
Abstract
SHetA2 is a promising, orally active small molecule with anticancer properties that target heat shock proteins. In this study, we aimed to investigate the pharmacodynamic (PD) effects of SHetA2 using preclinical in vitro and in vivo models of ovarian cancer and establish a physiologically based pharmacokinetic (PBPK)/PD model to describe their relationships with SHetA2 concentrations in mice. We found that daily oral administration of 60 mg/kg SHetA2 for 7 days resulted in consistent plasma PK and tissue distribution, achieving tumor drug concentrations required for growth inhibition in ovarian cancer cell lines. SHetA2 effectively induced cyclin D1 degradation in cancer cells in a dose-dependent manner, with up to 70% reduction observed and an IC50 of 4~5 µM. We identified cyclin D1 as a potential PD marker for SHetA2, based on a well-correlated time profile with SHetA2 PK. Additionally, we examined circulating levels of ccK18 as a non-invasive PD marker for SHetA2-induced apoptotic activity and found it unsuitable due to high variability. Using a PBPK/PD model, we depicted SHetA2 levels and their promoting effects on cyclin D1 degradation in tumors following multiple oral doses. The model suggested that twice-daily dosing regimens would be effective for sustained reduction in cyclin D1 protein. Our study provides valuable insights into the PK/PD of SHetA2, facilitating future clinical trial designs and dosing schedules.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
SHetA2, an orally active small molecule, targets three homologous heat shock protein A (HSPA) proteins (Grp78/HSPA5, hsc70/HSPA8, and mortalin/HSPA9), which function as chaperones through binding unfolded or misfolded client proteins and catalyzing their proper functional three-dimensional structures. SHetA2 has demonstrated promising anticancer activities against various cancers (1,2,3). It has also shown chemopreventive activity both in vitro and in vivo (4, 5), with no adverse effects observed in various genetic toxicology assays and toxicity tests conducted on animal models (5,6,7). With its promising anticancer activity and lack of toxicity in preclinical studies, SHetA2 is currently in a phase I clinical trial for patients with advanced or recurrent cancers (clinicaltrials.gov: NCT04928508).
SHetA2 induces G1 cell cycle arrest in cancer cells by reducing cyclin D1 levels (8). Cyclin D1, a key member of the cyclin family, controls cell division cycles in collaboration with cyclin-dependent kinases (CDKs). It forms complexes with CDK4 and 6, which phosphorylate the retinoblastoma protein (Rb). Phosphorylated Rb then dissociates from E2F transcription factor, facilitating S phase entry and accelerating cell proliferation. Cyclin D1 levels are often elevated in several cancer types, including breast cancer, non-small-cell lung cancer, and endometrial cancer, as its proteasomal degradation is often deregulated (9,10,11,12). Overexpression of cyclin D1 in ovarian cancer is associated with worse patient overall survival (13). SHetA2 promotes the degradation of cyclin D1 as predicted through releasing it from its binding with HSPA chaperone proteins, leading to its phosphorylation, ubiquitination, and eventual degradation by proteosomes. The expression of a non-degradable mutant cyclin D1 or the overexpression of regular cyclin D1 substantially impeded the induction of G1 cell cycle arrest by SHetA2 (8). Conversely, the SHetA2’s decrease in tumor growth is associated with a significant reduction in cyclin D1 levels within tumors (14). Additionally, cyclin D1 reduction is associated with the decrease in tumor incidence following SHetA2 treatment (15). These findings collectively support the functional significance of cyclin D1 degradation in the mechanism of SHetA2.
Apart from inducing G1 cell cycle arrest, SHetA2 also triggers intrinsic apoptosis by disrupting the interaction between mortalin and its client proteins like p66shc, p53, and Bcl-2 (16). This causes cancer cell–specific degradation of Bcl-2 and Bcl-xl, resulting in swelling and membrane potential loss in mitochondria and cleavage of caspases 9 and 3 in cancer, but not in healthy cells (1,2,3, 7). The mitochondrial damage stimulates excessive mitophagy and translocation of apoptosis inducing factor (AIF) from the mitochondria to the nucleus, which both contribute to the mechanism of SHetA2-induced cell death in cervical cancer cells (17) and endometrial cancer (Chandra et al. 2021). Additionally, SHetA2 enhances the expression of CHOP (CAAT/enhancer binding protein homologous protein) and death receptor 5 and represses nuclear factor kappa B and cellular FLICE-inhibitory protein (c-FLIP), which promotes extrinsic apoptosis mediated by death receptors (18).
