
Abstract
The importance of lipid-based formulations in addressing solubility and ultimately the bioavailability issues of the emerging drug entities is undeniable. Yet, there is scarcity of literature on lipid excipient chemistry and performance, notably in relation to oxidative stability. While not all lipid excipients are prone to oxidation, those with sensitive moieties offer drug delivery solutions that outweigh the manageable oxidative challenges they may present. For example, caprylocaproyl polyoxylglycerides help solubilize and deliver cancer drug to patients, lauroyl polyoxylglycerides enhance the delivery of cholesterol lowering drug, and sesame/soybean oils are critical part of parenteral nutrition. Ironically, excipients with far greater oxidative propensity are omnipresent in pharmaceutical products, a testament to the manageability of oxidative challenges in drug development. Successful formulation development requires awareness of what, where, and how formulation stability may be impacted, and accordingly taking appropriate steps to circumvent or meet the challenges ahead. Aiming to fill the information gap from a drug delivery scientist perspective, this review discusses oxidation pathways, prooxidants, antioxidants, and their complex interplay, which can paradoxically take opposite directions depending on the drug delivery system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Lipid formulations vary by the excipients and the technologies used in their development. The simplest example of a lipid formulation is that of a poorly soluble drug dissolved in fixed oil(s). Fixed oils are monographed vegetable oils (e.g., soybean, sesame), with well-defined composition in triglycerides and fatty acid distribution. Self (micro)-emulsifying drug delivery systems (SEDDS, SMEDDS) on the other hand may consist of fixed oils, partial glycerides, polyoxylglycerides, polysorbates, and/or propylene glycol esters combined with a polyethylene glycol (1, 2). Some preparations may be of liquid consistency, filled into capsules. Others may be developed as solid particles by dispersion, adsorption onto solid carriers (3, 4), or melt granulation/congealing techniques (5). Solid lipid nanoparticles (6,7,8), microemulsions (9, 10), and liposomal carriers should not be excluded from this non-exhaustive list of possibilities (11,12,13). Nevertheless, few publications offer pharmaceutically relevant and aggregated information on the oxidative stability of such complex lipid formulations (14,15,16,17).
Meanwhile, the core causes of oxidative instability in lipid-based formulations remain misunderstood due in part to the large number of parameters and antagonisms governing oxidative instability. As a result, the misperceptions that lipid-based formulations present a risk of failure during stress and stability testing continue to linger (18). There is no factual basis for this thinking because lipid formulations have for many decades been used in oral, topical, and parenteral applications. Ether-based polymers with far greater propensity to oxidation such as polyethylene glycols (macrogols) and polyoxyethylene glycol–polyoxypropylene glycol block copolymers (poloxamers) are commonly found in marketed drug products (19, 20). At minimum, these achievements are testament to the manageability of oxidative events in pharmaceutical products.
Combing the literature, we find confusing reviews, exemplified by an article (21) opining on lipids and SEDDS, citing them as complex and as carriers of reactive impurities. Yet, the examples of chemistries cited were solubilizers based on polyethylene oxide (PEO), polypropylene oxide (PPO), and polyethylene glycols (PEG). Also cited were the account of an “unstable” drug X in a SEDDS formulation of unidentified composition, or the peculiar case of drug Z reacting with its sole carrier, oleic acid — even though free fatty acids are known as strong prooxidants. Such anecdotal references emerge when fundamentals of formulation science are overlooked, and formulation attempts fail for the wrong reasons.
Undeniably, oxidative change may lead to loss of drug potency, diminished shelf life, and time to market (22, 23). Incomplete assessment of oxidative risks may translate to missed opportunities in applying antioxidants or a wrong antioxidant system, leading to acceleration of undesirable reactions (24, 25). However significant, oxidative challenges are not unsurmountable. Appropriate choices in excipient selection, formulation/process, and antioxidant selection are key to successful development. Apprised knowledge about oxidative pathways becomes an essential part of a new thinking about lipid-based drug delivery systems. The aim of this paper is to provide a consolidated review of the current understanding of oxidative events encountered in lipid formulations; to underline oxidative risks, where they lie; and offer insight on how they can be prevented or managed.
OXIDATION
The term autoxidation has long been used to describe oxidative events in biomolecules, reactions that self-propagate once initiated or which result from “spontaneous oxidation in air of a substance not requiring catalysts.” Important to note is that an uncatalyzed (unmediated) oxidation of biomolecules is an unlikely or rather a non-significant event when dioxygen is in its ground state. A sensitizer such as light and more commonly a transition metal catalyst must mediate the conversion of dioxygen into its highly reactive singlet 1+O2 state for oxidation to occur. Since biomolecule oxidations are largely catalyzed by metals, they cannot be considered autoxidation (26, 27). Despite this subtle differentiation, oxidation and autoxidation have become interchangeable terms in the cited literature.
Oxidative pathways are complex and can vary by a host of factors. Once initiated, the process involves a slow and steady formation of radicals, followed by peroxides and hydroperoxides, the primary by-products of oxidation. These highly reactive and unstable species can then go on propagating and eventually decomposing into secondary oxidation products like small organic acids, aldehydes, and ketones. The rate and extent of the reactions can also vary by the nature of the delivery system (liquid, solid, biphasic) and the concentration of the oxidation prone substrates (excipient, drug, and or antioxidants) (26, 28,29,30,31,32,33). The initiators, substrates, intermediaries, and end products of oxidation are short listed in Table I.
An unhindered chain of oxidative events has three distinct phases: initiation, followed by propagation and eventually termination. These critical phases of oxidation are further described below.
Initiation
Initiation is a radical producing event, triggered by light (hv) and oxygen or metal catalysts (Mn+) and oxygen (Table II), when a hydrogen breaks off the substrate, leaving what is universally described as a free radical (R•). The free radical may consist of an unsaturated fatty acid ester of a glyceride molecule, an excipient with polyethylene oxide groups, an oxidation prone drug, or an antioxidant (15, 30, 34, 35).
Initiation is generally slow and has limited impact on the integrity of the substrate or the finished product. It does not normally produce peroxyl radical (ROO•) except in the combined presence of light, oxygen, and transition metals.
Propagation
Propagation is characterized by the chain process of the peroxyl radical (ROO•) abstracting a hydrogen atom quite rapidly from another RH group (substrate) to form hydroperoxides (ROOH) (28, 30, 31). The process consumes oxygen to form more radicals, peroxides, and hydroperoxides (Table III). As the primary by-products of oxidation, peroxides and hydroperoxides are the dominant species during the early stages of propagation. From reaction schemes in Tables II and III, it becomes apparent that the rate of hydroperoxide formation during the early propagation is limited by the substrate concentration and that reaction between atmospheric oxygen and the organic substrate occurs indirectly, via formation of radicals. Generally, the radicals participate in reactions at rates that are retarded by antioxidants or accelerated by prooxidants (30, 35,36,37,38). Hydrogen abstraction is a rate-limiting step in the oxidation process because it depends on the substrate, and more specifically the strength of the RH bond being broken (28, 39), further discussed below. Also, hydrogen atom abstraction by lipid peroxides is a reversible process but may be irreversible for drug entities or other biochemicals.
The later stages of the propagation phase are associated with hydroperoxide decomposition. The decomposition of hydroperoxides into radical species requires high energy input which is mediated by metal catalysts. Hydroperoxide decomposition results in significant increase in the amounts of free radicals, peroxyl radical (ROO•), alkoxyl radical (RO•), and water, further propagating the oxidation cycle (19, 30, 40). Frequently unstable, hydroperoxides and other intermediaries may then degenerate into secondary compounds (aldehydes, ketones) and other reactive substructures (41). Eventually, the chain reactions terminate when substrate (drug, excipient, or the antioxidant) concentration diminishes and or when peroxides being in abundance, begin encountering each other to form inactive, stable by-products (42). Briefly, hydroperoxides participate in the chain reactions, as radicals in the system are formed and quenched concurrently. Therefore, the peroxide concentration measured at any given point in the process represents the balance between hydroperoxide formation and decomposition.
Wide-Angle Perspectives of Oxidation
A larger view of the consecutive phases, i.e., initiation, propagation, and termination in a lipid-based system, becomes apparent by tracing the peroxide concentration. Peroxide value, an official method for measuring hydroperoxide concentration, is commonly used to assess oxidation potential or the progression of oxidation in the test product, over time. The test involves an iodometric titration with sodium thiosulfate, with the results expressed as the amount of peroxide oxygen in milliequivalents per kilogram (mEq O2/kg) of the test substance.
At initiation, peroxide value is low but increases gradually to take an exponential rise during the propagation phase (Fig. 1). At propagation, the rate of hydroperoxide formation exceeds its rate of decomposition. When all the available oxygen is consumed and or the oxidizable substrate is depleted, the process reaches a steady state, indicated by a plateau in the peroxide concentration. At this point, the rate of hydroperoxide formation is equal to its decomposition rate. In the absence of an oxidizable substrate or oxygen, termination phase begins, where the rate of hydroperoxide decomposition exceeds its formation (28, 31, 35, 43).

Oxidation unhindered. Kinetic schemes for main events during initiation, propagation, and termination phases of oxidation. Dotted circle indicates tolerance space for peroxide and acid values in pharmaceutical excipients (adapted from Finley and DeMan 2018, Hovorka 2001)
Peroxide value takes meaning when plotted over time and against other developing parameters, like acid value which is expressed as mg of KOH required to neutralize a gram of the test material (mg KOH/g). In freshly produced glycerides such as highly refined oils or fractionated oils, acid value represents the level of unesterified (free) fatty acids. A rise in acid value in aged excipients may indicate fatty acids breaking off from their parent molecules. This applies to glycerides, polyoxylglycerides, polyethylene glycol esters, phosphatides, and polysorbates (17, 34, 44, 45). Acid value can also signify release of smaller carboxylic (formic, acetic) acids due to oxidative breakdown. Associated with oxidative stress but hydrolytic in nature, these reactions are catalyzed by metals, and further assisted by presence of heat, acid/alkali, or increased atmospheric pressure. Briefly, acid value is not a direct measure of oxidation; it is rather an indicator of a fall out effect of oxidative stress (46). Unlike peroxide value which rises and eventually drops, acid value continues to increase over the course of oxidation, the rise being pronounced in accelerated (heat, humidity) conditions (Fig. 1).
The picture gets more complex when an oxidation prone substrate is added to the picture. Typically, the concentration of an oxidation prone substrate (drug entity, excipient, or antioxidant) diminishes slowly at first at the initiation phase and drops at a faster rate during propagation (15, 44). Unhindered, the oxidative chain of events can go on producing other secondary by-products, non-volatiles followed by volatile species. The secondary by-products of oxidation like aldehydes are electrophiles, i.e., seek another atom or molecule containing an electron pair to bond. Hence, even at 10–20 μg/g (ppm) level aldehydes may serve as adduct to DNA, proteins, and other nucleophilic biomolecules or cause cross-linking in gelatin capsules (39).
From safety standpoint, the primary products of oxidation (Tables I–III), especially at low levels, are of little or no concern for most formulations. It is rather the secondary oxidation products such as aldehydes that are implicated in cytotoxicity and/or pathogenicity. From drug product stability standpoint, both hydroperoxide and acid value in the lipid system need to be at possible minimums. The smaller dotted circle in Figure 1 represents the limits tolerated for excipients in pharmaceutical applications, where secondary oxidation products are either absent or if present, only at significantly low quantities. For instance, hydroperoxide concentration limits imposed by compendia are typically <10 mEq O2 kg−1, while the actual values reported by excipient manufacturers are in the 0.1 to 1 mEq O2 kg−1 range.
Challenging the conventional view of oxidation, Schaich (24) proposed a more comprehensive depiction of the possible pathways. The proposed scheme (Fig. 2) integrates the classical hydrogen abstraction theory (in the center of the diagram) with other potential pathways, including potential participation of peroxyl radicals in multiple scission and cross-linking events. The diagram in Fig. 2 points to, among others, the by-products of thermal oxidation, i.e., dimers, polymers, and cyclic polymers of triglycerides that can form due to long (excessive) exposure to elements, including heat (37). The higher the temperature, the higher the rate of hydroperoxide decomposition and the higher the reactivity of the transition metal ions and the greater the rates for redox reactions in general. Throughout the concurrent and competing events, different reactions may dominate at different oxidation phases depending on the test conditions and the concentration of the reactants. However, the general pattern of degradation for oxidation prone substrates will over time result in loss of drug potency and or performance (32, 47).

