50 rpm) for 1 and 3 min respectively to avoid surface relief. In another experiment, the samples were dried at 30 °C for 7 days and vacuum impregnated with epoxy resin (Struers Epofix) following the procedure described in Wong et al. [36], prior to grinding and polishing. Four regions of interest (ROI) of 80 × 60 µm2, two in the carbonated zone and two in the non-carbonated zone, were analysed at a step size of 1 µm. The surface roughness (Sa) of the ROIs was measured as the average height difference between surface irregularities and the arithmetical mean height under SEM (Sect. 2.4) using the Hitachi map 3D software (v.7.4).
2.3.4 Prolonged laser irradiation during mapping
In another series of experiments, spot analysis was performed on calcite, unreacted cement and portlandite on the pre-conditioned sample blocks (33% RH, 21 °C) for six successively larger number of scan accumulations (1, 5, 10, 100, 500, and 1000) to evaluate the effect of prolonged laser irradiation at 0.55 mW and 4.15 mW. Five different locations on the sample surface were analysed for each phase. Furthermore, point-by-point mapping was performed on both as-cut and epoxy-impregnated polished surfaces to determine if closely-spaced laser irradiation over an extended area (120 × 90 µm2) would induce microstructural changes. A systematic study involving varying laser power (0.55, 0.89, 2.48, 4.15 mW), exposure time (0.1, 0.5, 1, 2 s) and step size (0.3, 1, 5, 10, 20 µm) was performed. While examining the effect of one parameter, the others were kept constant at 2 s exposure time, 4.15 mW laser power, and step size as 1 µm. Two ROIs (each in the carbonated and non-carbonated zone) were studied and duplicate accumulation was applied throughout. Immediately after Raman imaging, the same area was observed with BSE imaging to detect microstructural changes. Microcracks that existed before and after Raman imaging were extracted by manual tracing in Fiji [37] to calculate crack density (total crack length ÷ image area).
2.4 Backscattered electron microscopy
A Hitachi TM4000 Plus scanning electron microscope (SEM) equipped with an Oxford Instruments Aztec Advanced X-Act Compact energy dispersive X-ray detector (EDS) was used for microstructural characterisation and elemental analysis. The SEM was operated at 15 kV in the backscattered electron (BSE) mode and working distance was set to 7 mm to achieve optimal spatial resolution according to Monte Carlo simulations [38]. Images were captured at magnifications of 500 × and 1000 × at a scan format of 1280 × 960 pixels to give pixel sizes of 0.2 and 0.1 µm respectively.
3 Results
3.1 Effect of laser wavelength, fluorescence background and scan accumulation
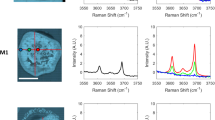
Figure 1 a-b show examples of Raman spectra for calcite (from carbonated zone), unreacted cement and portlandite obtained with 532 nm (4.15 mW) and 785 nm (4.03 mW) lasers before and after background subtraction respectively. The fluorescence background in the 532 nm spectra was markedly higher than in the 785 nm spectra (Fig. 1a). This is expected given that cement-based materials are prone to autofluorescence and that this effect is greater at short excitation wavelengths due to the higher excitation energy [39]. Nevertheless, the least squares polynomial subtraction was sufficient to remove the fluorescence background while retaining the Raman peaks of interest (Fig. 1b). The intensity of the Raman peaks for calcite, unreacted cement and portlandite were significantly higher in the 532 nm spectra. This is due to the fact that Raman scattering is inversely proportional to the fourth power of the excitation wavelength [40].

Example Raman spectra of calcite (from carbonated zone), unreacted cement and portlandite acquired with 532 and 758 nm lasers before (a) and after (b) background removal. Asterisks mark the photoluminescence bands
Another disadvantage of long excitation wavelength (785 nm) is the introduction of broad intense photoluminescence bands (asterisked in Fig. 1b) caused by the presence of trace rare earth impurities in the cement mineral lattice [41]. Such bands are intrinsic and cannot be corrected by background subtraction. Furthermore, these photoluminescence bands can overlap with certain phases of interest including alite, belite and calcite, obscuring their detectability. Similar observations were reported by Richardson et al. [39], Dyer et al. [42] and Bonen et al. [43] at higher laser wavelength of 1064 nm. This study shows that the use of 532 nm laser can avoid the formation of such photoluminescence bands and is thus deemed a better option for measuring cement-based materials. It is noted that previous studies have employed this wavelength for studying alkali-silica reaction [44] hydration kinetics [45], and for quantifying anhydrous cement [46].
Figure 2a shows the calculated SNR of the Raman peaks for calcite (1085 cm−1), alite + belite (835 to 864 cm−1), and portlandite (356 cm−1) after one scan accumulation. It can be seen that the SNR for all phases were higher when measured with the 532 nm laser compared to the 785 nm laser. Calcite gave the highest SNR, followed by portlandite, and unreacted cement (alite, belite), in line with their order of crystallinity [28]. It is also worth noting that the spectrum and SNR for calcite are similar regardless of whether the analysis was conducted in the carbonated or non-carbonated regions (from limestone filler in the cement).

