
ABSTRACT
Increased antibacterial resistance (ABR) and limited drug discovery warrant optimized use of available antibiotics. One option is to rationally combine two antibiotics (fixed dose combination (FDC)) that may delay or prevent emergence of ABR in notorious pathogen. Major concern with FDC is the mutual interaction of its components that might influence their pharmacokinetic (PK) profile, requiring reassessing of whole formulation (adding cost and time). The interaction can be identified by comparing PK profile of a drug present in FDC with its independent entity. An open-label, crossover, single-dose comparative PK study of FDC (ceftriaxone and sulbactam) with their individual reference formulations was performed in 24 healthy adult subjects. No mutual PK interactions between ceftriaxone and sulbactam were observed. Pharmacokinetic data was used to develop a population-PK model to understand between-subject variability (BSV). Pharmacokinetics of ceftriaxone/sulbactam was explained by one and two compartment models, respectively. The subject’s “weight” was identified as a covariate explaining BSV. Both internal and external validations (healthy/infected subjects) were done. The model-derived population-PK parameters of FDC’s active components in infected subjects were similar to literature reported values of individual components. Efficacies of various FDC dosage regimens over a range of minimum inhibitory concentrations (MICs) were assessed by Monte Carlo simulations using population-PK parameters of infected/healthy subjects. In infected subjects, 3 g FDC/24 h can treat bacteria with MIC ≤8 μg/mL, while for MIC 8–32 μg/mL, 3 g FDC/12 h is recommended. Lastly, the developed population-PK model was successfully used to predict drug exposure in pediatric population.
Similar content being viewed by others

INTRODUCTION
The rapid emergence of antibacterial resistance (ABR) imposes fresh challenges and creates serious public health threat around the world. Extended-spectrum β-lactamases (ESBLs) and metallo β-lactamases (MBLs) are major contributors of ABR (1). Rising trends of ESBLs are being observed all over the world including the USA. More than 90% ESBL cases are resistant to third/fourth generation cephalosporins. Carbapenems are the last resort for serious ESBL-bacterial infections and also, carbapenems are ineffective against MBL strains. The situation is worsening further due to dried pipeline of newer antibiotics (2,3). Recent research work has shown that the rational use of pharmacodynamically synergistic antibiotics through fixed dose combination (FDC) approach may delay or even prevent emergence of ABR (4). Antibiotic combination (pharmacodynamic synergism) choices and further using non-antibiotic adjuvant (NAA) have shown the synergistic effect to the level of ABR elimination. On these lines, a combination of ceftriaxone, sulbactam, and NAA ethylene diamine tetra acetate (EDTA; the concentration is so low that it qualifies as excipient) was developed. The FDC works on ABR by eradicating biofilm, inhibiting conjugal transfer, bacterial adhesion, and decreasing efflux transporters expression (5). The activity of FDC is similar to meropenem in ESBL strains and is active against MBL strains; thus, it can be used as carbapenem sparer drug (5–8).
All three molecules are approved individually by various regulatory agencies including USFDA. It is well known that the addition of different components in a FDC product may change the pharmacokinetic (PK) of the active components (ceftriaxone + sulbactam) and further the safety profile of the FDC product, which may affect the therapeutic outcome. Thus, in the current study, FDC of ceftriaxone, sulbactam, and disodium EDTA (2/1/0.037 w/w/w) (marketed as Elores®) was evaluated for PK parameters of active components (ceftriaxone and sulbactam). The PK profile of ceftriaxone and sulbactam of FDC was compared with that of individual formulation of ceftriaxone and sulbactam, respectively. The PK of individual drugs is well characterized across populations (9,10). however, the chances of variability in drug exposures within a population increase, when given in combination. The therapeutic potential of the FDC against various infections can be assessed from the understanding of population PK of active components of the FDC and the severity of infection. Population PK of EDTA (non-antibiotic adjuvant) will not add any relevant information as the concentration is very less in the FDC, but its influence on active component’s PK might be relevant; therefore, the population PK was focused on ceftriaxone and sulbactam only.
To understand the possible variability introduced by confounders (including age, weight, renal status), the population PK of active components of FDC was evaluated using a two-step approach. The first stage involved the assessment of individual PK parameter estimates in healthy subjects by utilizing individual’s concentration-time data (data-rich situation). The second stage involved identifying relationships between estimated parameters and covariates using regression and covariance analysis. On the basis of modeling approaches (using Pmetrics), a population PK model was created and external/internal validations were performed to evaluate the robustness of the model. In the external validations, the robustness of model was tested using data of active components (from FDC) in patient population (11) and data from stand-alone arms in healthy population (from the current study).
The use of FDC in pediatric population is another area with potential risk originating from variable exposures. Therefore, the utilizability of the model was extended to predict the PK parameters (and hence exposures) in pediatric population. The concentration-time data and estimated parameters for pediatrics were taken from the published study (12,13). The population PK model predicted parameter values in pediatrics were compared with that observed in published study, and thus, the reliability of the model was assessed for understanding exposures of FDC’s active components in pediatric population.