Reaction scheme that integrates alternate pathways of lipid oxidation with the traditional chain reactions driven by hydrogen abstractions (adapted from Schaich 2012)
Briefly, the schemes presented in Figs. 1 and 2 and Tables I–III are generalizations from a century of research, depicting a wildfire of possible oxidative pathways elucidated under forced experimental conditions. While necessary for understanding of the oxidative risks, these pathways are largely avoidable in pharmaceutical settings. Moreover, oxidative potential for the pharmaceutical grade lipid excipients is (must be) quite low or near zero, as noted by the dotted circle in Fig. 1.
Evaluating Oxidative Change
Since no single analytical method can provide a full picture of the potentially occurring events, it is “advisable to use at least two, if not more, different analytical approaches” (29). Simple routine testing for acid value, peroxides, and water content can help assess oxidative changes following critical processing and formulation steps. Tracing changes in pH, loss of drug solubility, or loss of dispersion capacity in emulsion systems are other general yet helpful measures when evaluating lipid formulation stability. If relevant, measuring rate of oxygen consumption, or loss of antioxidant concentration over time can provide important clues to the extent or the mitigation of oxidative reactions (48). Measuring secondary oxidation by-products such as aldehydes or formic acid may be more complex, but critical to biopharmaceutical molecules and/or gelatin-based capsules. Moreover, the stability testing conditions need to be relevant to the drug product formulation, mode of production, and storage before use.
For the specifics of various analytical methods, the reader may refer to the articles reviewed herein including others that take a deeper dive into the subject (23, 49,50,51,52,53,54).
OXIDATION PRONE MOIETIES IN LIPID FORMULATIONS
Biomolecules (lipid, protein, or RNA) are susceptible to oxidation through various mechanisms such as electron transfer, peroxide mediated reactions, or radical induced chain events. Such reactions may be triggered by “autoxidation” and or initiated by residual levels of impurities such as transition metals, peroxides, aldehydes, ketones, and free fatty acids, being both the source and by-products of oxidative chain reactions. For lipid excipients, two chemical groups stand out as potential substrates for oxidation: (i) unsaturated alkyl chains and (ii) polyethylene or polypropylene oxide groups in the lipids’ molecular scaffoldings.
Ethylene Oxide Chains
Much of the lipid formulation history is intertwined with the use of polymeric cosolvents like polyethylene glycol (PEG) and polyethylene oxide moieties (PEO). These may be added to a lipid formulation to boost drug solubility in microemulsions or SEDDS/SMEDDS, or incorporated into the molecular scaffolding of the lipid excipient in order to achieve higher solubilization capacity. Polyoxyethylene sorbitan esters and polyoxyl 40 hydrogenated castor oil are examples of excipients based on PEG/PEO chemistries (55). Polyether compounds are highly susceptible to degradation by molecular oxygen, due to the labile nature of protons at α-carbon atoms of the ether bond reacting at all temperature conditions (56).
The polyoxyethylene linkages in poloxamers, PEG, or PEG based lipids (e.g., polysorbates) are prime candidates for oxidation. The free radical mediated oxidation of the polyethylene oxide chains occurs at the ends, by a β-scission from the hydroperoxide, or a C-C cleavage in the “ethylene oxide” unit. The breakdown is preferentially occurring on the monoesters rather than on the diesters of PEG which are substituted at both ends. The shortening of the chains produces hydroperoxides and peroxide free radicals that will continue propagating oxidation. The peroxides emerging from this breakdown are known cause of oxidative breakdown in a host of drug actives (34). The phenomenon is accelerated by light, heat, and participation of pro-oxidants such as Cu2+ or Fe3+.
The secondary degradation by-products resulting from oxidation (breakage) of PEO/PEG chains include acetaldehyde, formaldehyde, PEG-aldehydes, and short-chain carboxylic acids like acetic and formic acids (45, 57, 58). Formaldehyde and formic acid are implicated in the degradation of several drugs, notably ones with amino and nucleic acid moieties (59, 60). Formaldehyde and formic acid are also responsible for cross-linking in the gelatin capsule shells (32, 42, 57, 59). It is worthwhile to note that PEGs can be a significant source of impurities like ethylene oxide, 1,4-dioxane or alkali catalysts such as sodium hydroxide, potassium hydroxide, or sodium acetate or lactate which are used to prepare low-molecular weight PEGs (48). Being not biodegradable, the use of PEGs is limited to molecular weights of <40 kDa, to avoid accumulation in human tissue (61).
Polysorbates
Polysorbates are fatty acid esters of polyoxyethylene sorbitan, with different alkyl chain lengths depending on the grade. Polysorbate 20 is differentiated from polysorbate 80 by fatty acid predominance, i.e. by lauric acid in polysorbate 20 and oleic acid in polysorbate 80. Polysorbates are notorious for undergoing oxidation and hydrolysis and their degradation pathways under pharmaceutically relevant conditions are relatively well known (62, 63). Polysorbate breakdown can occur in several ways: cleavage at the ethylene oxide subunits (previous section), hydrolysis of the fatty acid ester bond to form free fatty acids, and/or oxidation of the unsaturated hydrocarbon chains of their fatty acids. Mediated by oxygen and/or transition metals, oxidation of polysorbates involves free radical initiated chain reactions and scission of both C–O and C–C bonds, producing short-chain acids like formic and acetic acids (45). Formation of the free fatty acids and shortened polyoxyethylene chains may bring about change in the HLB value, therefore the emulsification capacity of the excipient. The POE-Fatty acid radical species can oxidize therapeutic proteins, for example, at methionine and tryptophan residues causing alterations of proteins or generation of oligomeric species. In the presence of water, the hydrolysis of fatty acid ester bonds in polysorbates depends on the temperature, surfactant concentration, and pH of their use. These degradations may occur in neat polysorbate, at ambient temperatures or slightly higher temperatures in the presence of light and atmospheric oxygen. Hence, storage below ambient temperature, under inert gas (e.g., nitrogen) is advised (20, 34, 45, 63).
Polyoxylglycerides
Little is published on polyoxylglycerides from oxidation potential perspective (48). However, many of the principles discussed in this review can easily apply to them. Polyoxylglycerides are lipid excipients obtained by reacting glycerides (fixed oils, partial glycerides, or fatty acids) with PEG. Hence, they inherit an improved solubilization capacity, but also the oxidative sensitivities and high levels of hydroperoxide and aldehyde impurities associated with PEGs. Interestingly, PEO and PEG, despite being core contributors of residual prooxidants in the lipid excipients, are not always acknowledged for their oxidative potential. The glyceride moieties of polyoxylglycerides on the other hand, may be prone to oxidation due to unsaturation (see below) or quite stable, depending on the choice of the glycerides in their scaffolding. The stability of polyoxylglycerides is drastically improved with the use of saturated fatty esters in their production. Examples include C8/C10 esters in Labrasol® ALF (caprylocaproyl polyoxyl-8 glycerides) and C18:0 esters in Gelucire 48/16 (Polyoxyl-32 stearate). In the case of unsaturated oils, preference may be given to the mono-unsaturated lipids, or those like corn oil which are rich in natural antioxidants.
Unsaturation of Fatty Acid Chains
Fatty acids are carboxylic acids, varying in hydrocarbon chain length (8–22 carbons) and degree of unsaturation (14). Fatty acids define the identity and phys-chem properties of lipid excipients. Fatty acids are presented in nature as triglycerides in fixed oils. In synthesized lipids (excipients), fatty acids may exist as mono-, di-, or tri-esters in fractionated and/or structured lipids (48, 64, 65). Moreover, the fatty acids may be located in different molecular positions and to different degrees in polysorbates (34), polyoxylglycerides(48) and phospholipids (64).
Unsaturated fatty acids have one or more double bonds along their hydrocarbon chains. Lipids with saturated fatty acids have excellent oxidative stability profiles. Examples in the latter category are medium chain glycerides (liquid, C8-C10), and solid excipients like glyceryl dibehenate (pellets and powder, C22), and glyceryl stearate (pellets). Figure 3 shows the five possible configurations for C18 fatty acids.

Configuration of the C18 fatty acids with 0, 1, 1OH, 2, and 3 unsaturated bonds
Lipid excipients consisting of unsaturated fatty acids are subject of oxidative vulnerability, depending on the degree of unsaturation, treatment temperature, and the nature of the formulation system. In the example of linoleic acid (C18:2), oxidation is initiated with hydrogen abstraction on its C-11, where the bis-allylic subunit R1–CH=CH–CH2–CH=CH–R2 is naturally homoconjugated. Loss of the hydrogen atom results in an intermediate and unstable lipid radical which then adds oxygen at either end of the subunit, shifting the double bond to the more stable adjacent carbons to produce C9- and C13-conjugated diene peroxyl radicals. These chain-carrying peroxyl radicals become key substrates and influencers of chain propagation as well as chain terminating rate constants (37, 66,67,68,69,70).
A direct correlation between degree of unsaturation and oxidative vulnerability in oils has been established by various studies focusing on oxidation rate constants (26, 31, 37, 66, 68, 69, 71). The exponential relationship between unsaturation and oxidative potential in oils can be observed from the relative oxidation rates for C18 fatty chains (Table IV). It must be noted that the values listed in Table IV were obtained at an elevated temperature of 100°C because oxidation of methyl oleate (C18:1) does not occur at ambient temperature, whereas methyl linoleate (C18:2) and linolinate (C18:3) can oxidize at ambient conditions. This is explained by the dissociation energy of the hydrogen of the allylic group in oleic acid (18:1) which is 10 Kcal/mol greater than that of the unsaturated fatty acids (18:2; 18:3) translating to a stability in the order of C18:0 ≫ C18:1 ≫ C18:2 > C18:3. The lower the degree of fatty acid unsaturation, the more stable the vegetable oil is.
The aforesaid rank relationship between degree of fatty acid unsaturation and oxidative vulnerability holds true in oils but not in emulsified systems. Demonstrating a direct correlation between oxidative vulnerability (measured by peroxide value, and the degree of unsaturation) in triglycerides is an earlier study by Miyashita(67), where unsaturated synthetic triglycerides of various unsaturated fatty acids were subjected to stability testing at 40°C/75% RH conditions. Figure 4 shows the results, where tri-linoleate (LLL) was significantly more stable to oxidation, relative to tri-linolinate (LnLnLn).

Hydroperoxide development in unsaturated triglycerides at 40°C, 75% relative humidity. L = linoleic (C18:2) and Ln = linolenic (C18:3) (Miyashita 1990)
A reverse trend is observed when the glycerides of unsaturated fatty acids are in micellar, liposomal, or microemulsion systems (72,73,74). In a later study, Miyashita (74) combined NMR, GC-MS, and digital modelling, to study the oxidative behavior of polyunsaturated fatty acids (PUFA) in micellar systems. The results (Fig. 5) depicted the reversal of oxidative vulnerability, where the highly sensitive PUFAs like docosahexaenoic acid (C22:6) or eicosapentaenoic acid (20:5) was incredibly stable in emulsion systems (74), relative to linoleic (C18:2), and linolenic (C18:3) fatty acids.

Oxidative stability of polyunsaturated fatty acids in micelles (Miyashita 2014)
One explanation for this unexpected stability of PUFAs in micellar systems is that the hydroperoxides formed from the oxidation of eicosapentaenoate (20:5) are more polar than those generated by linoleate (18:2). As such, they migrate out of the oil phase toward the polar interfacial region, thus being removed from the propagation reactions that occur at the core of the oil micelle droplet (75). A more plausible explanation of the PUFA stability in emulsions was provided by Miyashita (74) stipulating that the physical and stereochemical properties of the fatty acids and the tight packing conformations due to curvature near the double bonds (Fig. 3) making them less available may have a critical role to play in the inhibition of hydrogen abstraction from the bis-allylic positions.
KEY CONTRIBUTORS TO OXIDATIVE INSTABILITY
Transition Metals
Transition metals are ubiquitously present in nature and may be found in pharmaceutical excipients at trace levels (76). In the presence of trace (parts per billion) quantities of Cu+, Cu2+, Fe2+, or Fe3+, hydroperoxides can decompose to alkoxyl radicals and peroxyl radicals which in turn can trigger other events cited in the preceding section. Although copper is the most reactive element (Fig. 6), the prevalence of iron in nature makes it the most important oxidant in biochemistry (31, 77, 78).

Transition metals and their relative propensity to partake in oxidation of oils (Talbot 2016)
Due to their ability for electron redox cycling, transition metals are strong prooxidants and photosensitizers, responsible for formation of highly reactive species like hydroxyl radical (OH•) and singlet oxygen (1+O2). A direct reaction of dioxygen with the vast majority of biomolecules is spin-forbidden because ground-state dioxygen is a biradical (79). Differently put, the activation energy for a direct reaction between the acyl chain (LH) of a lipid and dioxygen is too high to occur in ground state because of the opposite spin directions between the two molecules (80). This suggests that the oxidation of biomolecules (lipids, proteins) is largely an occurrence mediated by sensitizers like light, and most commonly catalyzed by transition metals (22, 29,30,31). This point alone underlines the critical importance of adding a chelating agent to inhibit sensitizing metals, while minimizing exposure to air, during handling or processing.
Hydroperoxide Impurities
Propensity to oxidation for pharmaceutical preparations is frequently attributed to the presence of reactive impurities (peroxides, aldehydes, and trace transition metals) in excipients (20). Hydroperoxide levels, in particular, are cause for concern if the reaction involves an oxidation prone drug substance, and if the formulation is destined for a parenteral route of administration (81). Hydroperoxide impurities are not unique to lipids. They are present in commonly used excipients including povidone, lactose, HPMC, and polyethylene glycol (20, 22, 32, 36, 49). Table V offers a sampling of hydroperoxide concentration ranges for various excipients, including the variabilities noted among different manufacturers or batch-to-batch from the same manufacturer (22). In actuality, the hydroperoxide levels that are currently reported for most lipid excipients are negligible (<10 nmole/g), similar to that reported for medium chain glycerides in Table V.
In the absence of transition metals, lipid hydroperoxides are relatively stable at room temperature. Otherwise, they can readily decompose to peroxide, peroxyl, and alkoxyl radicals, in which case, other (secondary) reactive species such as aldehydes, ketones, acids, esters, alcohols, and short-chain hydrocarbons may begin to form (37). These can subsequently affect the quality of the excipient, drug stability, and the eventual integrity of the formulation (19, 36, 60).
To minimize potential for oxidative change, it is advisable to begin work with excipient(s) that have the lowest possible hydroperoxide impurities. In parallel, it is necessary to consider the sensitivity of the excipient(s) to the conditions of storage and use. One study investigated the effect of light, oxygen, and initial peroxide levels in polysorbate 80 and the peroxides formed during stability testing (34). As part of the study, 20% solutions of polysorbate 80 were stored at 40°C under various conditions: exposed to air under fluorescent light, exposed to air in the dark, or exposed to light without air exposure. The results shown in Fig. 7 demonstrate how exposure of polysorbate 80 to light alone, under vacuum (without oxygen), did not produce hydroperoxides, whereas the presence of air in the headspace in the dark led to significant increase in the hydroperoxides level. The most dramatic rise in the hydroperoxides was noted when the samples were exposed to light and air combined, where peroxide levels rose from 1 to 1300 mEq within a 5-week incubation period at 40°C, eight times higher than that observed under air, without light.

Formation of hydroperoxides in polysorbate 80 solutions incubated at 40°C under different light, air exposure conditions (Ha 2002)
As part of the same study, the stability of an oxidation-prone model protein, IL-2 mutein, was investigated in solutions made up of two different batches of polysorbate 80 — one with initially low and the other with high peroxide level. The samples of the initially low-peroxide (0.33 μEq O2 kg−1) and the stressed, high-peroxide (25 μEq O2 kg−1) polysorbate batches were incubated at 40°C under different light and air exposure conditions, alongside the control solution (without polysorbate 80). At the end of the 30-day incubation period, the study recorded a 5% drug loss from the control samples (Fig. 8), affirming the general sensitivity of the drug. Meanwhile, there were significant drug losses in the solutions consisting of polysorbate 80, where IL-2 mutein losses amounted to 35–45% in the high-peroxide and 10–13% in the low-peroxide batch of polysorbate 80. This study underlines the impact the initial hydroperoxide levels may have on drug stability.