Effect of laser wavelength on the signal-to-noise ratio (SNR) of calcite (from carbonation and limestone filler), unreacted cement and portlandite peaks after single scan (a) and multiple scan (b) accumulation. Analysis was carried out at 4.15 mW laser power and 1 s exposure time. Dotted lines show best-fit power law relations (R2 > 0.96). Error bars represent standard deviation
Figure 2b shows the effect of scan accumulation on the SNR of the Raman peaks. Irrespective of the laser line and the measured phase, SNR increased with increase in scan accumulation following a power law trend. An accumulation of 10 scans was observed to improve the SNR by a factor of 2–4 compared to the SNR of one scan. However, the acquisition time was also significantly prolonged and this makes large area mapping impractical. Nevertheless, it is noted that an accumulation of 2 scans with 532 nm laser was sufficient to achieve an SNR > 10 for all phases. This is adequate for spectral mapping subtraction (e.g. AFt and CH peaks in Fig. 4) and therefore deemed as a good balance for achieving high SNR at relatively short testing time.
3.2 Effect of laser power and exposure time
Figure 3 shows the effect of laser power and exposure time on the SNR of the Raman peaks. It can be seen that the SNR increased with exposure time and laser power, again following a power law trend (R2 > 0.95) regardless of the laser wavelength and analysed phase. Increasing the laser power was found to be more effective in improving SNR than increasing the exposure time. For example, a combination of an exposure time of 2 s and a laser power of 4.15 mW would provide an SNR comparable to that obtained with an exposure time of 10 s and a laser power of 2.48 mW, but with 80% less acquisition time. It is worth noting that the implementation of high laser power (10–20 mW) and long exposure time (20–60 s) in previous studies (e.g. [44, 47].

Effect of laser power and exposure time on the SNR of the main Raman peaks of calcite (from carbonation and limestone filler), alite + belite, portlandite and ettringite. Analysis was carried out with 532 nm laser at scan accumulation of 1. Dotted lines show best-fit power law relations (R2 > 0.95). Error bars represent standard deviation
The SNR for ettringite (988 cm−1) was much lower than for other phases (Fig. 3d) due to its presence as a minor phase intermixed with calcite (Fig. 4a). Ettringite is also known to dehydrate and lose part of its chemically combined water above 100 °C [48, 49]. Here, the SNR for ettringite was observed to increase with laser power and exposure time. This suggests that the largest applied laser power (4.15 mW) and exposure time (10 s) were safe and did not produce sufficient heat to cause degradation of ettringite. The effect of excessive laser exposure leading to microstructural changes and phase decomposition will be examined further in Sect. 3.4.

Effect of exposure time on the quality of Raman spectra for calcite (from carbonated zone) (a) and unreacted cement (from non-carbonated zone) (b). The SNR of the weak peaks of AFt and CH are shown. Analysis was carried out with 532 nm laser, 4.15 mW laser power and scan accumulation of 1
Figure 4 shows a series of spectra for calcite and unreacted cement from carbonated and non-carbonated zone respectively at increasing exposure time and a constant laser power of 4.15 mW. It can be clearly seen that a longer exposure time improves detectability of not only the target phases, but also the minor intermixed phases such as ettringite (AFt) and portlandite (CH). At short exposure time below 2 s, the background signal increases dramatically, and the detectability of weaker peaks diminishes.
3.3 Effect of surface moisture and roughness
Figure 5 shows the evolution of the main Raman peaks for calcite, alite + belite and portlandite with ambient drying of the freshly-cut surface at 45% RH and 33% RH, 21 °C. It can be seen that peak intensities for all phases increased with drying. This suggests that although the sample surface was towel-dried prior to analysis, there was still a small amount of residual free water present in the pore space obscuring the detection of the solid phases. Detectability of the solid phases improved once the residual surface water was removed, and the Raman spectra gradually converged towards a stable form. Figure 5d shows the evolution of the SNR of the Raman peaks as a function of drying time. The SNR improved significantly during the first 1 min of drying at 45% RH, 21 °C and stabilised after ~ 4 min. After ~ 9 min, the SNR is very similar to those obtained after long-term drying (1 month) at 33% RH, 21 °C (empty symbols) to mass equilibrium.

Evolution of the main Raman peaks (a-c) and SNR (d) of calcite, alite + belite and portlandite with ambient drying at ~ 45% RH, 21 °C (for less than 9 min) from a freshly cut surface and at 33% RH, 21 °C until mass equilibrium (for 4 weeks). Analysis was carried out with 532 nm laser, 4.15 mW laser power, 2 s exposure time and scan accumulation of 2
Figure 6 shows the effect of surface roughness on the Raman maps of alite + belite and calcite acquired from the carbonated zone. Note that the sample was dried at 33% RH, 21 °C and epoxy-impregnated prior to grinding and polishing. The measured surface roughness (Sa, mean surface height from SEM) decreased from 0.41 μm after SiC grinding at 1000 grit to 0.13 μm after diamond polishing at 1 μm showing a progressively finer surface. The Raman maps were produced based on the intensity of the main Raman peaks (1085 cm−1 for calcite, 835–864 cm−1 for alite + belite). Blue pixels indicate low intensity whereas red pixels indicate high intensity areas corresponding to the phase of interest. The same colour scales were used for different surface roughness for ease of comparison.

Colour-coded Raman maps of alite + belite and calcite from the carbonated zone at different surface roughness after grinding and polishing. Colour scale bar indicates peak intensities (a.u.). Optical and BSE images show the microstructure of the matching area after polishing. Analysis was carried out with 532 nm laser, 4.15 mW laser power, 2 s exposure time, scan accumulation of 2 and 1 μm step size. AH: unreacted cement; HP: hydration product
Figure 7 shows the average map intensity of alite + belite and calcite in the carbonated (A & B) and non-carbonated (C & D) regions plotted against surface roughness (Sa). The results were normalised to those of the SiC ground surface. Clearly, the Raman intensity improved with decreasing surface roughness, especially after diamond polishing. Results show that decreasing surface roughness from ~ 0.4 to 0.1 µm improved Raman peak intensities by 5 to 30%. This is because less interference from surface irregularities occur and thus more Raman scattering events are detectable [50]. As such, the spatial resolution of the maps also improved with decreasing surface roughness, enabling the detection of finer features.