The parameters obtained from the robust population PK (adult) model were further used to make predictions for changes in exposures corresponding to different dosage regimens of FDC. The exposures were further related to changes in pharmacodynamic (PD) responses characterized by a specific PK/PD index. Both components of the FDC are characterized by time-dependent antibacterial activity, and the percentage of the exposure time during which the free-drug concentration remains above the minimum inhibitory concentrations (MICs) (%T > MIC) is the PK/PD index that best correlates with efficacy. Monte Carlo simulations (MCSs) were performed to assess the efficacies of various dosage regimens of FDC in terms of the probability of target attainment (PTA) of PK/PD index 70%T > MICcomb (a value generally used for beta-lactam antibiotics as the desired PK/PD index for efficient bacterial killing and MICcomb is the combined MIC of FDC for a single pathogen) over a range of MICs (1–64 μg/mL). For MCS, 1000 subjects’ simulated data was generated using PK parameters derived from population PK model. The PTA was calculated for 70%T > MIC over a range of MICs and the dosage regimen showing ≥90%PTA was identified as an efficacious therapeutic option.
MATERIAL AND METHODS
Drugs
Test formulation CSE 1034 (Elores® FDC of ceftriaxone sodium USP equivalent to ceftriaxone 1000 mg + sulbactam sodium USP equivalent to sulbactam 500 mg + non-antibiotic adjuvant qualifying as excipient disodium EDTA 37.0 mg; batch no. 0FA0903, Exp: May/2012) was manufactured by Venus Remedies, India. The reference products, i.e., Rocephin® (containing ceftriaxone sodium equivalent to ceftriaxone 1000 mg, batch no. B3478B01, Exp. Dec/2012, reference 1) and Bact-S® (containing sulbactam sodium 0.5 g, batch no. DV0G1511, Exp. June/2012, reference 2) were manufactured by Hoffmann- La Roche Inc, France and Venus remedies, India, respectively.
Study Population and Design
The analysis was performed on the PK data of 24 patients from a randomized, cross-over, single-dose comparative pharmacokinetic study under fasting conditions. In brief, patients eligible for study were 22 years or older and healthy. Subjects with a history of serious disease or infection were excluded. The study was designed in accordance with good clinical practices and approved by an independent ethics committee (IEC) [protocol no. AZ/BE/P/038, dated 20 May 2010 approved on 13 July 2010]. A single dose was administered to study subjects as intravenous infusion over a period of 30 min in supine posture in each treatment period. The study was conducted at Azidus Laboratories Ltd., Vandalur, Chennai, India.
Blood Sampling and Drug Assay
Blood samples were collected from each subject at 0 h (pre-dose), 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 16, and 24 h following drug administration. The separated plasma samples were analyzed for ceftriaxone and sulbactam using a validated high-performance liquid chromatography (HPLC, 3 Agilent 1200 series, column Hypersil 250 × 4.6 mm, 5 μ) method. The mobile phase was a mixture of tetrabutyl ammonium hydroxide buffer (pH 7.0 ± 0.1) and acetonitrile (70:30). Aliquots (0.25 mL) of plasma samples were mixed with equal volume of chilled acetonitrile and were kept at 2–8°C for 5 min. Samples were then centrifuged (3500 rpm for 30 min) and supernatants were injected in HPLC system for analysis.
Pharmacokinetic Analysis
PK parameters analyzed in the study included maximum measured plasma concentration (C max), area under the plasma concentration vs. time curve (AUC), time of maximum measured plasma concentration (t max), elimination rate constant (K e), clearance (CL), and volume of distribution (V D).
Population Pharmacokinetic Analysis
Concentration-time data of ceftriaxone and sulbactam were used to perform non-parametric PK modeling in Pmetrics (version 0.40, Laboratory for Applied Pharmacokinetic, Los Angeles, CA, USA) (14). Non-parametric approach was used to better detect outliers patients and unexpected sub-populations. Both algebraic and ordinary differential equation solver were used to fit the model to the data.
Structure Model
One, two, and three compartment structure models with zero-order input and first-order/Michaelis-Mentis elimination were evaluated as potential PK models. The model selection was based on the Akaike information criterion (AIC) index, and the goodness-of-fit of both population- and individual-predicted vs. observed plots. Between-subject variability in ceftriaxone and sulbactam pharmacokinetics followed log normal distribution. For explaining residual variability, both the additive lambda and multiplicative gamma error models in Pmetrics were tested during model development. Each observation was weighted by 1/error2 and error was calculated as follows:
where SD was modeled with a polynomial equation, i.e., C 0 + C 1 * [obs]2 + C 2 * [obs]3 C 3 * [obs]4, where [obs] was the observed concentrations of ceftriaxone or sulbactam and C 0, C 1, C 2, and C 3 were assay error polynomial coefficients.
Covariate Analysis
Dependencies between PK parameters and covariates (age, weight, and creatinine CL) were evaluated using linear, power (allometric and population-mean-normalized) and exponential regression models. In allometric model, CL and VD were scaled to body size using coefficients of 0.75 and 1, respectively. Covariates were scaled to median population values and continuous covariates were extrapolated between observations. Forward/backward stepwise approach was used for covariate selection. Diagnostic plots, change in the objective function values (OFVs), and parameter variability were used to select covariates that improved the model prediction.