Well-designed preclinical in vitro and in vivo studies combined with predictive modeling approaches can provide critical information regarding optimal dose, schedule, and drug administration. We have previously conducted allometric scaling and pharmacokinetic (PK) modeling of SHetA2 in mice, rats, and dogs to determine the appropriate first human doses of the drug (19). We have also developed a physiologically based pharmacokinetic (PBPK) model for SHetA2 in tumor-bearing mice to describe its oral absorption kinetics and disposition in different tissues (20). Pharmacodynamic (PD) analysis of the biochemical and biological effects of anticancer drugs on specific targets in the cancer cell is an important component of early stage of clinical trials. Incorporating PD markers in early clinical trials may provide information on coverage of the target pathway in patients, which can inform the rational selection of biologically effective doses for further testing.
This study utilized experimental and computational methods to investigate PD effects for SHetA2 in preclinical models of ovarian cancer. As SHetA2 only affects the function of its target (mortalin) and not its cellular levels (21), we evaluated the reduced levels of the cyclin D1 protein in tumors as a measure of SHetA2-induced G1 cell cycle arrest. Additionally, we monitored circulating levels of caspase-cleaved cytokeratin 18 (ccK18) (22) as a measure of SHetA2-induced apoptosis. ccK18 represents the major intermediate filament protein and ccK18 is released by the action of caspases in apoptotic epithelial cells. The aims of this study were threefold: (1) to investigate the tissue distribution of SHetA2 in tumor-bearing mice after receiving multiple oral doses, (2) to analyze the changes in cyclin D1 and ccK18 levels induced by the drug during the treatment period, and (3) to develop a PBPK/PD model to characterize the relationships between SHetA2 tumor concentration and its PD effects.
Materials and Methods
Chemicals and Reagents
SHetA2 was supplied by the National Cancer Institute RAID Program for the animal models and synthesized by K. D Berlin, PhD (Oklahoma State University, Stillwater, OK) for cell culture studies. Internal standard, 2,3-diphenylquinoxaline, was purchased from Acros Organics (New Jersey), and HPLC grade acetonitrile (HPLC-JT9012) was procured from VWR (Radnor, PA). Kolliphor HS 15 was purchased from Sigma-Aldrich (St. Louis, MO), and sterile phosphate-buffered saline (PBS) was from Global Cell Solutions (Charlottesville, VA). Antibodies for anti-Cyclin D1, and anti-β-actin were purchased from Cell Signaling Technology (Boston, MA). PathScan® Total Cyclin D1 Sandwich ELISA Kit was purchased from Cell Signaling Technology (Boston, MA). Protein extraction buffer m-PER was purchased from Thermo-scientific (Waltham, MA). Pierce BCA protein assay kit was purchased from Thermo Fisher Scientific (Grand Island, NY). Protease inhibitor cocktail and phosphatase inhibitor cocktail were purchased from Roche (New York, NY). PVDF membrane and ECL reagent were purchased from Bio-Rad (Hercules, CA).
Cells
SKOV3, a human ovarian cancer cell line, stably expressing firefly luciferase (SKOV3-Luc) was obtained from MD Anderson Cancer Center, Houston, TX (a generous gift from Dr. Anil Sood). A2780, a human ovarian cancer cell line, was obtained from WPR Cancer Institute at University of Arkansas for Medican Sciences (a generous gift from Dr. Michael Birrer). The cells (A2780 and SKOV3) were cultured in RPMI 1640 medium (Mediatech, VA) supplemented with 10% fetal bovine serum (FBS) (Serum Source International, NC) and 1% antibiotic-antimycotic (Thermo Fisher Scientific, MA).