Effect of surface roughness (indicated by the measured mean surface height Sa from SEM) on the average map intensity of alite + belite and/or calcite at carbonated (A & B) and non-carbonated (C & D) locations
3.4 Effect of prolonged laser irradiation during mapping
Figure 8 plots the SNR, normalised to the first scan for comparison, against scan accumulation. The results showed large fluctuations at 0.55 mW laser power, presumably because the weak excitation produced insufficient signals and caused inconsistencies in the Raman spectra. The SNR at 4.15 mW laser power were more consistent, with variations no larger than 3.5%. Close inspection of the Raman spectra confirmed no occurrence of peak shift or formation of new peaks, indicating that the molecular structure and chemical composition of the phases remained unchanged.

Evolution of SNR of the main Raman peaks for calcite (from carbonation and limestone filler), alite + belite and portlandite as a function of scan accumulation by spot analysis, at 0.55 mW (filled symbol) and 4.15 mW (empty symbol) laser power. Analysis was carried out with 532 nm laser and 2 s exposure time. Error bars represent standard deviation
Figure 9 shows that extensive microcracking and pore coarsening appeared after Raman mapping, especially for the non-epoxy impregnated sample. The microcracks were generally < 1 μm wide, < 100 μm in length, and randomly distributed. These characteristics are reminiscent of microcracks induced by drying shrinkage [51, 52]. The crack density of the epoxy-impregnated sample after Raman mapping at 0.3 µm step size was 73% lower than the non-impregnated sample mapped at 1 µm step size.

Area-matching BSE images showing the appearance of microcracks on the as-cut (non-epoxy impregnated) surface and impregnated/polished surface before and after Raman mapping at step sizes of 1 and 0.3 µm. Both locations were from the carbonated zone. Microcracks were extracted for quantification via image analysis
Figure 10 shows that the crack density increased with increase in laser power and exposure time, and with smaller step size. Therefore, extended Raman mapping on a small area (120 × 90 µm2) can cause microstructural damage due to concentrated laser-induced heating. Carbonated areas were more susceptible to microcracking. This is perhaps unsurprising given that carbonation causes additional shrinkage, the effect of which is exacerbated by exposure to laser. However, epoxy-impregnation is very effective in protecting the microstructure. For example, Fig. 10c shows that crack density in the carbonated epoxy-impregnated sample was 95% lower than that of the non-impregnated sample at 1 µm step size. Microcracking in the carbonated epoxy-impregnated sample remained significantly lower than the non-impregnated sample even when the step size decreased to 0.3 µm.

Crack density observed in carbonated and non-carbonated areas (120 × 90 mm2) subjected to varying laser power (a), exposure time (b) and step size (c) with 532 nm laser and scan accumulation of 2
It is interesting to note that laser power as high as 20–400 mW has been used in previous studies, but the potential damaging effects have not been investigated [27, 53, 54]. Therefore, a further experiment was performed at 37 mW, which is the highest achievable laser power in the current setup, on the same epoxy-impregnated sample. The exposure time, scan accumulation and step size were set to 2 s, 2 and 1 µm respectively to scan an area of 256 × 192 µm2. The obtained Raman maps (not shown) were oversaturated with signals, rendering the maps unusable. A more striking observation however, was that the microstructure of the cement paste was significantly altered with formation of many large interconnected cavities, especially in the originally porous regions (Fig. 11a, b). A similar observation was made on pure hardened epoxy (Fig. 11c–d). This suggests that the severe damage in the cement paste was partly due to softening of the epoxy, which has a glass transition temperature of < 170 °C [55]. It should be noted that cement hydrates, especially C-S-H and ettringite, will undergo dehydroxylation at such temperature.

Area-matching BSE images showing epoxy-impregnated cement paste in the carbonated zone (a, b) and plain epoxy (c, d) before and after Raman mapping with high laser power of 37 mW (2 s exposure time, accumulation scan of 2 and 1 µm step size)
3.5 Comparison between BSE images and confocal Raman maps
Figure 12 compares BSE images and Raman maps acquired from carbonated and non-carbonated regions of the same epoxy-impregnated polished sample after carbonation. The Raman maps were acquired with 532 nm laser at 4.15 mW, 2 s exposure time and a step size of 0.3 µm over an area of 160 × 110 µm2 to be comparable with the BSE images. Both regions contained a large unreacted cement particle surrounded by a porous hydration product matrix (Fig. 12a, b). In the BSE images, hardly any differences can be detected between the non-carbonated and carbonated regions. With CRM however, the microstructures are clearly distinct between these two regions.