Model Validation
Internal Validation
Visual predictive check (VPC) and bootstrap analysis were performed to validate the predictability and stability of the model graphically as well as statistically. For VPC, 1000 data sets were simulated using final population model parameters. For the VPC, 2.5th, 50th, and 97.5th percentiles of the simulated concentrations were processed using R platform and observed concentration against time was plotted to see the boundaries of these data points against the 2.5th and 97.5th percentiles. For non-parametric bootstrap analysis, re-sampling with replacement was done 500 times and the 2.5th and 97.5th percentile of the parameter estimates were taken in order to build the 95% bootstrap confidence intervals (CIs). The values of estimated parameters from bootstrap analysis were compared with those estimated from the original data set. The entire bootstrap procedure was done in automated fashion using SPSS ver. 20 (IBM, SPSS 20).
External Validation
The population PK model of ceftriaxone and sulbactam was also evaluated on external validation data sets of (1) 24 healthy male subjects receiving Rocephin and Bact-S, respectively, and (2) 12 patients (complicated urinary tract infection) receiving the FDC (11). From the Bayesian prior model parameter joint density, Pmetrics calculated the Bayesian posterior joint density for each subject in the separate external validation data sets (health subjects and patients separately). The median marginal PK parameter values of each posterior density were used to calculate the predicted drug (ceftriaxone and sulbactam) concentrations, given individual dose and subject covariates. The PK parameters estimates of external data sets were then compared with the PK parameters of main data set (used for population PK model development).
Model Prediction in Pediatric Population
The predictive performance of the developed model was further evaluated in pediatric patients. The PK data of ceftriaxone and sulbactam in pediatric population was taken from literature (12,13). The individual concentrations were predicted and compared using Bayesian approach, as explained in external model validation; and associated PK parameters were compared with the reported values in the literature (12,13).
Monte Carlo Simulations
Monte Carlo simulations for 1000 adult subjects were performed to determine how likely the FDC dose of 0.75, 1.5, 3, 6, and 9 g would achieve 70%T > MICcomb at different values of MICcomb, i.e., 1, 4, 8, 16, 32, and 64 μg/mL. The population PK parameters (CL and K e obtained from final population-PK model) were utilized in simulations. The concentration-profiles of once-a-day and twice-a-day dosing regimens were generated using CL and K e of each simulated subjects, and %T > MICcomb was calculated for all simulated subjects for different exposures at all MIC values. The PTA (target PTA minimum 90%) was then defined as percentage of simulated subjects showing %T > MICcomb of more than 70.
RESULTS
Pharmacokinetic Analysis
The PK evaluations were done using data from 24 male subjects, 22–32 years old, with BMI in the range of 18.56–24.96 kg/m2, respectively (Table I). Table I has also shown the demographics of 12 cUTI patients, whose PK data was employed for external validation of final population PK model of ceftriaxone and sulbactam.
Comparative PK Profiles of Drugs Given Individually and in FDC
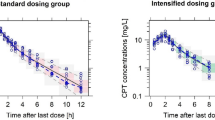
The comparison of PK profile of the FDC's active components (i.e. ceftriaxone and sulbactam) of FDC with their respective reference formulations were done graphically as well as statistically. The plasma concentration-time curves of ceftriaxone and sulbactam under fasting condition were super-imposable with that of reference drugs (Rocephin: ceftriaxone alone; Bact-S: sulbactam alone) within experimental errors (Fig. 1). The PK parameters of ceftriaxone and sulbactam in each formulation (FDC and reference drugs) were compared (Table II) using paired t-test; and the differences between the CL or VD values in two different IV formulations were observed statistically non-significant. Exposures to the body provided the same dose was given. No statistical significant difference was observed in ceftriaxone or sulbactam PK parameters when given in combination (FDC) or as individual drug (Rocephin and Bact-S) (Table II). These results confirms that overall drug exposure to the body do not change, irrespective of nature of formulation, i.e., FDC or drugs given separately.

Mean plasma concentrations (± SD) vs. time profiles of ceftriaxone a and sulbactam b in human healthy subjects (n = 24) after single-dose IV infusion of FDC [ceftriaxone/sulbactam (1 g/0.5 g)] and reference drugs (1 g Rocephin for ceftriaxone and 0.5 g Bact-S for sulbactam)
Population PK Modeling
Structural PK Model
The main impetus for study was to develop population PK model for the FDC of ceftriaxone and sulbactam. In total, 528 plasma concentrations (264 each of ceftriaxone and sulbactam) were used to develop the population PK model. The non-parametric pharmacokinetic modeling was performed using Pmetrics (described in “MATERIALS AND METHODS”).
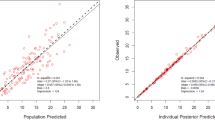
The lowest AIC value (2178) and good diagnostic plot (correlation coefficient of 0.85 for population-predicted vs. observed and 0.92 for individual-predicted vs. observed) of ceftriaxone was obtained in one compartment with gamma error (supplementary Table 1). It should be noted that similar values were obtained for all three compartments model, but diagnostic plots of one-compartment showed higher imprecision and bias. Moreover, the V D of ceftriaxone was reported as 7–12 L (15). which was best explained by one compartment model with first order elimination kinetics. The Michaelis-Menten equation showed poor diagnostic plots and PK parameter estimates. Thus, structural PK model for ceftriaxone was constructed using one compartment, zero order input, gamma error model and first order elimination.