Oxidation of IL-2 Mutein under different incubation conditions and different levels of initial hydroperoxides in polysorbate 80 solution at 40°C (Ha 2002)
PEGs tend to have varied but significant levels of hydroperoxide impurities. Additionally, their chains become substrates for formation of even higher levels of hydroperoxides during oxidation. PEGs of varying molecular weight are commonly used as solvents or cosolvents in lipid formulations. Lower molecular weight PEGs (≤400 Da) are viscous liquids, whereas those above 1000 Da are solids at ambient temperature. In a comparative study involving PEGs of different molecular size stored under identical conditions, there were significant increases in peroxide levels, 17-fold and 70-fold, respectively, for PEG 1450, and PEG 20000 (19). The differences in the rates of hydroperoxide formation were attributed to the higher fraction of water present in PEG 20000 solution. This water-related increase in oxidative instability has been explained in part by a higher amount of dissolved oxygen in the PEG system. Additionally, water may play a key role in the solubilization and distribution of transition metals in the aqueous media (19, 28, 47, 77, 82). The peroxides emerging from the aforesaid mechanisms are the known cause of oxidative breakdown for a host of drug actives (34).
Aldehydes and Short-Chain Acid Impurities
As indicated, the secondary byproducts like acetaldehyde, formaldehyde, acetic acid, and formic acid are formed during progression of oxidation (38, 81). The low-molecular-weight aldehydes (C1–C8), may also be present in neat excipients as trace impurities. They may also be generated due to exposure to heat or atmospheric oxygen following reception, storage, and most likely during handling and manufacture (81). Aldehydes have the capacity to then react with the drug substance, forming degradation by-product, and/or react with the gelatin capsule shells.
Exemplifying the contribution of formaldehyde to the formation of a drug degradant is a study by Nassar and colleagues (81) . The work involved injectable emulsions of a BMS drug in polysorbate 80 and PEG 300, where an unknown impurity was identified to be the drug-formaldehyde adduct being formed at temperatures of 5 to 25°C. Several formulations at 1, 10, and 100 mg/g of API were spiked with 0.1, 1.0, and 10 mg formaldehyde. The results established a clear correlation between formaldehyde levels in polysorbate 80 (16 ± 8.5 μg/g) and PEG 300 (41.4 ± 69.3 μg/g), compared to zero aldehydes in alcohol as control. As a result, aldehyde content was identified as critical material attribute to be closely monitored and controlled (81).
Actives with amino acids or nucleic acid moieties may be subject of cross-linking or chemical modification of the amino group of the N-terminal amino acid residue in the presence of formaldehyde, more so than with peroxides (59). The stability of O6-benzylguanine, a chemotherapeutic agent, for example was directly correlated with the presence of formaldehyde, which reacted with the drug to form a methylene-bridged product containing two O6-benzylguanine. In this work, the hydrolysis of the drug substance in 40% PEG 400 solutions was attributed to the significant rise from 0.5 μg/mL to 194 μg/mL between fresh and aged PEG 400. Ultimately, a stable formulation was possible by selecting fresh samples with aldehyde levels below 3 ppm, pH adjustment, and by minimizing exposure to heat and oxygen. Storing the fresh samples in refrigerated conditions under argon was helpful in maintaining low formaldehyde levels (59).
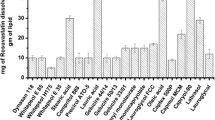
Aldehydes require complex detection/quantification methods. Applying gas chromatography coupled with mass spectrometry (GC–MS), Li et al (49) validated a method for analysis of C1–C8 aliphatic aldehydes. Method accuracy was verified by spiking excipient samples with known concentrations of aldehydes followed by analyzing the percent recovery of the analyte added to the samples. Subsequently, 30 excipients commonly used in oral capsules were tested. The aldehydes measured in more than a dozen lipid excipients ranged between 0 and 5 μg/g (Fig. 9). It is important to note that no aldehydes were detected in corn oil, medium chain glycerides, or propylene glycol esters of caprylate/caprate (C8/C10). This limited capacity for aldehyde formation may be explained in part by the saturated nature of the medium chain, C8/C10 fatty acid esters and in the case of corn oil, by the potential presence of natural antioxidants. Meanwhile, the aldehyde levels detected in most of the lipid excipients were by one to three orders of magnitude smaller than those determined for the PEGs (Fig. 10). Among the PEG’s, those with average molecular weights of 200, 400, and 600 had the highest aldehyde levels (49).

Total aldehydes detected in various lipid excipients (Li 2007)

Total aldehydes detected in liquid and solid polyethylene glycols (Li 2007)
Aldehydes, namely, acetaldehyde and formaldehyde, can oxidize respectively into acetic and formic acid which are known to cause cross-linking in gelatin capsules. Gelatin is sensitive to heat, humidity, and is susceptible to reactions with trace levels of formaldehyde and formic acid, impurities of the PEG/PEO based excipients. When stored in accelerated (high relative humidity) conditions, formaldehyde, and its by-product, formic acid cause cross-linking of the gelatin polypeptides. Cross-linking manifests itself with the formation of a water-soluble film, loss of plasticity, increased opacity, and brittleness of the shell. Due to the potential adverse effect on gelatin capsule integrity, it is necessary to monitor aldehyde levels as part of the standard excipient evaluation procedure (21, 49, 83).
Hemenway and colleagues (58) developed a 2,4-dinitrophenylhydrazine (DNPH) precolumn derivatization HPLC–UV method to quantify formaldehyde and acetaldehyde levels in PEG 400 solutions at 1:1 dilution with water. As part of the study, the group evaluated for up to 90 days at 40°C, the total PEG aldehydes emerging under various experimental conditions, like (i) increased acidity by addition of 0.012 M HCl or 2% citric acid, (ii) raising the incubation temperature from 40 to 50°C, or (iii) spiking of the samples with 100 ppm of Fe2+. In all cases, the formaldehyde levels increased within 21 days of the study. Meanwhile, formic acid levels rose continually and dramatically, reaching 10 times their initial values. Reported as total PEG aldehydes (Fig. 11), their findings appear to suggest that acidity and temperature had more prominent effects on total aldehydes formation, more so than with samples spiked with 100 ppm Fe2+. One may speculate that the PEG 400 solution spiked with Fe2+ (Fig. 11) had lower aldehyde levels in part because it may have sped the oxidation of aldehydes into acids.

Effect of lowered pH, increased temperature, and addition of Fe2+ on the total aldehyde levels in 50% solutions of PEG400 in water incubated at 40°C for up to 90 days (adapted from Hemenway, 2012)
Extreme cross-linking is known to produce brittle capsule shells, subject to breakage and leaking of the contents. Even if the capsule shells are not fully compromised, a small degree of crosslinking may lead to failure during the in vitro dissolution testing (49, 83). To determine whether a change in dissolution profile in vitro has relevance to in vivo conditions, official pharmacopoeia methods allow addition of digestive enzymes (bromelain or papain) to the in vitro dissolution media for gelatin capsules. Figure 12 shows the seemingly failing release profile of acetaminophen from cross-linked capsules (red dotted line) compared to dissolution in the media consisting of digestive enzymes (55, 84). In addition to the USP dissolution method of assessing cross-linking, a variety of capsule shell compositions have emerged. One example is that of compositions based on cellulosic polymers and or starch and carrageenan (Optishell).

Acetaminophen dissolution profiles from cross-linked gelatin capsules using the enzymes bromelain and papain in pH 5.0 acetate buffer (adapted from Gray 2014)
Formulation uniformity, dosage accuracy, and stability are of critical importance in both hard and soft gelatin capsules. Whereas hard gelatin capsules are amenable to solid and semi-solid formulations, soft gelatin capsules are mainly suited for liquid to viscous formulations. Some have suggested that soft gelatin capsules provide easier blending, better control of fill material, and inert encapsulation conditions that are conducive to oxidative stability (85, 86). Meanwhile, the two-piece hard shells are gaining a competitive advantage over the soft capsule, thanks to the introduction of new and less sensitive shell compositions. However, the overall perspectives are bound to change as new lipid formulations techniques such as amorphous solid dispersion, melt congealing, or high shear spheronization continue to emerge.
The capsule fill material is typically heated by 10–15°C above the highest melting point excipient in the formulation to ensure all excipients are well blended in molten free flowing fluid state prior to filling. Thus, the heating step may exceed 60°C (depending on the excipients) and the mixture may be held for 24 to 72 hours prior to filling into capsules. Attention is therefore needed to avoid exposure to air, mixing time, and addition of appropriate antioxidants to minimize oxidative reactions in the mixing tank.
Oxygen and Light
Generally, the rate of peroxide formation depends on oxygen supply, proportional to the concentration of the oxygen dissolved in the aqueous or oily phase of the formulation (47, 87). Exposing the product to air facilitates the dissolution of atmospheric O2 into the product. Thus, the oxygen consumption in stability testing can be an indicator of oxidative developments for samples that are stored in closed vessels. Cuvelier (88) measured the amount of oxygen dissolved in polar and apolar solvents as a function of temperature, between 5 to 50°C. Water served as the highly polar and dodecane as the apolar points of reference. The results (Fig. 13) confirmed that both sunflower oil glycerides and propylene glycol diesters of C8/C10 (MCM) can solubilize 4 to 5 times higher concentrations of oxygen compared to water. Another key observation was the drop in concentration of the dissolved oxygen with increasing temperature. This very interesting effect associated with oxidation kinetics suggests that oxidation may occur at a higher rate at refrigerated temperatures, compared to ambient or accelerated study conditions.

Temperature dependent solubility of oxygen in oils, esters, and water (Cuvelier 2016)
Many studies pointing to the quantum mechanics, thermodynamic, and kinetic aspects of reactions involving dioxygen indicate that it can become reactive due to sensitizers like light and metal catalysts (26, 79, 87, 89). The reactivity of singlet oxygen (1O2) is 1500 times greater than that of oxygen in ground state (44). In the presence of ultraviolet and visible light (300–800 nm range), singlet oxygen reacts directly with oxidation prone bonds (drug or excipient) to form hydroperoxides. This type of reaction skips radical formation and has no induction period (30, 32, 47, 71, 90, 91).
Therefore, ensuring low levels of transition metals while preventing exposure to oxygen are the first line of defense against oxidative degradation. Using inert gas in the overhead/headspace of containers or applying vacuum to the headspace of vials containing polysorbate solutions in order to remove trapped oxygen in liquid formulations is reported to significantly reduce the peroxide concentrations and to help prevent drug degradation. Decanting/pouring liquids onto the inner side of the receiving container is another way to reduce exposure to air.
Time length of the exposure to air, temperature, and or light can drastically change the rate of oxidative reactions. Progressively, oxidation takes place at a higher rate in the presence of moisture, metals (Fe, Cu), and process conditions like heat, mixing time, and mixing speed (92). Physically removing oxygen or peroxides is a last but possible option for highly sensitive (e.g., injectable) forms of protein and peptide formulations. Drying under vacuum (0.1 mm Hg, 48 h) at 25°C to remove residual peroxides from various PEG solutions has been reported to be a successful approach, reducing peroxide levels by up to 90% (19).
Free Fatty Acids
Free fatty acids are naturally present in the unrefined vegetable oils but are almost entirely removed during the refining process and manufacture of lipid excipients. The degree to which free fatty acids participate in the oxidative process depends on their degree of unsaturation, in the following order linolenic (C18:3) < linoleic (C18:2), < oleic (C18:1). Free fatty acids are more susceptible to oxidation than their corresponding esterified fatty acids (35); they speed up the rate of hydroperoxide decomposition, and cause hydrolysis of triglycerides into mono and di-glycerides (48).
Free fatty acids behave as prooxidants due to their capacity to influence the surface charge in emulsified dispersions, the properties of reverse micelles in the bulk oil, or by participation in the chain of oxidative events by oxidizing themselves (44). Addition of small amount of oleic acid to an oil-in-water emulsion, as low as 0.1% in the lipid phase has been shown to increase the hydroperoxide and volatile compound formation. Investigating the effect of free fatty acid concentration on the oxidative stability of oil-in-water (O/W) emulsions, Waraho (93) observed an increasing rate of oxidation as concentration of the free fatty acid (oleic acid ) increased (Fig. 14).

Formation of lipid hydroperoxides as a function of free fatty acid (oleic) concentration in soybean oil emulsions during storage at 15°C in the dark (Waraho 2009)
In dispersed (aqueous) systems, free fatty acid solubility drops by hydrocarbon chain length but increases with rising pH. At pH ranges above their pKa, free fatty acids become negatively charged and so, may impact the surface charge of the emulsion droplets. At pH ranges below their pKa however, free fatty acids may fall out of solution and form insoluble particles. This is an undesirable occurrence, especially in aqueous dispersions involving proteins or RNA, where non-ionic surfactants like polysorbates have been added to prevent protein aggregation. The free fatty acids released from polysorbates are cause for formation of insoluble complexes. Doshi and colleagues (62) investigated the solubilities of the main fatty acids found in polysorbate 20 namely lauric (C12), myristic (C14), or palmitic (C16) acids and their effect on particle formation in two monoclonal antibody formulations. Overall, particle formation was found to be a function of the individual fatty acids’ solubility limits, hence their concentrations and pH conditions. Increase of surfactant concentration and raised pH produced an opposite effect, i.e., increased the solubility limits of the free fatty acids.
Free carboxylic acids may also be responsible for loss of tocopherols, the natural defense of oils against oxidation. One study has demonstrated that complexation with free fatty acids renders tocopherols unavailable for antioxidant activity (43). This type of loss may well explain the loss of oxidative stability in modified oils, the inconsistencies reported in oxidative stability of fixed oils, or conversely the differences found in the efficacy of tocopherols as antioxidants. Hence, evaluation of tocopherols should ideally parallel an assay of free fatty acids in the individual excipients and their evolution in the formulation over time. Excipient manufacturers may provide information, whether appropriate process parameters are in place to ensure certain levels of tocopherol concentrations.
Water Activity and Relative Humidity
Water is naturally present in seed oils (~300 ppm) and is intentionally kept below 0.2% in the production of refined oils and lipid excipients. Several studies have reported that water at low levels (230–1000 ppm) has no effect on the oxidation of an oil despite the presence of a high interfacial tension. It is thought that at these levels, water is bound to the polar compounds in the oil, trapped in multilayer association colloids (41).
Among the few and the earliest works on the effect of relative humidity on lipid oxidation is a study by Maloney (94) which was later adapted for review by Nelson (68). This work involved methyl linoleate (C18:2) in a freeze-dried model, studied under different relative humidity (RH) conditions. Summarized in Fig. 15, the results for peroxide levels interestingly show significantly lower peroxide levels with the samples stored at 40% RH than 20% or 0.1% RH conditions. Since the solubility of oxygen in organic materials (oils) is about 5–10 times greater than in water, it has been proposed that the decrease in the rate of oxidation at higher moisture levels may result from dilution of catalysts and/or other unknown factors (68).