Comparison between BSE images (a, b), Raman maps of portlandite (c, d) and calcite (e, f) in carbonated and non-carbonated regions of the same sample after carbonation. Asterisks in (c, f) mark the spots analysed by EDS. Arrows mark the presence of portlandite (a) and calcite (e) in the non-carbonated region. Figures (g, h) show the reconstructed maps highlighting the segmented portlandite (green) and calcite (red). (532 nm laser, 4.15 mW laser power, 2 s exposure time and 0.3 μm step size)
In the non-carbonated region, CRM showed a substantial amount of portlandite surrounding the unreacted cement particles (Fig. 12c). On close inspection, some of the large portlandite precipitates were also visible in the BSE image (arrowed in Fig. 12a), with spatial distribution that matched the Raman map. This is further corroborated by EDS spot analysis showing a high level of Ca, but low levels of Si and C. The corresponding calcite map (Fig. 12e) shows two distinct spots with high Raman intensities (arrowed), which are remnants of limestone filler from the CEM I cement. The other areas show negligible calcite, confirming that the region was non-carbonated.
In contrast, the carbonated region showed no portlandite (Fig. 12d) suggesting that the phase had reacted completely. This is confirmed by the corresponding calcite map (Fig. 12f) showing widespread distribution throughout the outer hydration product matrix. The calcite precipitates are microscopic, inter-mixed with other hydration products and not readily detectable by BSE microscopy. However, EDS spot analysis shows elevated levels of Ca, C and O, which corroborates with Raman microscopy. It is also worth noting that the unreacted cement particle remained non-carbonated.
The BSE images and Raman maps were further analysed and combined to construct composite images showing the spatial distribution of the segmented phases (Fig. 12g, h). Unreacted cement and capillary pores were extracted from the BSE images using the Overflow method [56], whereas calcite and portlandite were extracted from the Raman maps by manual thresholding. Results show that the non-carbonated region had 7.9% portlandite and 0.1% calcite, while the carbonated region had 0% portlandite and 8.9% calcite. These findings demonstrate the potential of Raman microscopy to provide quantitative information (concentration and spatial distribution) on phases otherwise difficult to measure with BSE microscopy.
The spectra from all pixels of the Raman maps (Fig. 12) were summed and are presented in Fig. 13. The main identifiable Raman peaks are those of calcite, alite, belite and portlandite. As expected, the calcite peaks were significantly stronger in the carbonated region. However, neither aragonite nor vaterite were detected. It is known that these polymorphs are metastable and they form mainly from the carbonation of C-S-H under high CO2 levels [1]. The Raman band of C-S-H (660 to 680 cm−1) [29, 34] in the carbonated region was similar to that in the non-carbonated region, indicating that the C-S-H was not carbonated. A similar observation was made for alite + belite. Portlandite appeared to be the only phase to have undergone carbonation, as indicated by the absence of its Raman peaks in the carbonated region.

Comparison of the summed Raman spectra between non-carbonated and carbonated areas, obtained from the Raman maps shown in Fig. 12
4 Discussion
Results show that samples can be analysed as-is with Raman microscopy without requiring extensive drying. This makes the technique particularly suitable for analysis of samples that are sensitive to drying-induced changes or damage. For point-by-point mapping, however, the sample surface may carbonate during the scanning process due to the long acquisition time required. Our recent study showed that surface carbonation of hardened cementitious materials can take place within an hour of exposure to ambient CO2 level [57]. To prevent this from happening, it is recommended to keep the CO2 level in the microscope enclosure to ≤ 10 ppm by using soda lime.
This study optimises CRM for spectral mapping of cement-based materials focusing on calcite (from limestone filler or carbonation), portlandite, ettringite and unreacted cement. Calcite was selected because it is the most common CaCO3 polymorph to form in natural carbonation. However, other crystalline polymorphs involving aragonite and vaterite may also form during C-S-H carbonation [1]. These polymorphs are metastable and act as precursors to the precipitation of more stable calcite [58, 59]. In addition, amorphous calcium carbonate (ACC) and monohydrocalcite are two other possible CaCO3 products. ACC is known to form at early carbonation prior to crystalline CaCO3 [60] whereas monohydrocalcite can form in a high relative humidity (75–90%) environment [61]. All these CaCO3 polymorphs were not observed in this study, but they are readily detectable by CRM [60, 62, 63] and hence can be included for analysis if necessary.
Optimisation shows that the 532 nm laser is a better option than the 785 nm laser for testing cement-based materials because it can yield higher SNR in the collected spectrum at the same parametric input, and can avoid photoluminescence bands. Another option is the 633 nm laser, which was not tested in this study but shown in an earlier work to be sufficient in avoiding photoluminescence [39]. The SNR increases with increase in laser power, exposure time and accumulation scan following a power law. However, this comes at a cost of longer analysis time and greater risk of specimen damage induced by laser exposure. The results in Fig. 4 indicate that a SNR of at least 10 is adequate for retaining the weaker peaks during background subtraction. Guided by this, our optimisation work suggests that a laser power of 4.15 mW, exposure time of 2 s and scan accumulation of 2 is a good operating condition for Raman analysis of cement-based materials.
Prolonged laser irradiation during Raman mapping can induce localised heating, drying shrinkage and severe microcracking. To mitigate this, it is recommended to use a mapping step size of at least 20 µm, which is adequate for large-scale mapping (e.g. to determine carbonation depth). For high spatial resolution mapping (e.g. to resolve cement hydrates), a smaller step size of ~ 1 µm would be necessary. For such applications, it is recommended that the sample is epoxy-impregnated to protect the microstructure and flat polished to minimise signal interference from surface irregularities. Furthermore, the laser power should be kept as low possible. In this study, 4.15 mW was found to be sufficient to yield satisfactory Raman signals without causing degradation of moisture-sensitive phases such as ettringite.