Concentration- and Time-Dependent Changes in Cyclin D1 In Vitro
Ovarian cancer cell lines (A2780 and SKOV3) were seeded (0.5 × 106 cells per well of six-well plate) in RPMI media supplemented with 10% FBS and 1% antibiotics at 37°C and 5% CO2 overnight. SHetA2 was prepared in cell culture grade DMSO. The cell lines were treated with SHetA2 at various concentrations (0, 1, 2.5, 5, 7.5. 10, 15 µM) for 24 h. In SKOV3, treatment was also performed at different time points (2, 4, 6, 8, 12, 24 h) using 2.5 and 10 µM SHetA2. Additionally, the recovery of cyclin D1 after a 24-h exposure to 10 µM SHetA2 was evaluated at 4, 8, and 24 h following the removal of SHetA2, in comparison to during continuous drug exposure for 24 and 48 h.
Orthotopic SKOV3 Xenograft Mouse Model
Crl:NU(NCr)-Foxn1nu (homozygous; strain code 490) female athymic nude mice (6-to-8-week-old, 15–20 g) were purchased from Charles River Laboratories (Houston, TX). Mice were housed in a temperature-controlled room on a normal 12-h light/dark cycle, with free access to water and standard laboratory food. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Oklahoma Health Sciences Center. After acclimatization for 1 week, the animals were intraperintoneally (i.p.) injected with one million SKOV3-Luc cells per animal. For tumor incoculation, the cells were harvested, washed once with Hank’s balanced salt solution (HBSS, Ca2+, Mg2+, and phenol red-free), and re-suspended in HBSS (Thermo Fisher Scientific). Tumor development was monitored at weeks 1 and 3 by imaging the animals using Carestream In vivo Xtreme imaging system (Carestream Molecular Imaging, CT) following i.p. injection of 125 µL of D-luciferin (30 mg/mL; Caliper Life Sci. Inc., Hopkinton, MA). The net light intensity of the image was taken to derive tumor burden.
Drug Administration and Sample Collections (Fig. 1)
This study was designed to determine the tissue distribution of SHetA2 over the period, reaching to the steady state, and PD effects in plasma or tumor tissues which correlate with SHetA2 levels. For oral administration, an SHetA2 suspension was prepared in 30% Kolliphor HS 15 with a final concentration of 6 mg/mL to provide an oral dose of 60 mg/kg when administered with a dosing volume of 10 mL/kg. Three weeks after the SKOV3-luc cells injection, tumor-bearing mice that have successfully developed tumor were randomized into two groups based on mean luminal intensity. The treatment group (n = 33) received SHetA2 formulated in 30% Kolliphor HS 15 daily for 7 consecutive days. This group was subsequently divided into eleven subgroups, each consisting of three mice, for sample collection at pre-defined time points. At each time point (first dose at 1, 4, 6, 8, and 12 h) and troughs (immediately before daily subsequence doses; 24, 48, 72, 96, 120, and 144 h), 250 μL of blood was collected from three animals via saphenous vein, while they were under appropriate restraint. The subgroups were rotated in a staggering manner to avoid repeated bleeding of individual animals during the 7-day dosing period. After the seventh dose, non-survival blood was collected from the inferior vena cava while the animals were under deep anesthesia with isoflurane. The animals were then euthanized, and tumor and other tissue samples were collected at specific time points (0, 0.25, 0.5, 1, 2, 2.5, 4, 6, 8, 12, and 24 h after the last dosing on the seventh day).

In vivo study design. Female athymic nude mice were intraperitoneally injected with SKOV3 human ovarian cancer cell line. Tumor development was confirmed by imaging after approximately 3 weeks. The tumor-bearing animals were randomly assigned to receive either vehicle (n = 3) or 60 mg/kg SHetA2 (n = 33; 11 subgroups of three mice) orally once daily for 7 days. Blood samples were collected at various time points throughout the 7 days, including during the first dose, at troughs, after the last dose. Both blood and tissues samples were collected after the last dose. The blood and tissue samples were used for SHetA2 quantifications and PD effects
The control group (n = 3) received a vehicle (30% Kolliphor HS 15 in distilled water) once daily for 7 consecutive days. At each of trough time point (0, 24, 48, 72, 96, and 120 h after the start of the first dose), 100 µL of blood was collected from the saphenous vein to measure ccK18 levels. The animals were euthanized at 144 h (24 h since the 7th dose), and blood, tumor, and tissue samples were collected for further analysis.