Similar model development was done for sulbactam. The lower AIC value (1003) and good diagnostic plots (correlation coefficient of 0.78 and 0.89 for predicted vs. observed for population and individual, respectively) were obtained for two compartment with lambda error model. It must be noted that gamma error model have shown lower AIC values (367) vs. lambda error model (1003) for two compartment PK model, but diagnostic plots of higher correlation coefficient (0.89 vs. 0.85), lower bias (0.07 vs. −0.20) and lower imprecision (0.87 vs. 0.91) were obtained with lambda error model (supplementary Table 2). Additionally, more residual variability can be explained by inter-individual covariates in lambda model, improving the robustness and validity of the population-PK model. On comparing the one and two compartment models with lambda error model, similar AIC and diagnostic plots were obtained.
However, the estimated V D (closer to the reported values of 20–30 L) can be better explained by two-compartment model (16). Thus, the structural PK model for sulbactam was constructed with zero-order input, lambda error model, and second-order elimination.
Incorporation of Covariates
The effect of covariates on PK of ceftriaxone and sulbactam were investigated to compensate for between-subject variability (BSV) of the PK data and to improve/refine the structural PK model. All three variables (age, weight, and creatinine CL) were evaluated to explain the BSV of the population-PK model. Stepwise forward/backward approach was used to identify covariates which are highly correlated to PK parameters. The identified covariate-PK parameter relationship was modeled using linear, exponential, and power (allometric and normalized) equations to reduce the BSV and improve the model-fit.
For allometric scaled models, primary PK parameters were allometrically adjusted to body size using body weight and coefficient of 0.75 (for CL) and 1 (for V D) (17). For ceftriaxone, “K e” was weakly correlated with weight (abbreviated as "wt") of subjects, but the correlation was better than other covariates tested. The correlation of K e ~ wt was best explained by allometric-scaled power model normalized with median population weight (lower AIC [2175] and lower bias [−0.023] of diagnostic plot; supplementary Table 1). The “refined” model was slightly better than structural PK model of ceftriaxone (Fig. 2a). For sulbactam, it was observed that K e was correlated with "wt" and “age,” whereas V D was correlated with "wt". All possible covariate models were applied similar to ceftriaxone. However, Ke ~ wt improved the model fit more than any other covariates or their combinations (supplementary Table 2). The “improved” model had shown lower AIC value (901 vs. 1003) and better model-fit with lower imprecision (0.758 vs. 0.873) as compared to its structural PK model (Fig. 2b). Mention must be made that sulbactam PK analysis was based on PK data of 12 h because of its very short half–life, i.e., ~1 h (18).

Plot of observed vs. population-predicted concentrations (left) and observed vs. individual-predicted concentrations (right) of ceftriaxone a and sulbactam b, generated from the final pop-PK model. Circles represent the observed concentration. Each solid line indicates a non-parametric regression with robust linear fit
The final population PK model of ceftriaxone and sulbactam was explained by one-compartment (1C) model and two-compartment (2C) model, respectively. However, in both of the cases, weight was the main covariate impacting the elimination constant as per the following equation k e = k e0 * (70/wt)0.25, where "wt" was weight of subjects. For ceftriaxone, multiplicative gamma error model was used (gamma [g] = 5) with assay error polynomial coefficients of 0.02, 0.05, and −0.0002. In case of sulbactam, additive lamda error model was used (lamda [L] = 0.1) with assay polynomial coefficients, i.e., C1 and C2 of 0.1 and 0.1, respectively. For detailed equations of error models, please refer to “Population Pharmacokinetic Analysis” subsection of “MATERIAL AND METHOD” section. The residuals between observed and predicted observations were normalized by the standard deviations of the data (conditional weighted residuals) and plotted with predicted data as well as with time for both of the final models (data not shown). In both cases, no systematic trends were observed. The PK parameters of the final population-PK model are given in Table III.
Model Validation
Internal Validation
The ability of the final population-PK model to predict the observed data was evaluated graphically using VPC. One thousand data sets were simulated using final population model parameters. For a model in which random effects are well estimated, approximately 95% of the observed values are expected to be within the 2.5th and 97.5th prediction interval (19). In case of ceftriaxone, 3 and 4% of observed values were outside the 2.5th and 97.5th percentile, respectively. In case of sulbactam, 94.1% observed values were within the 2.5th and 97.5th prediction interval. These results validated the predictability of final pop-PK models of ceftriaxone and sulbactam (Fig. 3).

Plots of visual predictive checks obtained from the simulations of ceftriaxone 1 g OD a and sulbactam 0.5 g OD b using the final pop-PK model. Solid lines represent the median (middle one) and 95% confidence interval (side ones) obtained from the simulated data
Stability and performance of final model were also assessed by non-parametric bootstrap analysis with replacement and re-sampling. Re-sampling was performed 500 times, and the 2.5th and 97.5th percentile of the parameter estimates were taken in order to build the 95% bootstrap confidence intervals. For ceftriaxone, K e and V D were 0.083 (vs. 0.082 of final model) and 7.470 (vs.7.471), respectively (Table III). Similarly for sulbactam, the estimated K e and V D values were 0.650 (vs. 0.61 of final model) and 28.928 (vs. 29.71 of final model), respectively (Table III). The bootstrap-estimated values with 95% CI were in close proximity with those estimated from the final population-PK model (Table III), suggesting the reliability of the final population-PK models of both ceftriaxone and sulbactam.