This study demonstrates that Raman microscopy can be used to characterise the spatial distribution of CaCO3 in non-carbonated (limestone filler) and carbonated cement paste at the microstructural level. When combined with image analysis, precise quantification and profiling of carbonation can be performed. This is especially important for natural carbonation, where the precipitation of CaCO3 is a slow process and difficult to measure with conventional bulk analysis techniques such as XRD, TGA and IR at early stages of carbonation. Despite the fact that Raman imaging can be time consuming, mapping an area of 160 × 110 µm2 at a step size of ~ 1 µm takes 19 h with the optimised operating conditions. Further work on applying CRM for high-resolution quantification of carbonation following short-term natural exposure is ongoing and will be presented in a follow-up publication.
It is worth pointing out that some Raman microscopes such as the one used in this study have the capability to perform fast mapping by line scanning [31, 64], where the sample is illuminated with a line of laser, rather than an intense spot of laser. This enables large-area mapping in a shorter time without inducing damage. However, this function is usually proprietary and limited to specific instruments only. Moreover, the signals obtained tend to be weaker than those attainable with spot analysis especially for less Raman-active or low content phases. Nevertheless, line scanning remains useful for highly Raman-active phases such as CaCO3 polymorphs, which do not require intense laser power [31].
It is envisaged that the findings from this study can be applied to other minor phases in cementitious materials including C3A, C4AF, gypsum and monosulphate. Our ongoing research shows that the recommended settings are applicable to systems containing supplementary cementitious materials such as PFA and GGBS, in addition to CEM I. Furthermore, quartz is Raman active and has a fingerprint peak at 465 cm−1. The recommended settings are applicable to mortars containing siliceous aggregates, even though the quartz fines are sometimes mixed with other crystalline minerals such as calcite, feldspar and mica. In any case, the presence of these phases is not expected to interfere with the detectability of cement hydrates and clinker minerals in the cement paste matrix. For mortars and concretes containing high aggregate volume fraction however, a small mapping step size should be used corresponding to the thinner cement paste regions between aggregate particles. We endeavour to present these results in subsequent publications.
5 Conclusion
This study optimises confocal Raman microscopy (CRM) for spectral mapping of cement-based materials. The effect of sample moisture and surface roughness, and scanning parameters including laser wavelength (532, 785 nm), laser power (0.55, 0.72, 0.89, 2.48, 4.15 mW), exposure time (0.1, 0.5, 1, 2, 5, 10 s), scan accumulation (1, 2, 3, 5, 10) and mapping step size (0.3, 1, 5, 10, 20 µm) were investigated. The investigation was carried out in the naturally carbonated and non-carbonated regions of w/c 0.45 cement pastes, focusing on four main phases: calcite (from limestone filler or carbonation), portlandite, ettringite and unreacted cement. The effects of prolonged laser irradiation and mapping step size were also studied. The key findings are:
-
a)
The use of short laser wavelength (532 nm) is advantageous over 785 nm for cement-based materials as it yields higher signal-to-noise ratio (SNR) in the collected spectrum. Longer laser wavelength induces broad photoluminescence bands that are intrinsic and cannot be corrected by background correction. The photoluminescence bands obscure the peaks of certain phases of interest such as calcite and unreacted cement particles.
-
b)
The SNR of the collected spectrum increases with increase in laser power, exposure time and accumulation scan following a power law trend (R2 > 0.95). Our optimisation work shows that a 4.15 mW laser power, 2 s exposure time and scan accumulation of 2 with a 532 nm laser is a good operating condition for Raman analysis of cement-based materials. This yields an SNR of > 10 for all investigated phases at a relatively short testing time and low risk of laser-induced damage.
-
c)
Freshly cut cement-based specimens can be analysed as-is without drying, however the presence of residual surface moisture can obscure phase detection. This problem is mitigated by short gentle drying prior to Raman analysis. For example, drying at 45% RH, 21 °C for several minutes improves SNR significantly, achieving values that are equivalent to those obtained after long-term drying (1 month) at 33% RH.
-
d)
Surface grinding and polishing prior to analysis increases peak intensities, improves spatial resolution of Raman maps and enables detection of finer features of cement-based materials. For example, decreasing surface roughness from ~ 0.4 µm (after SiC grinding at 1000 grit) to 0.1 µm (after diamond polishing) improves Raman intensities by 5 to 30% due to the decreased interference from surface irregularities.
-
e)
Repeated spot laser irradiation up to 1000 accumulations at 4.5 mW did not cause detectable Raman peak shifts or formation of new peaks in cement-based materials, suggesting no changes to the chemical structure or composition of the analysed phases. However, closely spaced laser irradiation (< 20 µm step size) during Raman mapping can cause extensive microcracking in the hardened cement paste due to localised heating, especially for carbonated systems. This effect can be mitigated by using a step size > 20 µm or by epoxy impregnating the specimen prior to mapping.
-
f)
Confocal Raman microscopy has huge potential for high-resolution chemical mapping to characterise the composition and spatial distribution of phases in cement-based materials for various applications. For example, the spatial distribution of unreacted cement (alite, belite), portlandite and calcite can be mapped at micron resolution. The obtained maps can be further processed by image analysis to extract quantitative information to complement results from BSE microscopy.