Evolution of peroxide value in freeze-dried methyl linoleate as a function of relative humidity (RH) over time (Maloney 1966, Nelson 1992)
Another decade-old study into the inhibitory effect of water, i.e., its retardation effect on oxidation, involved testing in both the presence and absence of added metals (95). The latter work suggested that metal catalysts like Cu+, Cu2+, Fe2+, and Fe3+ are most active in the dry state. Based on the reaction kinetics of hydroperoxide decomposition, the authors proposed that water may have deactivated the metal catalysts by hydrating their coordination shells and also by hydrogen bonding with hydroperoxides, thus interfering with their bimolecular decomposition reactions (68, 94, 95).
As oxidation progresses to the propagation phases, the water/lipid phase boundary increases with increasing water activity; the polar ROOH groups move to the interface and become bonded to the water, and so they are effectively taken out of the reaction. The situation is reversed in dispersed systems such as O/W emulsions where factors like hydroperoxide concentration and hydration levels can change the mechanisms at play. In this case, increased water content diminishes the rate of hydroperoxide decomposition and reduces rate of free-radical formation (68, 96).
There exists a paradoxical relationship between water activity and the relative rate of oxidation. Further explanations were offered by an investigation of methyl linoleate stability under different water activity settings (96). To this effect, the oxidative stability of methyl linoleate was assessed by measuring the time needed for a gram of methyl linoleate to absorb 800 μL oxygen, i.e., to reach 1% oxidation as endpoint. The results (Fig. 16) show that samples having water activities of 0.3–0.6 took 1.5 to 2 times longer to reach the selected endpoint. The delay in oxidative activity may be due to the unavailability of hydroperoxides formed during propagation step as they are drawn to the oil-water interface; that held by hydrogen bonding, hydroperoxides will not engage in the oxidative process, not before they have saturated the oil-water interface (94). The diminishing oxidation rates observed in the low water activity range of 0.2–0.4 Aω is also explained by the unavailability of water molecules for catalytic effect of transition metals (68, 94,95,96).

Effect of humidification on the time for methyl linoleate to reach 1% oxidation (adapted from Maloney, 1966)
Figure 17 summarizes the relationship between water activity and rates of oxidation. It is likely that in the presence of water, the more polar radicals become mobilized at the oil-water interface to form non-radical species, and this occurs at a much faster rate than in dehydrated conditions. Also, water forms hydrogen bonds with the hydroperoxides as they are produced at the propagation step, thus effectively removing them out of the chain of reactions (41, 68, 96). This knowledge may be of interest to formulation scientists, particularly in the development of powdered or granulated amorphous solid dispersions involving lipids and SEDDS.

Reaction rates in lipid peroxidation as a function of water activity (Nelson 1992)
A significant correlation is established also between water activity and oxidative breakdown products in relation to amino acids, peptides and proteins found in the gelatin capsule shells (96). Due to exposure to hydroperoxides, free radicals are formed in the proteins of gelatin causing scission and degradation of amino acids (histidine, tryptophan, and lysine). This decomposition is strongly correlated with water activity levels. It is proposed that water may contribute to the disappearance of protein radicals by proton donation, or by “quenching” protein free radicals by promoting radical-radical recombination, ultimately leading to the crosslinking of the gelatin (96). In summary, the relationships between lipid oxidation and water activity and relative humidity are complex and their impacts need to be validated and controlled for the formulation at hand.
ANTIOXIDANTS
Antioxidants are a convenient and effective approach to stabilization of complex pharmaceutical dosage forms (90). Table VI provides a list of antioxidants reported in marketed drug products — by route of administration, amount per unit dose, and maximum daily exposure. Antioxidants are applied in very low concentrations largely due to safety of use limits but also for loss of efficacy at higher use levels. As a counter measure, synergistic combinations of antioxidants are commonly explored.
Antioxidants may be classified by different attributes and most appropriately for this review, tabulated by functional mechanisms (Table VII).
Primary Antioxidants
Primary antioxidants are phenolic compounds (Fig. 18) consisting of monohydroxy or polyhydroxy phenol groups with ring substitutions that interrupt the oxidative chain reactions by scavenging free radical species. Among the phenolic antioxidants, butylated hydroxytoluene (BHT), butylated hydroxy anisole (BHA), propyl gallate (PG), and tertiary butyl hydroquinone (TBHQ) are synthetic, whereas tocopherols fall in the natural antioxidant category. By the virtue of their solubility and affinity for the oil phase, phenolic antioxidants can protect the oxidation-prone substrates (drug or excipient) against oxidative stress in lipid-based systems. In emulsion systems, however, other criteria such as partitioning properties of the antioxidants between lipid and aqueous phases may supersede the solubility effects (41, 90, 97, 98).

Chemical structures of phenolic chain-breaking antioxidants
Phenolic antioxidants have very low activation energies for donating hydrogen atom(s). By readily transferring hydrogen atoms to the alkyl, alkoxyl, and peroxyl radicals, they render them more stable, less radical, or non-radical. The antioxidant effect of phenolic compounds may be described by their interactions with radicals as two distinct functions: quenching and termination (Table VIII) (29, 30). Quenching occurs when the antioxidant donates a hydrogen atom to a first radical. Termination occurs when the yielding antioxidant radical (A•) from the first reaction interacts with another radical to form a stable complex, effectively terminating the radical chain. As such, phenolic antioxidants can block oxidative chain reactions both in early (initiation) and also in the later (propagation) stages of the oxidation process (99).
The concentration dependent efficacy of phenolic antioxidants has been well documented, pointing to the key challenge which is establishing the optimal concentration for the formulation at hand. Ideally, the best antioxidant of choice is that which is effective in the lowest possible concentration, enough to counter the oxidative species. Above the optimal concentration, there is a “cut-off” or “inversion” effect, where the antioxidant activity may plateau, diminish, or even reverse due to prooxidant effects. The effect is explained in part by the fate of the antioxidant radical (A•). In excess concentrations, the antioxidant radical may participate in hydrogen abstraction, and chain-transfer reactions, notably in the presence of transition metals. (28,29,30).
To be effective, the selected antioxidant must carry its activity throughout the handling, processing, and packaging of the drug product. Attention is needed to the temperatures at which the antioxidant may degrade. Phenolic antioxidants are especially sensitive to temperature, degrading, and/or vaporizing at elevated temperatures. The thermal resistance of synthetic antioxidants obtained under dynamic thermogravimetric analysis has shown to be in the order of PG > TBHQ > BHA > BHT. When subjected to isothermal conditions (110°C), BHA and BHT exhibited total mass losses after 90 and 155 min, respectively, whereas propyl gallate breakdown occurred at temperatures >146°C. The authors suggested that thermal stabilities of TBHQ, BHA, and BHT may need evaluation at lower temperature conditions (85).
Tocopherols
Tocopherols are natural antioxidants present in most vegetable oils in quantities ranging from 200 to 1200 ppm (14). They protect unsaturated fats and oils by reacting with lipid peroxyl radicals at a much faster rate (104 to 109 M−1 s−1) than those occurring between unsaturated fats and peroxyl radicals (10 to 60 M−1 s−1). One tocopherol molecule can protect about 103 to 108 polyunsaturated fatty acid molecules under low peroxide value conditions. The optimal concentration range for tocopherols in vitro happens to follow the antioxidant-substrate molar ratios of roughly 1:103 to 1:104 as found in nature (33, 53).
Alpha-tocopherol, and its homologues β-, γ-, and δ-tocopherols are the alcohol forms of vitamin E. The antioxidant activity of tocopherol homologues may vary by type of formulation and temperature conditions. At elevated temperature, the antioxidant activity of tocopherols in oils is in the order of δ- > γ- > β- > α-. This order reverses to α- > β- > γ- > δ- at low or mild (<40°C) temperatures. In vivo, α-tocopherol provides a higher antioxidant activity than γ-tocopherol, thus the most studied form among tocopherols (33, 37, 43). Successful incorporation of α-tocopherol in two commercial lipid emulsions (Lipofundin® and Trivé 1000 ®) may explain in part its popularity, as commonly selected antioxidant additive (100).
Like other phenolic compounds, tocopherols have a concentration dependent efficacy limit. A recent comprehensive review summarizes the literature on tocopherols’ antioxidant properties as abundant, contradictory, and strongly system dependent (101). It concludes that it is impossible to predict the precise effectiveness of tocopherols in lipid dispersion system or in bulk oil, because the fate of the tocopherol radical (A•) depends on the chemistry and/or configuration of other molecules in its proximity. Above the optimal range, the excess tocopherols may induce a prooxidant effect at ambient temperatures. Interestingly, this prooxidant activity is diminished at elevated temperatures due possibly to reduced solubility of oxygen in the excipient/formulation system. However, any positive effect of increased temperature on tocopherols’ activity at high concentrations is contrasted with the potential negative effects of heat, which leads to increased rate of hydroperoxide decomposition, increased reactivity of the transition metal ions, and the greater oxidation rates in general. Thus, suppressing oxygen supply to the system may be the more practical approach (33, 53)
Tocopherols are known to degrade gradually upon prolonged storage and much more rapidly on exposure to light (20). They may lose activity due to complexation with free carboxylic acids (43), eliminated during high-temperature refinement of fixed oils, or stripped during fractionation and synthesis of structured lipids (102). It is therefore important to establish at the onset whether the excipient being used has residual levels of antioxidants and if yes, whether their concentrations are consistently monitored.
When evaluating antioxidant efficacy, it is necessary to first identify the substrate(s) that are potentially vulnerable to oxidation. Some general indicators of oxidative processes have been mentioned in the earlier section, “Evaluating oxidative change.” Selecting a suitable endpoint for oxidation and finding the concentration limit for the antioxidant’s efficacy are equally important. The results may be interpreted differently, depending on the parameters being tested (35). The absolutely safe limit to avoid prooxidant activity by tocopherols is ~ 200 ppm according to the latest review (101).
Secondary Antioxidants
Among the examples of secondary antioxidants (Fig. 19), reducing and chelating agents have a crucial role to play in the inhibition of oxidative reactions. Their functions include scavenging of oxygen, sequestering of transition metals, reducing radicals, and preventing the depletion of primary antioxidants by replenishing their lost hydrogen atoms (30, 47, 90).

Reducing and chelating agents
Reducing agents are water-soluble and can protect lipid-soluble antioxidants in a complementary fashion, especially in complex matrix systems. With lower redox potential relative to the substrates that need oxidative protection, reducing agents sacrificially react with oxygen and other reactive species. Their use, however, requires caution because their function may be adversely affected by pH, temperature, and most importantly by presence of transition metals (30).
Ascorbic Acid
Ascorbic acid (vitamin C) is a water-soluble reducing agent that functions by several mechanisms: primarily scavenging (quenching) various forms of oxygen, reducing free radicals, and regeneration of primary antioxidants. The antioxidant efficacy of ascorbic acid is attributed to the ease in which it donates an electron, hence reducing the reactive species in the free radical-mediated chain of events. In the presence of ascorbic acid, tocopherols can readily regenerate from their tocopherol radicals (37, 99).
However, the efficacy of ascorbic acid as antioxidant holds true, so long as trace metals like free iron ions are not prevalent. Otherwise, ascorbic acid can reduce transition metals, such as Fe3+ to Fe2+ boosting the pro-oxidant chemistry of these metals. Since transition metals and their reactions with dioxygen are not spin restricted, the autoxidation of ascorbic acid in the presence of transition metals, is likely to produce O2−, hydrogen peroxide, and hydroxyl radicals (26, 79).
Ascorbyl Palmitate
Ascorbyl palmitate is an ester of ascorbic acid and palmitic acid. It is fully soluble in liquid and solid lipids and alcohols. It inhibits oxidation by donating hydrogen to radical species, hence very effective in minimizing oxidation in oils. Ascorbyl palmitate is considered safe because after intake it can be hydrolyzed to its parts, and so, found in infant formula as a line of defense for protection of polyunsaturated fatty acid chains against oxidation (103). The efficacy of ascorbyl palmitate in protecting cottonseed and olive oils at 400 ppm has been demonstrated (104). Its effectiveness in quenching oxygen in gamma irradiated oils was demonstrated by Lee et al (105). The authors went on to suggest that ascorbyl palmitate, being a colorless product, may be indispensable for protecting oxidative stability of oil soluble vitamins, carotenoids, and tocopherols. Another aspect of ascorbyl palmitate is its relative polarity, thus ability to interact with the oil/water emulsion interface, further discussed below, in the section on “Formulation Considerations”.
Metabisulfites
Sodium and potassium salts of metabisulfite may be used as antiperoxides in solutions and suspensions intended for injectable, oral, ocular, and dermal formulations. By dissociation in water metabisulfites form sulfur dioxide and sodium bisulfite, which can then react with hydroperoxides or form adducts with aldehydes(58). However, due to potential adverse reactions to sulfites, drug products containing sulfiting agents may require a warning on the label. The capacity of Na metabisulfite at different concentrations (10−7 to 10−2 M) to inhibit hydroperoxides formation in vitro, and in vivo were evaluated by Lavoie (106). This study cautions that Na metabisulfite’s antioxidant activity is concentration dependant, because reduction of hydroperoxides may transform Na metabisulfite into a sulfite radical. Hence, under specific conditions, it can contribute to the generation of toxic oxidants.
Edetates
The critical role of transition metals in catalyzing and initiating oxidation is underscored throughout the literature. Since a complete removal of metals from the excipient or the formulation is not a practical solution, addition of substances that sequester metal ions by complexation (chelating agents) can be a reasonable solution. Chelating agents are important additives especially for aqueous dispersions of lipids in micellar solutions or emulsions, where the oxidation prone substrates (lipids and/or poorly soluble drug) come in close proximity with the transition metals of the aqueous phase, i.e., at the surfaces of the dispersed droplets (29). Among the commonly used chelating agents in pharmaceutical dosage forms are citric acid, and the salts of edetate (ethylenediamine tetra acetic acid, or EDTA). Edetates and their salts are quite effective in engaging and essentially removing oxidation catalysts such as Cu+, Cu2+, Fe2+, and Fe3+. Due to their positive charge and solubility, chelating agents can engage with metals in the aqueous phase to form non-reactive complex ions. The complexation may be ionic and/or by covalent bonds, with a net effect of reduced propensity for reactions between the metals and hydroperoxides (31, 77).
Citric acid can act as sequestrant at 50 ppm when added to fixed oils during the final refining stages. Edetates may be used in a similar fashion (~30 ppm), though it may be subject to regulatory hurdles in some countries (31). However, effectiveness of edetate is dependent on its concentration vis-à-vis the metal catalyst. When an equimolar ratio (1:1) of EDTA: Fe2+ were added to a fish oil emulsion, EDTA promoted oxidation (Fig. 20) whereas at higher (2:1 and 4:1) molar ratios, the excess EDTA effectively inhibited hydroperoxide formation(35). Sodium or calcium salts of edetate are more soluble than their edetic acid counterpart, with the latter being subject of decarboxylation if heated to 150°C. Subjecting chelating agents to a temperature of 110°C for prolonged periods can result in thermal instabilities in the order of: ascorbic acid > citric acid > EDTA (85).