External Validation of Population PK Model Using Data from Healthy Subjects
The population-PK models of ceftriaxone and sulbactam were also evaluated on another data set of same 24 healthy male subjects receiving reference formulations Rocephin® and Bact-S®, respectively. The Bayesian estimated concentrations were highly correlated with observed concentrations (r 2 = 0.92 for ceftriaxone and r 2 = 0.87 for sulbactam) (Fig. 4). On comparing population-PK-derived primary PK parameters of reference drugs (Rocephin® and Bact- S®) with corresponding active components of FDC, no significant changes were observed (Table III). This result further substantiates our initial claim that the PK parameter of ceftriaxone and sulbactam remains unaffected when given in the FDC of ceftriaxone and sulbactam along with EDTA.

Plot of observed vs. population-predicted concentrations (left) and observed vs.individual-predicted concentrations (right) of ceftriaxone a and sulbactam b, obtained from external validation of the final pop-PK model [Rocephin data was used for Ceftriaxone 1 g “external” data; Bact-S data was used for sulbactam 0.5 g external data]. Circles represent the observed concentration. Each solid line indicates a non-parametric regression with robust linear fit
External Validation of Population PK Model Using Data from cUTI Patients
Another validation of population PK model was carried out using PK data of 12 cUTI patients after the administration of FDC ceftriaxone/sulbactam (2/1) 3.0 g. Similar to above external validation, the Bayesian estimated concentrations were highly correlated with observed concentrations (r 2 = 0.91 for ceftriaxone and r 2 = 0.97 for Sulbactam) (Fig. 5). On comparing the PK parameters estimates, i.e., K e and V D of external data sets of ceftriaxone and sulbactam with the FDC of ceftriaxone and sulbactam, only marginal differences (statistically non-significant) in median of both K e (ceftriaxone 0.12 vs. 0.08; sulbactam 0.42 vs. 0.61) and V D (ceftriaxone 7.87 vs. 7.47; sulbactam 30.43 vs. 29.71) were observed (Table III). This supports the applicability of the developed pop-PK model of both drugs in cUTI patients.

Plot of observed vs. population-predicted concentrations (left) and observed vs.individual-predicted concentrations (right) of ceftriaxone a and sulbactam b, obtained from external validation of the final pop-PK model [PK data of ceftriaxone 2 g and sulbactam 1 g was taken from 12 cUTI patients]. Circles represent the observed concentration. Each solid line indicates a non-parametric regression with robust linear fit
Utilizability of Developed Population PK Model: Pediatric Extrapolation (Concerns Related to Special Population)
The utilizability of the model was extrapolated to predict the PK parameters (and hence exposures) in pediatric data sets extracted from literature (12,13). The demographics of pediatric subjects used in the ceftriaxone and sulbactam study are given in supplementary Table 3. Focusing on ceftriaxone population-PK model, one modifications was made, i.e., multiplicative gamma error value was increased from 5 to 10 for better model-fit of pediatric data. No statistical differences in diagnostic plots (r 2 value 0.90 for g = 10 vs. 0.91 for g = 5; bias 0.40 for g = 10 vs. 0.41 for g = 5) and PK parameters (K e and V D values 0.08 h−1 and 7.5 L, respectively, for both g = 10 and 5) were observed in the population-PK model having gamma error value of 5 and 10 (supplementary Table 1).
Model validity in pediatrics was evaluated graphically using the diagnostic plot between observed and individual posterior predicted values, which showed high correlation (r 2 = 0.89) and lower bias (0.888) (Fig. 5a). Lower AIC of 653 was obtained in the analysis. The estimated medians of K e and V D were 0.10 h−1 and 2.51 L, which were statistically in close agreement with observed values (reported in the literature from where pediatric data was extracted) of 0.12 h−1 and 3.54 L, respectively (Table III and Fig. 6). For sulbactam, the population-PK model adequately described pediatric data confirmed by diagnostic plots of high correlation (r 2 = 0.91) and lower bias (−0.423) (Fig. 6). Lower AIC of 409 was obtained in the analysis (Supplementary Table 2). The estimated PK parameters, i.e., K e and V D median values were 1.54 h−1 and 5.85 L, which were statistically in close agreement with reported values of 1.49 h−1 and 8.52 L, respectively (Table III). Thus, population-PK models of ceftriaxone and sulbactam could be extrapolated to pediatric population and hence exposures can be easily determined for dose modifications.