References
Morandeau A, Thiéry M, Dangla P (2014) Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem Concr Res 56:153–170. https://doi.org/10.1016/j.cemconres.2013.11.015
Bentur A, Berke N, & Diamond S (1997). Steel corrosion in concrete: fundamentals and civil engineering practi. E&FN Spon. https://www.routledge.com/Steel-Corrosion-in-Concrete-Fundamentals-and-civil-engineering-practice/Bentur-Berke-Diamond/p/book/9780367863715
Chang CF, Chen JW (2006) The experimental investigation of concrete carbonation depth. Cem Concr Res 36(9):1760–1767. https://doi.org/10.1016/j.cemconres.2004.07.025
Matsushita F, Aono Y, Shibata S (2000) Carbonation degree of autoclaved aerated concrete. Cem Concr Res 30(11):1741–1745. https://doi.org/10.1016/S0008-8846(00)00424-5
Shah V, Scrivener K, Bhattacharjee B, Bishnoi S (2018) Changes in microstructure characteristics of cement paste on carbonation. Cem Concr Res 109(April):184–197. https://doi.org/10.1016/j.cemconres.2018.04.016
Villain G, Thiery M, Platret G (2007) Measurement methods of carbonation profiles in concrete: thermogravimetry, chemical analysis and gammadensimetry. Cem Concr Res 37(8):1182–1192. https://doi.org/10.1016/j.cemconres.2007.04.015
Poole AB, Sims I (2016) Concrete petrography: a handbook of investigative techniques, 2nd edn. CRC Press, London
Walker HN, Lane DS, & Stutzman PE (2006). Petrographic methods of examining hardened concrete: a petrographic manual. Revised 2004. https://doi.org/10.21949/1404099
Wong HS, Poole AB, Wells B, Eden M, Barnes R, Ferrari J, Fox R, Yio MHN, Copuroglu O, Guðmundsson G, Hardie R, Jakobsen UH, Makoubi K, Mitchinson A, Raybould P, Strongman J, Buenfeld NR (2020) Microscopy techniques for determining water–cement (w/c) ratio in hardened concrete: a round-robin assessment. Mater Struct 53(25):1–19. https://doi.org/10.1617/S11527-020-1458-2/TABLES/5
Çopuroğlu O (2016) Revealing the dark side of portlandite clusters in cement paste by circular polarization microscopy. Materials. https://doi.org/10.3390/ma9030176
Erlin B, & Stark D (1990). Petrography applied to concrete and concrete aggregates–STP 1061. In: Cement and concrete composites (Vol. 13, Issue 1). https://doi.org/10.1016/0958-9465(91)90048-m
Sahu S, Beyene M, & Meininger R (2019). Characterization of carbonated calcium silicate cement-based concrete. 17th EMABM, 214–219.
Stutzman P (2004) Scanning electron microscopy imaging of hydraulic cement microstructure. Cement Concr Compos 26(8):957–966. https://doi.org/10.1016/j.cemconcomp.2004.02.043
Branch JL, Kosson DS, Garrabrants AC, He PJ (2016) The impact of carbonation on the microstructure and solubility of major constituents in microconcrete materials with varying alkalinities due to fly ash replacement of ordinary Portland cement. Cem Concr Res 89:297–309. https://doi.org/10.1016/j.cemconres.2016.08.019
Kashef-Haghighi S, Ghoshal S (2013) Physico-chemical processes limiting CO2 uptake in concrete during accelerated carbonation curing. Ind Eng Chem Res 52(16):5529–5537. https://doi.org/10.1021/ie303275e
Bensted J (1976) Uses of Raman spectroscopy in cement chemistry. J Am Ceram Soc 59(3–4):140–143. https://doi.org/10.1111/j.1151-2916.1976.tb09451.x
Torréns-Martín D, Fernández-Carrasco L, Martínez-Ramírez S, Ibáñez J, Artús L, Matschei T (2013) Raman spectroscopy of anhydrous and hydrated calcium aluminates and sulfoaluminates. J Am Ceram Soc 96(11):3589–3595. https://doi.org/10.1111/jace.12535
Guedes A, Valentim B, Prieto AC, Sanz A, Flores D, Noronha F (2008) Characterization of fly ash from a power plant and surroundings by micro-Raman spectroscopy. Int J Coal Geol 73(3–4):359–370. https://doi.org/10.1016/J.COAL.2007.09.001
Yue Y, Wang JJ, Bai Y (2018) Tracing the status of silica fume in cementitious materials with Raman microscope. Constr Build Mater 159:610–616. https://doi.org/10.1016/J.CONBUILDMAT.2017.11.015
Caggiani MC, Coccato A, Barone G, Finocchiaro C, Fugazzotto M, Lanzafame G, Occhipinti R, Stroscio A, Mazzoleni P (2021) Raman spectroscopy potentiality in the study of geopolymers reaction degree. J Raman Spectrosc. https://doi.org/10.1002/JRS.6167
Criado M, Martínez-Ramirez S, Bastidas JM (2015) A Raman spectroscopy study of steel corrosion products in activated fly ash mortar containing chlorides. Constr Build Mater 96:383–390. https://doi.org/10.1016/J.CONBUILDMAT.2015.08.034
Dang VH, François R, L’Hostis V, Meinel D (2015) Propagation of corrosion in pre-cracked carbonated reinforced mortar. Mater Struct 48:2575–2595. https://doi.org/10.1617/S11527-014-0338-Z/FIGURES/18
Larroumet D, Greenfield D, Akid R, Yarwood J (2007) Raman spectroscopic studies of the corrosion of model iron electrodes in sodium chloride solution. J Raman Spectrosc 38(12):1577–1585. https://doi.org/10.1002/JRS.1810