Influence of EDTA on the formation of peroxides in fish oil emulsion (Frankel 2005)
Lastly, carotenoids are cited in the literature for their ability to offer protection against light-induced oxidation or photooxidation. Unlike tocopherols which react with singlet oxygen in a non-reversible manner, carotenoids can absorb the excess energy of singlet oxygen, and convert it to ground state, followed by releasing the energy in the form of heat (30). Currently, β-carotene is referenced in the FDA IID for topical formulations. Carotenoids are lipid soluble antioxidants, naturally occurring in oils. β-carotene is most studied of carotenoids, and is known to slow down oil oxidation by light filtering, singlet oxygen quenching, sensitizer inactivation, and free radical scavenging due to its polyunsaturated hydrocarbon chain. One mole of β-carotene can quench 250 to 1000 molecules of 1O2 at a rate of 1.3 × 1010M−1 s−1 (37, 107).
FORMULATION CONSIDERATIONS
Oxidative Stability in the Presence of Polyethers and PEGs
Polyethers may constitute the molecular scaffolding of the lipid excipients (e.g., polyoxylglycerides, poloxamers, polysorbates) and or used in combination with lipid excipients in a formulation. As indicated, they can be significant source of hydroperoxides. Moreover, their oxidative degradation results in the formation of secondary impurities such as formaldehyde and formic acid, largely responsible for cross linking in gelatin capsules. Formic acid is frequently reported to be responsible for the formation of N-formyl impurities with protein biopharmaceutics or drugs consisting of primary or secondary amino moieties (56). There is little published on the oxidation pathways or the stabilization of polyethers. Hemenway and colleagues (58) examined the impact of various formulation variables (discussed above) and also the effect of antioxidants on the evolution of aldehydes in PEG 400 solutions. The evolution of the total aldehydes in 50% solutions of PEG 400 were examined following addition of ascorbic acid, BHA, BHT, propyl gallate, or Na metabisulfite. As shown in Fig. 21, the total aldehyde levels reduced in all the cases but the most significant results for aldehyde reduction were associated with use of 1% Na metabisulfite, followed by 0.1% propyl gallate, and 0.02% of BHA or BHT. The significant effect of Na metabisulfite on aldehyde reduction in PEG 400 was explained by the dissociation in water of sodium metabisulfite into sulfur dioxide and sodium bisulfite, with the latter reacting with aldehydes to form bisulfite adducts, and with oxygen to form sodium sulfate.

Effect of antioxidants on the total aldehyde levels in 50% solutions of PEG400 in water incubated at 40°C for up to 90 days (adapted from Hemenway, 2012)
It is worthwhile to note that included in the aforesaid study was D-alpha tocopheryl polyethylene glycol-1000 succinate (TPGS), an excipient known for surfactive properties and not as an antioxidant (58). Interestingly, the aldehyde levels with TPGS (not shown) were similar to that obtained with propyl gallate in Fig. 21. Since the tocopherol moiety in the surfactant (TPGS) is bound, i.e., not free to engage in hydrogen atom transfer, it is likely that the protective effect of TPGS was manifested by a different mechanism, such as formation of micellar particles that physically entrap and immobilize the reactive species.
Inhibition of Oxidation in Fixed Oils and Partial Glycerides
Fixed oils like soybean oil are commonly used in parenteral emulsions (108), ocular drops, nasal sprays, oral capsules, and topical emulsions. Hence, their oxidative stability is critical to understand and to prevent. Until early 1990s, oxidation was thought to occur at the air-oil interface (43, 97). Contrasting this notion is scientific evidence that tiny microstructures, described as association colloids exist in bulk/fixed oils as finely dispersed structures, acting as the site of oxidative events – as put forth by Brimberg (109, 110). The concept has been corroborated by other scientists (29, 31, 41, 102, 111, 112), stipulating that water, in trace amounts (<0.1%) occupies the core of the reverse micelles, which are delineated from the continuous oil phase by amphiphilic and trace quantities of phospholipids, mono- and di-, diglycerides and free fatty acids. Figure 22 depicts the proposed scheme for a reverse micelle dispersed in the bulk oil, where hydroperoxides and other amphiphiles aggregate to form reverse micelles. Hydrophilic moieties orient their hydrocarbon tails toward the continuous hydrophobic phase, with their polar heads extending to the hydrophilic region in the micelle exterior. Free fatty acids formed during propagation can also gradually interact with the reverse micelles, or existing microstructures by accumulating at the emulsion interface according to their solubility characteristics. Oleic acid for example is insoluble in water and is therefore likely to position itself at the oil phase of the interfacial region where hydroperoxides would accumulate. This proximity may explain acid catalyzed hydroperoxide decomposition and attraction of transition metals to the water rich regions and elucidates the role of free fatty acids as prooxidants (102).

Depiction of reverse micelles in bulk oil (adapted from Budilarto 2015)
Lipid oxidation mechanisms can vary by the reactive species being formed, their substrates, and the physicochemical environment in which it occurs (111). Oxidation in oils is initially slow, characterized by a pseudo-first order build-up of hydroperoxides. The time needed for the oil to reach the second and rapid oxidation rate phase is called lag phase, or induction period (Fig. 23) when reactions occur between hydroperoxides and unsaturated fatty acids. With the latter being of negligible concentration, the reaction rate can be described as zero order. As the reactions progress, the hydroperoxide levels rise to reach a critical micelle concentration (CMC) at which time the lag phase ends, and the reaction rates take an exponential order (31, 41, 102, 109, 110).

Peroxide value evolution during the autoxidation of flaxseed oil at 40°C, 75% RH over 30 days (Budilarto 2015, Talbot 2016)
A dated proposition for stabilization of oils against oxidation is one that suggests polar antioxidants (ascorbyl palmitate, tocopherols) may be most effective in apolar systems (oils) and those with lipophilic/phenolic nature perform better in emulsified systems. This proposition has been challenged by newer works (24, 47, 90) stipulating that the concept may apply only to limited scenarios in a narrow range of polarity; that antioxidant polarity and surface activity are important but not decisive determinants of activity in vegetable oils. Confirming the new perspective, is a study of possible relationship between antioxidant activity and antioxidant polarity in menhaden fish oil (98). Fish oil, rich in polyunsaturated fatty acids (PUFA), is typically dosed into gelatin capsules for nutraceutical end use, therefore an excellent model for evaluating antioxidants. The surface activity of structurally related antioxidants, having similar radical scavenging properties in menhaden oil, was as follows: δ-tocopherol > α-tocopherol > BHT. Overall, δ-tocopherol was most effective in reducing interfacial tension and outperformed α-tocopherol in the stabilization of both menhaden oil and menhaden oil emulsions. BHT on the other hand, being the least polar among the said antioxidants, was even more effective than δ-tocopherol in the samples incubated at 37°C (Fig. 24). Hence, surface activity and polarity could not be the sole basis for predicting antioxidant performance.

Influence of BHT and tocopherols (2.8 mmol kg−1) on delaying lipid hydroperoxides formation in menhaden oil at 37°C in the dark (adapted from Chaiyasit 2005)
An earlier work exploring the efficacy of various antioxidants in fish oil, involved stability testing at ambient temperature for 28 weeks, conditions that were deemed relevant to the process and use conditions for the oil (113). At the completion of the experiment, it was concluded that a ternary combination of δ-tocopherol (2% by weight) with ascorbyl palmitate (0.1%) and lecithin (0.5%) was unusually effective in preserving the integrity of the PUFA rich fish oil. These results contradicted previous reports in several ways: (i) much higher levels of tocopherol were effective than previously reported and (ii) protection increased with higher concentrations of ϒ/ δ-tocopherol but not with α-tocopherol. Explanations for these observations are found in the much later work by Evans (114) involving soybean oil. The aim of the latter group was to establish optimal tocopherol concentrations, or the cut-off point rather, beyond which tocopherols would behave as prooxidants. These were found to be ~100 ppm for α-tocopherol and ~300 ppm for ϒ-tocopherol, but no limit could be established for δ-tocopherol. The activity of δ-tocopherol increased with increasing concentration up to 1900 ppm levels tested. The authors ranked the antioxidant efficacy of tocopherol as α > ϒ > δ, an inference based on the low optimal concentration of α-tocopherol (100 ppm), beyond which it can act as prooxidant (37).
In fixed oils, the key role of the phenolic (primary) antioxidants and synergists is to stabilize the contents of the reverse micelles (Figs. 22 and 23) where they can be most effective during the initiation and early propagation phases (33, 53). Antioxidants can assume different roles in the fixed oil, depending on the phase of the oxidative chain reactions. In the early, initiation phase, an antioxidant may act as stabilizer of reverse micelles loaded with water, hydroperoxides, and other amphiphiles, but then act as scavenger of radicals during the propagation phase. This means polar antioxidants become important when moisture, hydroperoxides, and free fatty acids are all present. For instance, the presence of phosphatides in the bulk lipids enhances the antioxidant activity of α-tocopherol by forming aggregates and therefore improves proximity between tocopherol and the site of oxidation. The net effect of this is a better partitioning in the aqueous (polar) phase of reversed micelles where radicals are formed and trapped. Therefore, an antioxidant’s function can be chemical, e.g., hydrogen atom transfer as well as physical in nature, e.g., partitioning and orientation in its surrounding media (41, 111). Since antioxidant function is system dependent, Shahidi (90) offered that in bulk/fixed oils, the effect of antioxidant solubility in the oil phase dominates (is more important) over its contribution to interfacial phenomena, the latter having importance in micellar or emulsion systems.
Inhibition of Oxidation in Emulsions
Lipid-based emulsions are predominantly oil-in-water (O/W) systems that facilitate incorporation of both hydrophilic and lipophilic drugs. In an emulsion, the various pro-oxidant and antioxidant components distribute themselves according to their respective solubility characteristics and surface activities. The nature of the distribution is sensitive to a host of variables, such as the nature and concentration of the emulsifier(s), aqueous phase, oil phase, prooxidants, drug, and antioxidant, to name a few. As a result, there is little agreement amongst publications on the role of emulsion droplet size, or antioxidant polarity on oxidative stability. However, the current understanding strongly supports the hypothesis that the oil-water interfaces are critically involved in oxidative events (80, 92). Therefore, antioxidant molecules should ideally be found (or positioned) in the proximity of the water-oil interface of the emulsion. For this to occur, the antioxidant’s molecular size, and hydrophilic/lipophilic balance become significant.
The mechanism of oxidation or its inhibition at the oil-water interface is schematically presented in Fig. 25. Being surface active, hydroperoxides are likely to gravitate towards the oil-water interface, where they come in contact with the water-soluble metal ions dissolved in the aqueous phase (29, 77, 115, 116). The reactions that follow include formation of peroxyl (HO•) and alkoxyl (RO•) radicals in their vicinity, interacting with various substrates, including the alkyl chains in the glycerides located in the interior of the oil droplet. Fig. 25 also depicts how radicals can be inactivated by antioxidants capable of positioning at the water/oil interface, e.g., by ascorbyl palmitate, or propagated by reduction of iron due to a water-soluble reducing agent in the aqueous phase such as ascorbic acid (73, 117).