Plot of observed vs. individual posterior-predicted concentrations of ceftriaxone a and sulbactam b, generated from extrapolation of the final pop-PK model to pediatric population. Circles represent the observed concentration. Each solid line indicates a nonparametric regression with robust linear fit
Monte Carlo Simulations
Monte Carlo simulations of concentration-time profiles were performed using population PK parameters estimated from both populations (healthy and infected) combined. The population PK parameters utilized in simulations were CL of 0.75 ± 0.2 and 16.52 ± 4.63 L/h for ceftriaxone and sulbactam, respectively. The K e was set to 0.09 and 0.51 for ceftriaxone and sulbactam, respectively. The FDC in dose range of 0.75 to 9 g as OD and BD regimen were used to calculate PTA over a range of MICcomb (1–64 μg/mL) (Fig. 7). The FDC was noted to be very effective with PTA of ~100% at dose strength ≥1.5 g OD and ≥0.75 g BD for the strains corresponding to MIC of 1 and 4 μg/mL. For MIC 8 μg/mL, doses less than 3 g were not effective. Even 3 g OD achieved PTA ~80%, which can be improved to >95% when the same exposure was divided into two doses (1.5 g BD). Further increasing the dose to 3 g BD maximized the PTA to 100%. For MIC 16 μg/mL, OD regimen was ineffective as the maximum PTA achieved at highest 9 g OD dose was only 35%. The FDC was ineffective at all drug exposures (PTA ~0–5 for the MIC of 32 and 64 μg/mL).

Probability of target attainment of FDC, for two dosage regimens, i.e., once-a-day a and twice-a-day b, plotted as a function of dose and MIC. It was determined that how likely the FDC dose of 0.75, 1.5, 3, 6, and 9 g would achieve 70%T > MICcomb at different values of MIC, i.e., 1, 4, 8, 16, 3, and 64 μg/mL
DISCUSSION
Previous studies have proven the superiority of ceftriaxone (beta-lactam antibiotic) over other antibiotics in various infections (20–22). Inclusion of sulbactam (beta lactamase inhibitor) and disodium EDTA with ceftriaxone further enhances the antibacterial spectrum against difficult-to-treat ESBL-/MBL-positive infections, due to synergistic effect of the FDC, yielding better clinical outcomes (23,24). Carbapenems are the last resort for ESBL infections and are ineffective against MBLs. The FDC thus can be used as a carbapenem sparer. In addition, FDC also prevents ABR by multiple mechanisms (downregulation of efflux transporters, inhibition of conjugal transfer, biofilm eradication to name a few) (24). However, controlled exposures can keep a continuous check on ABR. The exposures can generally be estimated from PK parameters derived from a population but the information related to population PK of FDC antibiotics is limited. In this study, we created a generalized population PK model for active components (ceftriaxone and sulbactam) of FDC. The created model was further used to predict exposures of ceftriaxone and sulbactam in adult patients (urinary tract infection) and in pediatric population. The model predictions were robust across age groups and the population derived PK parameters were used to assess the efficacies at various dosage regimens of FDC over a range of MICs using probabilistic determinations of achievement of pharmacodynamic targets. These targets are generally linked to PK/PD indices (AUC/MIC ratio, C max/MIC ratio, and % T > MIC), routinely used for explaining the therapeutic efficacy in terms of achieving MIC. For beta-lactams, %T > MIC = 70% for beta lactam antibiotics is considered optimal for bacterial killing (in further discussion MICcomb will be used instead of MIC as the MIC of FDC is the combined effects of active ingredients)(25). Monte Carlo simulations were performed to estimate the PTA and further dosage regimens giving desired PTA were selected and clinical breakpoints for the FDC were identified.
The major hurdle in dose optimization of FDC is the variability of the drug exposure induced by potential interaction of the components of FDC. Therefore, PK profile comparison (graphical and statistical) between the FDC and their individual counterparts at similar doses (Fig. 1 and Table II) were done; and similar concentration-time profiles and PK parameters (CL, VD, AUCs, t1/2, Ke, tmax, Cmax) were observed. In addition, the PK parameters estimated in the study (Table II) were in concordance with the values mentioned in literature (15,18). Similarity in PK profile suggested similar systemic exposure, which would be beneficial to patients as two drugs can be given together to reduce pill burden to patients along with other benefits (cost reduction, pharmacodynamic synergism just to name few). It must be mentioned that EDTA was a part of the FDC formulation and super-imposable PK profiles were obtained for both ceftriaxone and sulbactam. This suggests that EDTA did not alter the PK parameters of the active components of the FDC.
The major focus of the present study was to develop population PK model for the FDC of ceftriaxone and sulbactam. The population-PK model assists in rationally optimizing the antimicrobial dose as per the individual physiology. Model selection was based on the AIC and the goodness-of-fit of both population and individual predicted vs. observed plots. In short, the finally developed population-PK model of FDC was explained by one and two compartments for ceftriaxone and sulbactam, respectively. Weight was the main covariate, impacting the elimination rate constant K e and explaining the between-subject variability of the data. The residual variability in the models was explained by error models. The model underwent procedures of internal and external validations for testing model robustness in different scenarios and hence to gauge the general utilizability of model in predicting exposures. Internal validation of model were done by VPC and bootstrap analysis and the estimated values with 95% CI for PK parameters CL and V D were similar to that obtained from the population PK model. External validations were done using data from healthy volunteers [taking ceftriaxone and sulbactam alone] and the data from cUTI patients [taking the FDC]. The model dependent CL and V D estimations were in line with reported values in the literature for both the populations, advocating the robustness and reliability of the population-PK model in predicting exposures.