Marcotte TD, Hansson CM (2007) Corrosion products that form on steel within cement paste. Mater Struct 40:325–340. https://doi.org/10.1617/S11527-006-9170-4
Schmid T, Dariz P (2013) Determination and imaging of binder remnants and aggregates in historic cement stone by Raman microscopy. J Raman Spectrosc 44(6):882–891. https://doi.org/10.1002/JRS.4296
Higl J, Köhler M, Lindén M (2016) Confocal Raman microscopy as a non-destructive tool to study microstructure of hydrating cementitious materials. Cem Concr Res 88:136–143. https://doi.org/10.1016/J.CEMCONRES.2016.07.005
Mikhailova O, del Campo A, Rovnanik P, Fernández JF, Torres-Carrasco M (2019) In situ characterization of main reaction products in alkali-activated slag materials by confocal Raman microscopy. Cement Concr Compos 99:32–39. https://doi.org/10.1016/J.CEMCONCOMP.2019.02.004
Torres-Carrasco M, Campo A, Rubia MA, Reyes E, Moragues A, Fernández JF (2019) In situ full view of the Portland cement hydration by confocal Raman microscopy. J Raman Spectrosc 50(5):720–730. https://doi.org/10.1002/jrs.5574
Torres-Carrasco M, del Campo A, de la Rubia MA, Reyes E, Moragues A, Fernández JF (2017) New insights in weathering analysis of anhydrous cements by using high spectral and spatial resolution confocal Raman microscopy. Cem Concr Res 100:119–128. https://doi.org/10.1016/j.cemconres.2017.06.003
Ling TC, Balachandran C, Munoz JF, Youtcheff J (2018) Chemical evolution of alkali–silicate reaction (ASR) products: a Raman spectroscopic investigation. Mater Struct. https://doi.org/10.1617/S11527-018-1151-X/FIGURES/9
Corvisier J, Brunet F, Fabbri A, Bernard S, Findling N, Rimmelé G, Barlet-Gouédard V, Beyssac O, Goffé B (2010) Raman mapping and numerical simulation of calcium carbonates distribution in experimentally carbonated Portland-cement cores. Eur J Mineral 22(1):63–74. https://doi.org/10.1127/0935-1221/2010/0022-1977
Mi T, Li Y, Liu W, Li W, Long W, Dong Z, Gong Q, Xing F, Wang Y (2021) Quantitative evaluation of cement paste carbonation using Raman spectroscopy. Npj Mater Degrad 5(35):1–7. https://doi.org/10.1038/s41529-021-00181-6
Ševčík R, Mácová P, Sotiriadis K, Pérez-Estébanez M, Viani A, Šašek P (2016) Micro-Raman spectroscopy investigation of the carbonation reaction in a lime paste produced with a traditional technology. J Raman Spectrosc 47(12):1452–1457. https://doi.org/10.1002/jrs.4929
Liu F, Sun Z, Qi C (2015) Raman spectroscopy study on the hydration behaviors of portland cement pastes during setting. J Mater Civ Eng 27(8):1–10. https://doi.org/10.1061/(ASCE)MT.1943-5533.0001189
ASTM E579–04. (2015). Standard test method for limit of detection of fluorescence of quinine sulfate in solution. https://compass.astm.org/EDIT/html_annot.cgi?E579+04(2015)
Wong HS, Zobel M, Buenfeld NR, Zimmerman RW (2009) Influence of the interfacial transition zone and microcracking on the diffusivity, permeability and sorptivity of cement-based materials after drying. Mag Concr Res 61(8):571–589. https://doi.org/10.1680/macr.2008.61.8.571
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682. https://doi.org/10.1038/nmeth.2019
Yio MHN, Wong HS, Buenfeld NR (2016) 3D Monte Carlo simulation of backscattered electron signal variation across pore-solid boundaries in cement-based materials. Cem Concr Res 89:320–331. https://doi.org/10.1016/J.CEMCONRES.2016.09.008
Richardson IG, Skibsted J, Black L, Kirkpatrick RJ (2010) Characterisation of cement hydrate phases by TEM, NMR and Raman spectroscopy. Adv Cem Res 22(4):233–248. https://doi.org/10.1680/adcr.2010.22.4.233
Gierlinger N, Keplinger T, Harrington M (2012) Imaging of plant cell walls by confocal Raman microscopy. Nat Protoc 7(9):1694–1708. https://doi.org/10.1038/nprot.2012.092
Dyer C, Smith BJE (1995) Application of continuous extended scanning techniques to the simultaneous detection of Raman scattering and photoluminescence from calcium disilicates using visible and near-infrared excitation. J Raman Spectrosc 26(8–9):777–785. https://doi.org/10.1002/JRS.1250260827
Dyer CD, Hendra PJ, Forsling W (1993) The Raman spectroscopy of cement minerals under 1064 nm excitation. Spectrochim Acta, Part A 49(5–6):715–722. https://doi.org/10.1016/0584-8539(93)80094-Q
Bonen D, Johnson TJ, Sarkar SL (1994) Characterization of principal clinker minerals by FT-Raman microspectroscopy. Cem Concr Res 24(5):959–965. https://doi.org/10.1016/0008-8846(94)90016-7
Leemann A (2017) Raman microscopy of alkali-silica reaction (ASR) products formed in concrete. Cem Concr Res 102:41–47. https://doi.org/10.1016/J.CEMCONRES.2017.08.014
Loh H-C, Kim H-J, Ulm F-J, Masic A (2021) Time-space-resolved chemical deconvolution of cementitious colloidal systems using raman spectroscopy. Langmuir 37(23):7019–7031. https://doi.org/10.1021/ACS.LANGMUIR.1C00609