Schematic representation of oil-in-water droplet (not to scale) with potential reactions of peroxyl radical, hydroperoxides, and antioxidants (adapted from McClements 2018; Waraho 2011 A; Goddard 2012)
Antioxidant use levels need to be justified and optimized for safety and regulatory reasons, but also to avoid efficacy cut off effects. As indicated in the primary antioxidant section, the concentration dependent inversion of an antioxidant efficacy is in part explained by chemical reactions, where the antioxidant radicals, when in excess, may behave as prooxidants. Another plausible explanation lies with the partitioning, positioning, and mobility of the antioxidants in the multi-phase system (90). The latter is supported by experiments showing that polar antioxidants, though unable to decrease the surface tension between the air and the product, could in fact decrease the interfacial tension, at the boundary between oil and water (98, 102, 111). Several studies suggest that the ability of the antioxidant to partition at the oil–water interface is more effective in limiting oxidation in the oil phase than direct addition of the antioxidant to the oil phase of emulsions (35, 113, 118). For example, ascorbyl palmitate may behave as antioxidant in the presence of phospholipids, due to the ability of the latter to interact with the ascorbyl radical (113). Depending on the choice of surfactant, co-surfactant, or oil phase and combinations thereof, there are an unlimited number of possible conformations to exist. Ultimately, the stereochemistry, and partitioning of an antioxidant within the oil phase, the aqueous phase, and the oil-water interface can influence oxidative stability (15, 74, 92, 117).
Should a surfactant concentration exceed its’ CMC, there will be tendency for the unadsorbed fraction to disperse and to rearrange into new formations such as micelles. Likewise, polar (amphiphilic) antioxidants (tocopherols, ascorbyl palmitate) can self-assemble into micelles, lamellar structures, and other association colloids (98). Competition for self assembly may help explain the cut-off effect of antioxidant effectiveness vis-à-vis its concentration. Interactions between the emulsifier and polar antioxidants may lead to reduced space for the antioxidant molecules to occupy at the emulsion interface where antioxidant effect is needed (Fig. 26). The antioxidant molecules may become trapped in the excess emulsifier micelles, or be literally “taken out of circulation,” taken to the aqueous phase where they can’t be efficient (41, 112, 119).

Excess amphiphilic antioxidant, or excess emulsifier may compete for positioning around the oil droplets or form their own respective micelles (Laguerre, 2015)
It is said that oxidation of encapsulated bioactive compounds in emulsions is triggered by permeation of free radicals generated at the oil-water interface. Considering the continuous movement and competition between the polar and apolar components for positioning within the oil and aqueous phases, McClements (29) hypothesized that lipid oxidation in emulsions may be “limited by the speed at which free radicals, hydroperoxides, or lipids can diffuse from one region to another within a droplet.” Others have also pointed to the constant exchange of positions between the interior and exterior of the oil droplets due to Brownian motion (92, 120).
To establish a correlation between the movement of the free radicals within the droplets and the overall oxidative stability of the emulsion system, Pan et al (118) used curcumin, as model drug, and fluorescence techniques to trace the movement of the radicals in two emulsion systems consisting of Tween 20 or lecithin. The permeation rate from the aqueous phase to the oil phase for peroxyl radicals was significantly higher (p<0.05) for emulsions stabilized with Tween 20 and oxidized lecithin compared to that of native lecithin. The results suggested an inverse relationship between permeation rate of the radicals and the stability of curcumin. This work also found the curcumin stability to be higher in the lecithin-based emulsions, possibly due to the negatively charged headgroups of the phospholipids in lecithin attracting the positively charged metal ions, thus inhibiting their engagement in oxidative events. A similar effect of phospholipids is reported by another work where liposomal suspensions were subjected to forced oxidation under varying concentrations of ferrous sulfate, and tested for stability for 14 days at 40°C (121). The liposome suspensions spiked with 0.2- or 1-mM iron had hydroperoxide concentrations of < 3 mmol kg−1 oil, whereas those spiked with higher levels of iron (10 or 48 mM) formed higher amounts of primary and secondary lipid oxidation products (Fig. 27). It was proposed that liposomal phospholipids may resist, or possibly delay the iron-catalyzed oxidation only until a certain iron concentration, above which they oxidize rapidly. These, and other studies discussed below indicate that the type and concentration of the emulsifiers at the water-oil interface can strongly influence the oxidative stability of the system (80, 121,122,123).

Hydroperoxide concentration of iron-loaded liposome suspensions containing different levels of iron, for a fixed phospholipid concentration, incubated at 40°C (Cengiz 2021)
To evaluate the effect of surfactant concentration, Berton and colleagues (122) developed a method for measuring the excess (unadsorbed) emulsifiers in the aqueous phase of O/W emulsions. The method consisted of direct aqueous transesterification of surfactants in the aqueous phase, followed by detection by gas chromatography. As part of a subsequent study (123) Berton investigated the specific role of the adsorbed emulsifiers on lipid oxidation of O/W emulsions. Emulsions of similar droplet size distribution were prepared with polysorbate 20, one at low concentration (4 g L−1) and another in excess concentration (35 g L−1). The samples were subjected to oxidation at 25°C by addition of 200 μM of 1:1 FeSO4-EDTA complex. Oxygen uptake, conjugated dienes, and volatile compound formation in the incubated samples were monitored. Figure 28 shows part of the study findings, where oxygen consumption of the polysorbate 20 emulsion was significantly reduced (oxidative stability improved) with the higher concentration (excess) polysorbate 20 emulsions during an incubation period of 70 hours at 25°C. These findings and other works (80) strongly suggest that the stability of the O/W dispersions is a critical parameter affected by adequate packing density of the emulsifier molecules at the oil-water interface, providing a barrier effect against lipid oxidation, or conversely modulating the accessibility of the lipid phase to water-soluble prooxidants. There are a number of established methods for evaluating emulsion/dispersion size stability. The list includes centrifugation, freeze thaw cycles, and accelerated stability conditions to establish phase separation. More specific tests could be monitoring of conductivity, dispersion size (photon correlation spectroscopy), refractive index (measure of polydispersity), and zeta potential which is a measure of surface charge and potential changes over time.

Effect of excess emulsifier in the aqueous phase at 25°C in the dark (Berton, 2011)
As indicated above, there exists a continuous competition between the polar and apolar components for positioning within an emulsion. Also, the speed at which free radicals, hydroperoxides, or lipids can diffuse from one region to the next within a droplet may influence the oxidative stability of the system. If the movement of radicals from droplet core to the interface area is a critical variable, droplet size should theoretically play a key role in the prooxidant or antioxidant events. In theory, a decrease in emulsion droplet size is equivalent to an increase in the droplet surface area, therefore a larger interface between the aqueous and oil phases. One would expect that the smaller the droplet size, the shorter would be the distance between its core and the interface, requiring less time for hydroperoxide molecules to travel across (116).
Since lipid oxidation in O/W systems generally proceeds from the interface to the interior of the oil droplet, some studies have focused on the role of droplet size, and others investigated the droplet interface. However, studies focusing on correlations between emulsion droplet size have produced mixed results. In a recent publication, the effect of droplet size on the oxidative stability of curcumin was evaluated in O/W emulsions consisting of medium chain triglycerides and a natural emulsifier from soapbark tree (120). Applying different mixing speed, time, and pressure during homogenization, emulsions with mean droplet diameters of approximately 80, 260, 2500, or 21,000 nm were obtained without adjusting the emulsifier content. Following stability testing at 55°C for 3 weeks, curcumin losses of ~9% with the largest droplets and up to 70% loss in the smallest droplets were reported. Pointing to the higher surface area provided by the smaller droplets facilitating interaction of curcumin with the aqueous phase, the authors proposed that droplet size had a critical role in the oxidative stability (or degradation) of curcumin (Fig. 29). Even though the degree of curcumin exposure to the aqueous phase may be a plausible variable, the findings do not support the role of particle size. More specifically, the study does not account for the higher levels of energy input during mixing for the finer emulsions, the impact of phase separation within days of the emulsion preparations, or the potential presence of transition metals introduced by the natural surfactant.

Oxidative degradation of curcumin in emulsions with identical emulsifier content but obtained by greater energy input to create the smaller droplet sizes (adapted from Kharat 2020)
The effect of droplet size on the oxidative stability of emulsions consisting of soybean oil having 53% linoleic acid or fish oil consisting of 37% docosahexaenoic acid (DHA) glycerides were evaluated by Azuma and colleagues (124). During forced degradation experiments, oxygen consumption in the solution, peroxide formation, and unoxidized PUFAs concentrations were measured. The results were mixed, depending on the oil type, which differed mainly in fatty acid composition. Whereas the oxidative stability of fish triglycerides increased with decreasing the droplet size, the reverse effect of the droplet size was observed on the oxidation of soybean oil triglycerides. These findings indicate that fatty acid conformations (Fig. 3) can have as significant a role in emulsion stability as droplet size.
Despite variations in mixing speed and lipid concentrations (10% and 30%), Osborn and colleagues found no significant effect of droplet size on oxidative stability of emulsions made of structured lipids derived from canola oil and caprylic acid interesterification (65). Their work was retrospectively challenged by Nakaya (116) citing that the interesterified (structured) lipids study by Osborn was not protected against metal induced oxidation; that addition of a chelating agent to offset the effect of transition metals may have produced a different outcome. In their work, Nakaya (116) found the oxidative stability of O/W emulsions obtained from three different oil types to increase with decreasing droplet size. The study involved monitoring of hydroperoxide content, hexanal formation, and residual oxygen in the headspace of the sample vials containing emulsions having small, medium, and large droplet sizes (0.8 μ; 3.3 μ or 10.7 μ, respectively). The emulsions with the smallest droplet size distribution (0.8 μ) had significantly lower residual oxygen in the headspace, lowest peroxide levels, and hexanal formation compared to the emulsions having larger droplet size. Nakaya hypothesized that the emulsifier molecules, as they orient their hydrophobic arms inward toward the interior of the droplet, immobilize the triglycerides in the tight space, that the larger droplets offer far less restriction to mobility of the oil located in the interior of the droplets. Hence the positioning of the emulsifier molecules at the oil-water interface improves the oxidative stability of the oils. Others have hypothesized that surface active molecules, by wedging themselves onto the interfacial region of oil droplets (Fig. 26), may immobilize the oil droplet contents and so, provide oxidative stability (73, 74, 77, 112, 116, 117). Whereas emulsifiers stabilize droplets against aggregation, the introduction of the co-emulsifier to the system may help further inhibit lipid oxidation in emulsions (92).
The concentration dependent efficacy (cut-off effect) applies to lipophilic (phenolic) as well as water soluble antioxidants. Water-in-oil emulsions with polysorbate 80, consisting of refined sunflower oil mixed with polyglycerol esters as the continuous (oil) phase were prepared (87). The antioxidant effect of ascorbic acid, added to the aqueous phase at 5 ppm or 100 ppm levels was compared to that obtained with TBHQ as antioxidant in the oil phase at 200 ppm. The results (Fig. 30) point to prominent efficacy of TBHQ at the level tested relative to that with 5 ppm ascorbic acid. Higher level of ascorbic acid (100 ppm) did not provide additional benefit.

Effect of ascorbic acid and TBHQ on oxidative stability (peroxide formation) of water-in-oil emulsions at 35°C (adapted from Rege 2015)
Effect of Interfacial Charge and pH on Oxidative Stability of Emulsions
The influence of pH on the oxidative stability of ionizable drugs is well documented (23). As for the effect of pH on the oxidative stability of lipid formulations, it is difficult to find confluence in the literature (77). A noticeable drop in pH of emulsions over time may indicate oxidative change due to liberation (or formation) of acidic moieties, including small carboxylic acids. Tamilvanan and colleagues (12) monitored the pH of castor oil-based nanoemulsions incubated at 4°C, 25°C, or 37°C for up to 6 months. The findings summarized in Fig. 31 indicated that pH began to drop within a month of storage at 25°C or 37°C, and eventually by 3–4 units by the end of the 6-month incubation. Under the conditions of the study, the pH of the nanoemulsions stored at 4°C was most stable, dropping by one unit after 6 months.