The developed model’s applicability was further tested on pediatric population. This special group of population is known to have differences in drug exposures due to variability in CL and V D. The inappropriate changes in exposures might result in sub-optimal efficacy and/or compromised safety. The developed model with weight as covariate was used to predict the PK parameters (and hence exposures) in pediatric data sets extracted from literature (12,13). Interestingly, it was observed that the developed PK model, after proper refinement, can be extrapolated to pediatric population. As per the model, the estimated CL and V D were similar to that reported in the literature (12,13). further reiterating the generalizablity and utilizability of the developed population PK model.
The model-derived parameters (CL and K e) from the combined healthy and infected subjects’ data were then used to simulate data for another 1000 subject. It should be mentioned that CL, K e, and V D of both populations were not statistically different (Table III). The populations were combined to increase the variability in the simulated concentration-time profiles and making it closer to a real situation. Monte Carlo simulations of 1000 subjects were done to determine how likely the different exposures determined by different dosage regimens (0.75 to 9 g OD/BD) of FDC would achieve the target %T > MICcomb of 70% over a range of MICs 1–64 μg/mL. Focusing on PTA of %T > MICcomb ~70%, the FDC has achieved 100% PTA for almost all doses against the strains for which it exhibits MIC of 1–4 μg/mL. Also for MIC ≤8 μg/mL, the lowest dose that can achieve the PTA of more than 90% is 1.5 g BD. Further increase in MIC, i.e., 16 μg/mL lowers the PTA, requiring higher doses to revive the PTA back to 90% (large amount of FDC dose, i.e., >6 g BD). However, higher doses might also compromise the safety of FDC. All doses were ineffective for strains corresponding to MIC 32 and 64 μg/mL. Thus, the strains corresponding to the MIC of ≤8, 8–16, and ≥32 μg/mL were susceptible, intermediate, and resistant, respectively to the FDC. The categorization of bacterial strains against a drug, in terms of MICs, evaluates the therapeutic potential of the drug. These clinical breakpoints provide an advantage over infections wherein ceftriaxone alone is showing therapeutic limitations. For example, the susceptibility for FDC is ≤8 μg/mL, which is four-fold higher than that of ceftriaxone resistant strains (MIC ≥2 μg/mL). In summary, a high PTA (≥90%) for a target 70%T > MIC with MIC ≤8 μg/mL was observed with 1.5 g BD dose; with same total exposure in OD dose (3 g), ~80% PTA was attained. Dosage regimen of 3 g BD showing PTA of 100% indicates the improvement in response, if required, in cases of severe infections. This claim was further supported from the clinical data of cUTI patients (data used for external validation purpose), where >90% cure was achieved with 3 g BD FDC dose (11).
It should be mentioned that the study also suffered from a few limitations. Firstly, ceftriaxone and sulbactam, total (free + protein bound) concentrations were measured, while only free drug was used to calculate PK/PD index and further antibacterial efficacy. Secondly, the study was conducted in healthy subjects and extrapolated to small pool of infected subjects (UTI); however, it would be difficult to extrapolate the findings to severely ill patients. The reason is differences in CL and V D, which will affect the plasma concentrations and the associated PK/PD index. Therefore, further studies in severely ill patients should be conducted to assess the antibacterial efficacies of various dosage regimens.
CONCLUSION
The present study demonstrated that the PK profile of ceftriaxone and sulbactam did not alter, when given in FDC, from their original PK profile, i.e., when given individually. In other words, there is no pharmacokinetic interaction between ceftriaxone, sulbactam, and EDTA, when given simultaneously in a FDC. The second section was focused on developing a population PK model using the plasma concentration-time data of 24 healthy adult subjects. Model validity was established internally using VPC and boot strap method. The developed model was validated externally in healthy as well as patient population and further the model was used to estimate exposure in pediatrics. Finally, 1000 patient MCS was performed using population PK model-based parameters to predict attainment of pharmacodynamic targets (PK/PD index; an efficacy driver) at various FDC doses over range of MICs. The optimal dosage regimens of 1.5 g BD/3 g OD/3 g BD had a likelihood of attaining target exposures. To conclude, in-depth population PK modeling of FDC containing ceftriaxone and sulbactam was done and the model-based parameters were used to estimate exposure-dependent attainment of efficacy.
AUC: area under curve; FDC: fixed dose combination; MIC: minimum inhibitory concentrations; PK: pharmacokinetics; PD: pharmacodynamics; pop-PK: population pharmacokinetic; VPC: visual predictive check; V D: volume of distribution
REFERENCES
Shaikh S, Fatima J, Shakil S, Rizvi SMD, Kamal MA. Antibiotic resistance and extended spectrum beta-lactamases: types, epidemiology and treatment. Saudi J Biol Sci. 2015;22(1):90–501.
Appelbaum PC. 2012 and beyond: potential for the start of a second pre-antibiotic era? J Antimicrob Chemother. 2012;67(9):2062–8.
Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12.
Tamma PD, Cosgrove SE, Maragakis LL. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev. 2012;25(3):450–70.