Polavaram KC, Garg N (2021) Enabling phase quantification of anhydrous cements via Raman imaging. Cem Concr Res 150:106592. https://doi.org/10.1016/J.CEMCONRES.2021.106592
Schmida T, Dariz P (2014) Shedding light onto the spectra of lime: Raman and luminescence bands of CaO, Ca(OH)2 and CaCO3. J Raman Spectrosc 46(1):141–146. https://doi.org/10.1002/JRS.4622/FORMAT/PDF
Pourchez J, Valdivieso F, Grosseau P, Govin A, Guilhot B (2006) Kinetic modelling of the thermal decomposition of ettringite into metaettringite. Cem Concr Res 36(11):2054–2060. https://doi.org/10.1016/J.CEMCONRES.2006.06.007
Zhou Q, Glasser FP (2001) Thermal stability and decomposition mechanisms of ettringite at <120°C. Cem Concr Res 31(9):1333–1339. https://doi.org/10.1016/S0008-8846(01)00558-0
Giridhar G, Manepalli RRKN, & Apparao G (2017). Confocal Raman spectroscopy. In: S. Thomas, R. Thomas, A. K. Zachariah, & R. Kumar Mishra (Eds.), Spectroscopic methods for nanomaterials characterization (Vol. 2, pp. 141–161). Elsevier. https://doi.org/10.1016/B978-0-323-46140-5.00007-8
Wu Z, Wong HS, Buenfeld NR (2017) Transport properties of concrete after drying-wetting regimes to elucidate the effects of moisture content, hysteresis and microcracking. Cement Concr Res 98:136–154. https://doi.org/10.1016/j.cemconres.2017.04.006
Wu Z, Wong HS, Chen C, Buenfeld NR (2019) Anomalous water absorption in cement-based materials caused by drying shrinkage induced microcracks. Cem Concr Res 115:90–104. https://doi.org/10.1016/j.cemconres.2018.10.006
Garbev K, Stemmermann P, Black L, Breen C, Yarwood J, Gasharova B (2007) Structural features of C-S-H(I) and its carbonation in air-a Raman spectroscopic study. Part I: fresh phases. J Am Ceram Soc 90(3):900–907. https://doi.org/10.1111/j.1551-2916.2006.01428.x
Tarrida M, Madon M, Le Rolland B, Colombet P (1995) An in-situ Raman spectroscopy study of the hydration of tricalcium silicate. Adv Cem Based Mater 2(1):15–20. https://doi.org/10.1016/1065-7355(95)90035-7
Allaoui A, El Bounia N-E (2009) How carbon nanotubes affect the cure kinetics and glass transition temperature of their epoxy composites?-a review. Express Polym Lett 3(9):588–594. https://doi.org/10.3144/expresspolymlett.2009.73ï
Wong HS, Head MK, Buenfeld NR (2006) Pore segmentation of cement-based materials from backscattered electron images. Cem Concr Res 36(6):1083–1090. https://doi.org/10.1016/j.cemconres.2005.10.006
Zhang K, Yio M, Wong H, Buenfeld N (2022) Real-time monitoring of carbonation of hardened cement pastes using Raman microscopy. J Microscopy 268:126
Al Omari MMH, Rashid IS, Qinna NA, Jaber A M, & Badwan AA (2016). Calcium carbonate. In: Harry G. Brittain (Ed.), Profiles of drug substances, Excipients and related methodology (41, p. 31–132). Academic Press.
Ogino T, Suzuki T, Sawada K (1987) The formation and transformation mechanism of calcium carbonate in water. Geochim Cosmochim Acta 51(10):2757–2767
Black L, Breen C, Yarwood J (2007) Structural features of C-S–H(I) and its carbonation in air—a raman spectroscopic study part II: carbonated phases. J Am Ceramic Soc 917(21881):908–917. https://doi.org/10.1111/j.1551-2916.2006.01429.x
López-Arce P, Gómez-Villalba LS, Martínez-Ramírez S, Álvarez de Buergo M, Fort R (2011) Influence of relative humidity on the carbonation of calcium hydroxide nanoparticles and the formation of calcium carbonate polymorphs. Powder Technol 205(1–3):263–269. https://doi.org/10.1016/J.POWTEC.2010.09.026
Coleyshaw EE, Crump G, Griffith WP (2003) Vibrational spectra of the hydrated carbonate minerals ikaite, monohydrocalcite, lansfordite and nesquehonite. Spectrochim Acta Part A Mol Biomol Spectrosc 59(10):2231–2239. https://doi.org/10.1016/S1386-1425(03)00067-2
Ševčík R, Mácová P (2018) Localized quantification of anhydrous calcium carbonate polymorphs using micro-Raman spectroscopy. Vib Spectrosc 95:1–6. https://doi.org/10.1016/J.VIBSPEC.2017.12.005
Bernard S, Beyssac O, Benzerara K (2008) Raman mapping using advanced line-scanning systems: geological applications. Appl Spec 62(11):1180–1188
Acknowledgements
K. Zhang acknowledges the funding from the China Scholarship Council (CSC). The research leading to this publication benefitted from EPSRC funding under grant No. EP/R010161/1 and from support from the UKCRIC Coordination Node, EPSRC grant number EP/R017727/1, which funds UKCRIC’s ongoing coordination.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, K., Yio, M.H.N., Wong, H.S. et al. Optimising confocal Raman microscopy for spectral mapping of cement-based materials. Mater Struct 55, 131 (2022). https://doi.org/10.1617/s11527-022-01979-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-022-01979-9