Drop in pH of castor oil-based nanoemulsion over months of storage at 4°C, 25°C, and 37°C (Tamilvanan 2010)
Generally, the non-ionic surfactants tend to be better facilitators for the transfer of antioxidant molecules between micelles and the oil-water interface. Should the surfactant carry a charge, it can influence the oxidative behavior of its formulation, especially in relation to transition metal ions. The repulsion or attraction of ferrous iron to the surface charge of droplets having different surfactants were evaluated by Mancuso (125). Highly unsaturated salmon oil emulsions were obtained with the following surfactants: anionic sodium dodecyl sulfate (SDS), non-ionic polysorbate 20, and cationic dodecyl trimethylammonium bromide surfactants (DTAB). The samples were spiked with 15 μL of ferrous iron (0.072 M) prior to incubation. The highest oxidative instability was observed with SDS followed by polysorbate 20, and DTAB. This ranking of oxidative stability confirmed the hypothesis that the positively charged metal ions were being attracted to the negatively charged droplet surfaces in the case of SDS, or conversely repelled by the positively charged droplets in DTAB, resulting in retardation of oxidation in the lipids situated in the interior of the droplets. Addition of EDTA (0.5–4.0 mM) or reducing the pH of the aqueous phase (7.0 to 3.0) in the aforesaid emulsions produced the opposite effect. Since oxidation was effectively inhibited by the addition of EDTA, it was proposed that iron is the key cause of oxidation in salmon oil O/W emulsions. Tracing the iron concentration levels in the aqueous phase, the authors also demonstrated that metal ions can, at reduced pH conditions, partition into the continuous aqueous phase, away from the oil-water interface. (125).
An association between lowered pH and oxidative stability relative to free fatty acid concentrations was put into evidence by Waraho and colleagues (73, 93, 117). Oxidative stability of soybean oil emulsions adjusted to pH 2, 4, 6, or 8 was tested with addition of 1% oleic acid as oxidant or incorporation of EDTA as antioxidant. The surface charge of the emulsion droplets, measured as zeta potential revealed a significant drop in charge (increasingly negative) with increasing pH, indicative of the fatty acids becoming negatively charged at pH values above their respective isoelectric points, hence becoming more soluble in the aqueous phase. A dramatic reduction in oxidation of soybean oil emulsions at pH values of <6. Moreover, addition of EDTA (200 μM) strongly inhibited lipid oxidations with samples spiked with 1% oleic acid. The results indicated that negatively charged free fatty acids can strongly attract transition metals onto the interfacial region of the emulsion droplets, where they can participate in the oxidative events (93).
Synergies
Synergistic use of antioxidants becomes important when the antioxidant concentration is at maximum allowed (safety) limit; and/or when antioxidant concentration is at the edge of its optimal range, beyond which efficacy is reversed. Combination of a metal chelator (e.g., EDTA) and a free radical scavenger (e.g., tocopherol) has demonstrated antioxidant activities that couldn’t be observed with either type used alone. Tocopherols work synergistically with ascorbic acid and phospholipids thanks to their ability to regenerate and to recycle their very own radicals as intermediates (30, 35, 37). Primary (phenolic) antioxidants are often combined with reducing and/or chelating agents for synergistic effect. The strategy helps diminish metal catalyzed oxidation, thus fewer free radicals for the primary antioxidant to tackle. Combined use of phenolic antioxidants such as BHA with BHT or BHA with PG has produced synergistic effects, explained by steric hindrance effects (47). Moreover, antioxidants can show synergy by physically associating with the colloidal interfaces according to their position (location). Hence, their function may be synergistic from both chemical, i.e., scavenging radicals via hydrogen donation, and physical, i.e., by partitioning at the oil-water interfaces.
SUMMARY
Drug-excipient interactions are often attributed to the presence of unwanted impurities like transition metals, hydroperoxides, and aldehydes. A broader examination of the failing formulations reported in the literature points to often inadequate choices in excipient type or quality, failure in compatibility screening, and missed opportunities to prevent oxidative stress. From drug development perspective, the overall goal is the application of lipid-based excipients in new and improved drugs, without compromising drug stability and efficacy. The key to managing oxidative stability however lies in understanding and identifying the root causes of instability, followed by protective measures against oxidation. In this context, propensity to oxidation should be questioned from different but complementary perspectives: (i) whether catalysts or prooxidant impurities are present within the formulation system; (ii) which component(s) within the drug or excipients are potential substrates for oxidative change; (iii) if or how does the change may compromise formulation efficacy and/or stability; (iv) whether a change in the selection of raw materials, formulation, process, or addition of antioxidants can help mitigate oxidative challenges.
Oxidation of biomolecules, including lipids is due largely to the catalytic role of transition metals and their ability to initiate oxidation by transforming dioxygen to its reactive singlet oxygen state. Light can also initiate the process but easier to manage relative to transition metals. Past initiation, radicals, and hydroperoxides are produced which proceed to propagate the chain reactions. If propagation is allowed to continue, a host of secondary by-products can come to form. Hence, oxidation is best managed by predictive and pre-emptive measures.
The antioxidant strategy has for long been employed for quality preservation of pharmaceutical and food products. Antioxidants can interfere with critical oxidative phases by chemical intervention, thus inhibiting onset of reactions, or by significantly reducing oxidative reaction rates, thereby protecting the dosage form integrity and drug stability. Chelating agents can be a first line of defense against metal assisted initiation of oxidation. A complementary and often necessary approach is to add phenolic antioxidants to scavenge radicals and to retard the propagation reactions. Synergistic combinations of reducing, chelating, and radical scavenging molecules are also recommended. Ideally, antioxidant selection should include evaluation of solubility in the intended formulation phase, more specifically where the oxidation-prone substrate (e.g., drug active) is likely to be located.
Lipid excipients have shelf stability of two to three years if kept under the manufacturer’s recommended settings or in their unopened original package. To avoid introducing unnecessary oxidative stress use of aged samples, especially leftover material from an already opened package should be avoided. Flushing of the headspace with an inert gas like nitrogen is a must. Since an excipient may be used in different ways and for different functionalities, it is important to delineate the excipient parameters that are relevant to the formulation/process at hand. For example, lowered pH due to rising free acids may have a greater influence on formulation stability/performance, than hydroperoxide levels in the <30 mEq O2 kg−1 range.
Overall, formulation design and appropriate process parameters can have greater impact on drug product stability, more than addition of antioxidants which come with limitations of their own, be for safety levels or reversal of antioxidant effects beyond the optimal concentrations. The challenge with the antioxidant strategy is to find the right combination and use levels for drug products. The current knowledge is largely based on research into vegetable oils and food systems that may not be relevant to drug product development settings. Research is needed to better understand the physic-chemical interactions between antioxidants and the oil-water interfaces found in pharmaceutical emulsions, microemulsions, and liposomal formations.
Change history
14 June 2022
A Correction to this paper has been published: https://doi.org/10.1208/s12249-022-02318-5
References
Bala V, Rao S, Bateman E, Keefe D, Wang S, Prestidge CA. Enabling oral SN38-based chemotherapy with a combined lipophilic prodrug and self-microemulsifying drug delivery system. Mol Pharm. 2016;13(10):3518–25.
Saxena S, Singh HN, Agrawal VK, Chaturvedi S. Lipid excipients in self emulsifying drug delivery systems. Asian Journal of Biomedical and Pharmaceutical Sciences. 2013;3(22):16–22.
Khames A. Investigation of the effect of solubility increase at the main absorption site on bioavailability of BCS class II drug (risperidone) using liquisolid technique. Drug Deliv. 2017;24(1):328–38.
Lam M, Ghafourian T, Nokhodchi A. Liqui-pellet: the emerging next-generation oral dosage form which stems from liquisolid concept in combination with pelletization technology. AAPS PharmSciTech. 2019;20(6):231.
Lo JB, Appel LE, Herbig SM, McCray SB, Thombre AG. Formulation design and pharmaceutical development of a novel controlled release form of azithromycin for single-dose therapy. Drug Dev Ind Pharm. 2009;00(00):090730035508060–8.
Dumont C, Bourgeois S, Fessi H, Jannin V. Lipid-based nanosuspensions for oral delivery of peptides, a critical review. Int J Pharm. 2018;541(1-2):117–35.
Thanki K, Date T, Jain S. Enabling oral amphotericin B delivery by merging the benefits of prodrug approach and nanocarrier-mediated drug delivery. ACS Biomater Sci Eng. 2021.
Dumont C, Bourgeois S, Fessi H, Dugas PY, Jannin V. In-vitro evaluation of solid lipid nanoparticles: ability to encapsulate, release and ensure effective protection of peptides in the gastrointestinal tract. Int J Pharm. 2019;565:409–18.
Hung WH, Chen PK, Fang CW, Lin YC, Wu PC. Preparation and evaluation of azelaic acid topical microemulsion formulation: in vitro and in vivo study. Pharmaceutics. 2021;13(3).
Seguy L, Groo AC, Goux D, Hennequin D, Malzert-Freon A. Design of non-haemolytic nanoemulsions for intravenous administration of hydrophobic APIs. Pharmaceutics. 2020;12(12).
Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Williams HD, Pouton CW et al. 50 years of oral lipid-based formulations: provenance, progress and future perspectives. Adv Drug Deliv Rev. 2016;101:167–94.
Tamilvanan S, Kumar BA, Senthilkumar SR, Baskar R, Sekharan TR. Stability assessment of injectable castor oil-based nano-sized emulsion containing cationic droplets stabilized by poloxamer-chitosan emulsifier films. AAPS PharmSciTech. 2010;11(2):904–9.
Barber BW, Dumont C, Caisse P, Simon GP, Boyd BJ. A 3D-printed polymer–lipid-hybrid tablet towards the development of bespoke SMEDDS formulations. Pharmaceutics. 2021;13(12).
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–46.
Hovorka SW, Schöneich C. Oxidative degradation of pharmaceuticals: theory, mechanisms and inhibition. J Pharm Sci. 2001;90(3):253–69.
Tomlinson A, Demeule B, Lin B, Yadav S. Polysorbate 20 degradation in biopharmaceutical formulations: quantification of free fatty acids, characterization of particulates, and insights into the degradation mechanism. Mol Pharm. 2015;12(11):3805–15.
Kishore RS, Pappenberger A, Dauphin IB, Ross A, Buergi B, Staempfli A, et al. Degradation of polysorbates 20 and 80: studies on thermal autoxidation and hydrolysis. J Pharm Sci. 2011;100(2):721–31.
Holm R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv Drug Deliv Rev. 2019;142:118–27.
Kumar V, Kalonia DS. Removal of peroxides in polyethylene glycols by vacuum drying: implications in the stability of biotech and pharmaceutical formulations. AAPS PharmSciTech. 2006;7(3):62.
Darji MA, Lalge RM, Marathe SP, Mulay TD, Fatima T, Alshammari A, et al. Excipient stability in oral solid dosage forms: a review. AAPS PharmSciTech. 2018;19(1):12–26.
Stella VJ. Chemical drug stability in lipids, modified lipids, and polyethylene oxide-containing formulations. Pharm Res. 2013;30(12):3018–28.
Wasylaschuk WR, Harmon PA, Wagner G, Harman AB, Templeton AC, Xu H, et al. Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci. 2007;96(1):106–16.
Waterman KC, Adami RC, Alsante KM, Hong J, Landis MS, Lombardo F, et al. Stabilization of pharmaceuticals to oxidative degradation. Pharm Dev Technol. 2002;7(1):1–32.
Schaich KM. Thinking outside the classical chain reaction box of lipid oxidation. Lipid Technol. 2012;24(3):55–8.
Khanum R, Thevanayagam H. Lipid peroxidation: Its effects on the formulation and use of pharmaceutical emulsions. Asian J Pharm Sci. 2017;12(5):401–11.
Miller D, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med. 1990;8(1):95–108.
Deliang Z. Understanding physicochemical properties for pharmaceutical product development and manufacturing II: physical and chemical stability and excipient compatibility. Journal of Validation Technology. 2009;15(3).
Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30(4):277–90.
McClements DJ, Decker EA. Lipid oxidation in oil-in-water emulsions: impact of molecular environment on chemical reactions in heterogeneous food systems. J Food Sci. 2000;65(8):1270–82.
Shahidi F, Zhong Y. Lipid oxidation and improving the oxidative stability. Chem Soc Rev. 2010;39(11):4067–79.
Talbot G. The stability and shelf life of fats and oils. the stability and shelf life of food 2016. p. 461-503.
Baertschi SW, Jansen PJ, Alsante KM. Stress testing: a predictive tool. In: Baertschi SW, Alsante KM, Reed RA, editors. Pharmaceutical stress testing: predicting drug degradation. Drugs and the Pharmaceutical Sciences: Informa Healthcare; 2011. p. 10–49.
Kamal-Eldin A, Appelqvist LA. The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids. 1996;31(7):671–701.
Ha E, Wang W, Wang YJ. Peroxide formation in polysorbate 80 and protein stability. J Pharm Sci. 2002;91(10):2252–64.
Frankel EN. Lipid Oxidation [EPub]: Elsevier; 2005.
Crowley P, Martini LG. Excipients for pharmaceutical dosage forms. Pharmaceutical Technology. 2001.
Choe E, Min DB. Mechanisms and factors for edible oil oxidation. Compr Rev Food Sci Food Saf. 2006;5(4):169–86.
Cao J, Deng L, Zhu XM, Fan Y, Hu JN, Li J, et al. Novel approach to evaluate the oxidation state of vegetable oils using characteristic oxidation indicators. J Agric Food Chem. 2014;62(52):12545–52.
Zielinski ZA, Pratt DA. Lipid peroxidation: kinetics, mechanisms, and products. J Organomet Chem. 2017;82(6):2817–25.
Ha E-S, Lee S-K, Choi DH, Jeong SH, Hwang S-J, Kim M-S. Application of diethylene glycol monoethyl ether in solubilization of poorly water-soluble drugs. Journal of Pharmaceutical Investigation. 2019;50(3):231–50.
Budilarto ES, Kamal-Eldin A. The supramolecular chemistry of lipid oxidation and antioxidation in bulk oils. Eur J Lipid Sci Technol. 2015;117(8):1095–137.