Chaudhary M, Payasi A. Role of EDTA and CSE1034 in curli formation and biofilm eradication of Klebsiella pneumoniae: a comparison with other drugs. J Antibiot (Tokyo). 2012;65(12):631–3.
Hall MJ, Westmacott D, Wong-Kai-In P. Comparative in-vitro activity and mode of action of ceftriaxone (Ro 13–9904), a new highly potent cephalosporin. J Antimicrob Chemother. 1981;8(3):193–203.
Bush K. Beta-lactamase inhibitors from laboratory to clinic. Clin Microbiol Rev. 1988;1(1):109–23.
Wenzler S, Schmidt-Eisenlohr E, Daschner F. In vitro activity of penicillin G/sulbactam compared with penicillin and other antibiotics against common organisms causing ear, nose and throat (ENT) infections. J Antimicrob Chemother. 2003;51(5):1312–4.
Garot D, Respaud R, Lanotte P, Simon N, Mercier E, Ehrmann S, et al. Population pharmacokinetics of ceftriaxone in critically ill septic patients: a reappraisal. Br J Clin Pharmacol. 2011;72(5):758–67.
Soto E, Shoji S, Muto C, Tomono Y, Marshall S. Population pharmacokinetics of ampicillin and sulbactam in patients with community-acquired pneumonia: evaluation of the impact of renal impairment. Br J Clin Pharmacol. 2014;77(3):509–21.
Attili VSS, Chaudhary M. Pharmacokinetics and pharmacodynamics of Elores in complicated urinary tract infections caused by extended spectrum beta-lacatamase strains. Int J Pharm Sci Res. 2014;6(6):2569–78.
Schaad UB, Guenin K, Straehl P. Single-dose pharmacokinetics of intravenous sulbactam in pediatric patients. Rev Infect Dis. 1986;8 Suppl 5:S512–7.
Schaad UB, Stoeckel K. Single-dose pharmacokinetics of ceftriaxone in infants and young children. Antimicrob Agents Chemother. 1982;21(2):248–53.
Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34(4):467–76.
Patel IH, Chen S, Parsonnet M, Hackman MR, Brooks MA, Konikoff J, et al. Pharmacokinetics of ceftriaxone in humans. Antimicrob Agents Chemother. 1981;20(5):634–41.
Meyers BR, Wilkinson P, Mendelson MH, Walsh S, et al. Pharmacokinetics of ampicillin-sulbactam in healthy elderly patients and young volunteers. Antimicrob Agents Chemother. 1991;35(10):2098–101.
Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32.
Foulds G, Stankewich JP, Marshall DC, O'Brien MM, Hayes SL, Weidler DJ, et al. Pharmacokinetics of sulbactam in humans. Antimicrob Agents Chemother. 1983;23(5):692–9.
Zhao W, Zhang D, Fakhoury M, Fahd M, Duquesne F, Storme T, et al. Population pharmacokinetics and dosing optimization of vancomycin in children with malignant hematological disease. Antimicrob Agents Chemother. 2014;58(6):3191–9.
Dietrich ES, Bieser U, Frank U, Schwarzer G, Daschner FD. Ceftriaxone versus other cephalosporins for perioperative antibiotic prophylaxis: a meta-analysis of 43 randomized controlled trials. Chemotherapy. 2002;48(1):49–56.
Esposito S, Noviello S, Vanasia A, Venturino P. Ceftriaxone versus other antibiotics for surgical prophylaxis : a meta-analysis. Clin Drug Investig. 2004;24(1):29–39.
Beskid G, Christenson JG, Cleeland R, DeLorenzo W, Trown PW. In vivo activity of ceftriaxone (Ro 13–9904), a new broad-spectrum semisynthetic cephalosporin. Antimicrob Agents Chemother. 1981;20(2):159–67.
Nazarudheen S, Dey S, Kandhwal K, Arora R, Reyar S, Khuroo AH, et al. Combining benefits of an adrenergic and a muscarinic blocker in a single formulation—a pharmacokinetic evaluation. Regul Toxicol Pharmacol. 2013;67(2):226–31.
Chaudhary M, Payasi A. Clinical, microbial efficacy and tolerability of Elores, a novel antibiotic adjuvant entity in ESBL producing pathogens: prospective randomized controlled clinical trial. J Pharm Res. 2013;7(4):275–80.
Kays MB. Comparison of five beta-lactam antibiotics against common noscomial pathogens using the time above MIC at different creatinine clearances. Pharmacotherapy. 1999;19(12):1392–9.
ACKNOWLEDGMENTS
This research is funded by Venus Remedies Ltd. Special thanks are extended to Azidus laboratories for carrying out this comparative pharmacokinetic study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was designed in accordance with good clinical practices and approved by an independent ethics committee (IEC) [protocol no. AZ/BE/P/038, dated 20 May 2010 approved on 13 July 2010].
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 119 kb)
Rights and permissions
About this article

Cite this article
Sharma, V.D., Singla, A., Chaudhary, M. et al. Population Pharmacokinetics of Fixed Dose Combination of Ceftriaxone and Sulbactam in Healthy and Infected Subjects. AAPS PharmSciTech 17, 1192–1203 (2016). https://doi.org/10.1208/s12249-015-0454-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0454-2