Harmon P, Boccardi G. Oxidative susceptibility testing. 2011. In: Pharmaceutical stress testing : predicting drug degradation [Internet]. Informa Healthcare. Second Edition. Drugs and the pharmaceutical sciences series; [168-92].
Hamam F, Shahidi F. Acidolysis reactions lead to esterification of endogenous tocopherols and compromised oxidative stability of modified oils. J Agric Food Chem. 2006;54(19):7319–23.
Finley JW, deMan JM. Lipids. Principles of food chemistry. Food Science Text Series2018. p. 39-116.
Kerwin BA. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: structure and degradation pathways. J Pharm Sci. 2008;97(8):2924–35.
Pokorny J. Stabilization of fats by phenolic antioxidants. Canadian Institute of Food Technology Journal. 1971;4(2):68–74.
Loftsson T. Drug stability for pharmaceutical scientists: Elsevier; 2014.
Jannin V, Rodier JD, Musakhanian J. Polyoxylglycerides and glycerides: effects of manufacturing parameters on API stability, excipient functionality and processing. Int J Pharm. 2014;466(1-2):109–21.
Li Z, Kozlowski BM, Chang EP. Analysis of aldehydes in excipients used in liquid/semi-solid formulations by gas chromatography-negative chemical ionization mass spectrometry. J Chromatogr A. 2007;1160(1-2):299–305.
Tian K, Dasgupta PK. Determination of oxidative stability of oils and fats. Anal Chem. 1999;71(9):1692–8.
del Barrio MA, Hu J, Zhou P, Cauchon N. Simultaneous determination of formic acid and formaldehyde in pharmaceutical excipients using headspace GC/MS. J Pharm Biomed Anal. 2006;41(3):738–43.
Niki E, Omata Y, Fukuhara A, Saito Y, Yoshida Y. Assessment of radical scavenging capacity and lipid peroxidation inhibiting capacity of antioxidant. J Agric Food Chem. 2008;56(18):8255–60.
Kamal-Eldin A, Pokorny J. Analysis of lipid oxidation. AOCS. 2008.
Pinchuk I, Shoval H, Dotan Y, Lichtenberg D. Evaluation of antioxidants: scope, limitations and relevance of assays. Chem Phys Lipids. 2012;165(6):638–47.
Damian F, Harati M, Schwartzenhauer J, Van Cauwenberghe O, Wettig SD. Challenges of dissolution methods development for soft gelatin capsules. Pharmaceutics. 2021;13(2).
Robnik B, Naumoska K, Casar Z. A novel testing approach for oxidative degradation dependent incompatibility of amine moiety containing drugs with PEGs in solid-state. Pharmaceutics. 2020;12(1).
Yang L, Heatley F, Blease TG, Thompson RIG. A study of the mechanism of the oxidative thermal degradation of poly(ethylene oxide) and poly(propylene oxide) using 1H- and 13C-NMR. Eur Polym J. 1996;32(5):535–47.
Hemenway JN, Carvalho TC, Rao VM, Wu Y, Levons JK, Narang AS, et al. Formation of reactive impurities in aqueous and neat polyethylene glycol 400 and effects of antioxidants and oxidation inducers. J Pharm Sci. 2012;101(9):3305–18.
Bindra DS, Williams TD, Stella VJ. Degradation of O6-benzylguanine in aqueous polyethylene glycol 400 (PEG 400) solutions: concerns with formaldehyde in PEG 400. Pharm Res. 1994;11(7):1060–4.
McGinity JW, Patel TR, Naqvi AH, Hill JA. Implications of peroxide formation in lotion and ointment dosage forms containing polyethylene glycols. Drug Development Communications. 1976;2(6):505–19.
Herzberger J, Niederer K, Pohlit H, Seiwert J, Worm M, Wurm FR, et al. Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: synthesis, novel polymer architectures, and bioconjugation. Chem Rev. 2016;116(4):2170–243.
Doshi N, Demeule B, Yadav S. Understanding particle formation: solubility of free fatty acids as polysorbate 20 degradation byproducts in therapeutic monoclonal antibody formulations. Mol Pharm. 2015;12(11):3792–804.
Larson NR, Wei Y, Prajapati I, Chakraborty A, Peters B, Kalonia C, et al. Comparison of polysorbate 80 hydrolysis and oxidation on the aggregation of a monoclonal antibody. J Pharm Sci. 2020;109(1):633–9.
Martin D, Reglero G, Señoráns FJ. Oxidative stability of structured lipids. Eur Food Res Technol. 2010;231(5):635–53.
Osborn HT, Akoh CC. Effect of emulsifier type, droplet size, and oil concentration on lipid oxidation in structured lipid-based oil-in-water emulsions. Food Chem. 2004;84(3):451–6.
Howard JA, Ingold KU. Absolute rate constants for hydrocarbon autoxidation. VI. Alkyl aromatic and olefinic hydrocarbons. Can J Chem. 1967;45(8):793–802.
Miyashita K, Frankel EN, Neff WE, Awl RA. Autoxidation of polyunsaturated triacylglycerols. III. Synthetic triacylglycerols containing linoleate and linolenate. Lipids. 1990;25(1):48–53.
Nelson KA, Labuza TP, editors. Relationship between water and lipid oxidation rates water activity and glass transition theory. American Chemical Society: Washington, DC,: ACS Symposium Series; American Chemical Society; 1992.
Xu L, Davis TA, Porter NA. Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J Am Chem Soc. 2009;131(36):13037–44.
Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc Chem Res. 2011;44(6):458–67.
Dominguez R, Pateiro M, Gagaoua M, Barba FJ, Zhang W, Lorenzo JM. A comprehensive review on lipid oxidation in meat and meat products. Antioxidants (Basel). 2019;8(10).
Kobayashi H, Yoshida M, Miyashita K. Comparative study of the product components of lipid oxidation in aqueous and organic systems. Chem Phys Lipids. 2003;126(1):111–20.
Waraho T, McClements DJ, Decker EA. Impact of free fatty acid concentration and structure on lipid oxidation in oil-in-water emulsions. Food Chem. 2011;129(3):854–9.
Miyashita K. Paradox of omega-3 PUFA oxidation. Eur J Lipid Sci Technol. 2014;116(10):1268–79.
Yazu K, Yamamoto Y, Niki E, Miki K, Ukegawa K. Mechanism of lower oxidizability of eicosapentaenoate than linoleate in aqueous micelles. II Effect of antioxidants. Lipids. 1998;33(6):597–600.
Wu Y, Levons J, Narang AS, Raghavan K, Rao VM. Reactive impurities in excipients: profiling, identification and mitigation of drug-excipient incompatibility. AAPS PharmSciTech. 2011;12(4):1248–63.
Goddard JM, McClements DJ, Decker EA. Innovative technologies in the control of lipid oxidation. Lipid Technol. 2012;24(12):275–7.
Mozuraityte R, Rustad T, Storro I. The role of iron in peroxidation of polyunsaturated fatty acids in liposomes. J Agric Food Chem. 2008;56(2):537–43.
Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145(5):532–41.
Berton-Carabin CC, Ropers M-H, Genot C. Lipid oxidation in oil-in-water emulsions: involvement of the interfacial layer. Compr Rev Food Sci Food Saf. 2014;13(5):945–77.
Nassar MN, Nesarikar VN, Lozano R, Parker WL, Huang Y, Palaniswamy V, et al. Influence of formaldehyde impurity in polysorbate 80 and PEG-300 on the stability of a parenteral formulation of BMS-204352: identification and control of the degradation product. Pharm Dev Technol. 2004;9(2):189–95.
Collin F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci. 2019;20(10).
Digenis GA, Gold TB, Shah VP. Cross-linking of gelatin capsules and its relevance to their in vitro-in vivo performance. J Pharm Sci. 1994;83(7):915–21.
Gray VA, Marques MRC, Cole E, JMD RT, Ghidorsi L, Guo J-H, et al. Use of enzymes in the dissolution testing of gelatin capsules and gelatin-coated tablets-revisions to dissolution <711> and disintegration and dissolution of dietary supplements <2040>. Dissolution Technologies. 2014;21(4):6–18.
Santos NA, Cordeiro AMTM, Damasceno SS, Aguiar RT, Rosenhaim R, Carvalho Filho JR, et al. Commercial antioxidants and thermal stability evaluations. Fuel. 2012;97:638–43.
Giuffrida F, Destaillats F, Egart MH, Hug B, Golay P-A, Skibsted LH, et al. Activity and thermal stability of antioxidants by differential scanning calorimetry and electron spin resonance spectroscopy. Food Chem. 2007;101(3):1108–14.
Rege S, Momin S, Bhowmick D. Effect of ascorbic acid on the oxidative stability of water-in-oil emulsion in the presence of lipophilic antioxidants. Int J Food Prop. 2014;18(2):259–65.
Cuvelier M-E, Soto P, Courtois F, Broyart B, Bonazzi C. Oxygen solubility measured in aqueous or oily media by a method using a non-invasive sensor. Food Control. 2017;73:1466–73.
Poon JF, Pratt DA. Recent insights on hydrogen atom transfer in the inhibition of hydrocarbon autoxidation. Acc Chem Res. 2018;51(9):1996–2005.
Shahidi F, Zhong Y. Revisiting the polar paradox theory: a critical overview. J Agric Food Chem. 2011;59(8):3499–504.
van Aardt M, Duncan SE, Long TE, O'Keefe SF, Marcy JE, Sims SR. Effect of antioxidants on oxidative stability of edible fats and oils: thermogravimetric analysis. J Agric Food Chem. 2004;52(3):587–91.
McClements DJ, Decker E. Interfacial antioxidants: a review of natural and synthetic emulsifiers and coemulsifiers that can inhibit lipid oxidation. J Agric Food Chem. 2018;66(1):20–35.
Waraho T, Cardenia V, Rodriguez-Estrada MT, McClements DJ, Decker EA. Prooxidant mechanisms of free fatty acids in stripped soybean oil-in-water emulsions. J Agric Food Chem. 2009;57(15):7112–7.
Maloney JF, Labuza TP, Wallace DH, Karel M. Autoxidation of methyl linoleate in freeze-dried model systems. I. Effect of water on the autocatalyzed oxidation. J Food Sci. 1966;31(6):878–84.
Labuza TP, Maloney JF, Karel M. Autoxidation of methyl linoleate in freeze-dried model systems. II. Effect of water on cobalt-catalyzed oxidation. J Food Sci. 1966;31(6):885–91.
Karel M. Lipid oxidation, secondary reactions, and water activity of foods. Autoxidation in Food and Biological Systems. 1980:191–206.
Frankel EN. Interfacial lipid oxidation and antioxidation. Journal of Oleo Science. 2001;50(5):387–91.
Chaiyasit W, McClements DJ, Decker EA. The relationship between the physicochemical properties of antioxidants and their ability to inhibit lipid oxidation in bulk oil and oil-in-water emulsions. J Agric Food Chem. 2005;53(12):4982–8.
Choe E, Min DB. Mechanisms of antioxidants in the oxidation of foods. Compr Rev Food Sci Food Saf. 2009;8(4):345–58.
Floyd AG. Top ten considerations in the development of parenteral emulsions. Pharm Sci Technol Today. 1999;4(2):134–43.
Barouh N, Bourlieu-Lacanal C, Figueroa-Espinoza MC, Durand E, Villeneuve P. Tocopherols as antioxidants in lipid-based systems: the combination of chemical and physicochemical interactions determines their efficiency. Compr Rev Food Sci Food Saf. 2021.
Chaiyasit W, McClements DJ, Weiss J, Decker EA. Impact of surface-active compounds on physicochemical and oxidative properties of edible oil. J Agric Food Chem. 2008;56(2):550–6.
Madhavi DL, Salunkhe DK. Food additive toxicology: Marcel Dekker. New York; 1995. 89-178 p.
Javidipour I, Tufenk R, Basturk A. Effect of ascorbyl palmitate on oxidative stability of chemically interesterified cottonseed and olive oils. J Food Sci Technol. 2015;52(2):876–84.
Lee KH, Yook HS, Lee JW, Park WJ, Kim KS, Byun MW. Quenching mechanism and kinetics of ascorbyl palmitate for the reduction of the gamma irradiation-induced oxidation of oils. J Am Oil Chem Soc. 1999;76(8).
Lavoie J-C, Lachance C, Chessex P. Antiperoxide activity of sodium metabisulfite. Biochem Pharmacol. 1994;47(5):871–6.
Liang R, Liu Y, Fu LM, Ai XC, Zhang JP, Skibsted LH. Antioxidants and physical integrity of lipid bilayers under oxidative stress. J Agric Food Chem. 2012;60(41):10331–6.
Mitrus O, Zuraw M, Losada-Barreiro S, Bravo-Diaz C, Paiva-Martins F. Targeting antioxidants to interfaces: control of the oxidative stability of lipid-based emulsions. J Agric Food Chem. 2019;67(11):3266–74.
Brimberg UI. On the kinetics of the autoxidation of fats. J Am Oil Chem Soc. 1993;70(3):249–54.
Brimberg UI. On the kinetics of the autoxidation of fats. II. Monounsaturated substrates. Journal of the American Oil Chemists Society. 1993;70(11):1063–7.
Chaiyasit W, Elias RJ, McClements DJ, Decker EA. Role of physical structures in bulk oils on lipid oxidation. Crit Rev Food Sci Nutr. 2007;47(3):299–317.
Laguerre M, Bayrasy C, Panya A, Weiss J, McClements DJ, Lecomte J, et al. What makes good antioxidants in lipid-based systems? The next theories beyond the polar paradox. Crit Rev Food Sci Nutr. 2015;55(2):183–201.
Hamilton RJ, Kalu C, McNeill GP, Padley FB, Pierce JH. Effects of tocopherols, ascorbyl palmitate, and lecithin on autoxidation of fish oil. J Am Oil Chem Soc. 1998;75(7):813–22.
Evans JC, Kodali DR, Addis PB. Optimal tocopherol concentrations to inhibit soybean oil oxidation. J Am Oil Chem Soc. 2002;79(1):47–51.
Wang C, Sun C, Lu W, Gul K, Mata A, Fang Y. Emulsion structure design for improving the oxidative stability of polyunsaturated fatty acids. Compr Rev Food Sci Food Saf. 2020;19(6):2955–71.
Nakaya K, Ushio H, Matsukawa S, Shimizu M, Ohshima T. Effects of droplet size on the oxidative stability of oil-in-water emulsions. Lipids. 2005;40(5):501–7.
Waraho T, McClements DJ, Decker EA. Mechanisms of lipid oxidation in food dispersions. Trends Food Sci Technol. 2011;22(1):3–13.
Pan Y, Tikekar RV, Nitin N. Effect of antioxidant properties of lecithin emulsifier on oxidative stability of encapsulated bioactive compounds. Int J Pharm. 2013;450(1-2):129–37.
Laguerre M, Giraldo LJ, Lecomte J, Figueroa-Espinoza MC, Barea B, Weiss J, et al. Chain length affects antioxidant properties of chlorogenate esters in emulsion: the cutoff theory behind the polar paradox. J Agric Food Chem. 2009;57(23):11335–42.
Kharat M, Aberg J, Dai T, McClements DJ. Comparison of emulsion and nanoemulsion delivery systems: the chemical stability of curcumin decreases as oil droplet size decreases. J Agric Food Chem. 2020;68(34):9205–12.
Cengiz A, Schroen K, Berton-Carabin C. Towards oxidatively stable emulsions containing iron-loaded liposomes: the key role of phospholipid-to-iron ratio. Foods. 2021;10(6).
Berton C, Genot C, Ropers MH. Quantification of unadsorbed protein and surfactant emulsifiers in oil-in-water emulsions. J Colloid Interface Sci. 2011;354(2):739–48.
Berton C, Ropers MH, Viau M, Genot C. Contribution of the interfacial layer to the protection of emulsified lipids against oxidation. J Agric Food Chem. 2011;59(9):5052–61.
Azuma G, Kimura N, Hosokawa M, Miyashita K. Effect of droplet size on the oxidative stability of soybean oil TAG and fish oil TAG in oil-in-water emulsion. J Oleo Sci. 2009;58(6):329–38.
Mancuso JR, McClements DJ, Decker EA. The effects of surfactant type, pH, and chelators on the oxidation of salmon oil-in-water emulsions. J Agric Food Chem. 1999;47(10):4112–6.
Funding
Open access funding was provided by the Gattefossé Group.
Author information
Authors and Affiliations
Contributions
Masumi Dave, Application Laboratory Manager, USA, contributed to the data collection review, accuracy of the data, writing review, and approval of the manuscript. Jean-David Rodier, R&D Director Oleochemistry, France, provided in-depth knowledge on the subject, was instrumental in the data collection review, accuracy of data and writing review, visualization, completion, and approval of the final version. Jasmine Musakhanian, Scientific and Marketing Director, USA, contributed to the conceptualization, data collection and curation, visualization, supervision of data accuracy and integrity, the redaction, and final review of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Musakhanian, J., Rodier, JD. & Dave, M. Oxidative Stability in Lipid Formulations: a Review of the Mechanisms, Drivers, and Inhibitors of Oxidation. AAPS PharmSciTech 23, 151 (2022). https://doi.org/10.1208/s12249-022-02282-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02282-0