Additional plasma and tumor samples from our previous single-dose studies (20) were used to investigate the PD effects. The plasma samples included nine time points (0, 0.08, 0.25, 0.5, 1, 2, 4, 12, 18, and 24 h) after a single intravenous (IV) dose of SHetA2 10 mg/kg for ccK18 quantification. We also used one time-point tumor samples (24-h postdosing) from a single oral dose of SHetA2 60 mg/kg for the cyclin D1 assay.
SHetA2 Quantitation in Tissues and Plasma
SHetA2 concentration was determined using the HPLC-UV method that had been previously developed and validated in our laboratory, as described in elsewhere (23). Briefly, 90 µL of plasma samples was mixed with 10 µL of internal standard (5 µg/mL) in an amber Eppendorf tube while kept in ice, which was then vortexed vigorously for 1 min. The mixture was subsequently precipitated by adding 80 µL of chilled 100% acetonitrile, vortexed vigorously for 10 min, and centrifuged at 21,381× g for 15 min at 4°C. The supernatant was collected and filtered using a Captiva filtration system (Agilent Technologies) by applying a vacuum of not more than 16,900 Pa. The filtrate was then injected (70 µL) and analyzed using the HPLC-UV (Agilent 1260 HPLC system). For tissue samples, the collected tissues were thawed under ice in glass tubes and homogenized (Kinematica Polytron TM 1600 EW benchtop homogenizer, Kinematica) with 3 mL/g of chilled phosphate buffer (pH 7.4) submerged in an ice water bath. Similarly to the plasma sample preparation, 90 µL of these tissue homogenate was mixed with 10 µL of internal standard (5 µg/mL), and the mixture was processed as described above. The lower limit of quantitation is 5 ng/mL, the intra-day accuracy ranged from 100.5 to 110.2%, interday accuracy ranged from 96.9 to 101.2%, and the precision is from 3.4 to 13.4 described by standard deviation (%RSD) of each calculated concentration.
Cyclin D1 Quantification by ELISA
A commercially available ELISA kit was used for detection of total cyclin D1 (PathScan® Total Cyclin D1 Sandwich ELISA Kit) in SHetA2 treated ovarian cancer cell lines (A2780 and SKOV3) and in tumor tissue obtained from xenografts. The ELISA was performed following the manufacturer’s instructions in an antigen-coated 96-well plate, and all the samples and negative controls were tested in duplicate in each plate. In brief, 100 μL of protein samples (5 µg/sample) was added in each well and incubated for overnight at 4°C. After the wash step, 100 μL of detection antibody was added and plate was incubated at 37°C for 1 h. The unbounded detection antibodies were washed out, and the wells were incubated in the presence of 100 μL of HRP-linked secondary antibody for 30 min at 37°C followed by washing. Subsequently, 100 μL of TMB (3,3′, 5,5″-tetramethylbenzidine) substrate was added and, after 10 min incubation at 37°C, the reaction was stopped by adding 100 μL of STOP solution, and the optical density (OD) was measured in Synergy™ H1 microplate reader (BioTek, Winooski, VT) at 450 nm. The assay kit sensitivity ranges from 0.014 to 0.45 mg/mL.
Western Blot Analysis of Cyclin D1
Total proteins from cells were isolated using M-PER protein extraction solution (Thermo Fisher Scientific, Waltham, MA) containing protease inhibitor cocktail and phosphatase inhibitor cocktail. The homogenates were centrifuged at 14,000 × g for 15 min at 4°C, and the supernatant containing proteins was collected. For in vivo samples, tumor tissue obtained from xenografts were homogenized in ice-cold T-PER protein extraction solution (Thermo Fisher Scientific, Waltham, MA) containing protease inhibitor cocktail and phosphatase inhibitor cocktail using a mortar-pestle with liquid nitrogen. The homogenate was incubated on ice for 1 h with occasional shaking followed by centrifugation at 14,000 × g for 15 min at 4°C, and the supernatant containing proteins was collected. Protein concentrations were determined using a BCA assay (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions.
Equal amounts of protein (20 μg for cell lysates and 35 μg for xenograft tumor tissues) were separated by gel electrophoresis and then transferred to an Immuno-Blot™ PVDF membrane. The membrane was blocked with 5% skimmed milk and then incubated with primary antibody to cyclin D1 (# 2922, Cell Signaling Technology, Danvers, MA) at 1:500 dilution overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase-conjugated antibody for 1 h. Antibody binding was detected using enhanced chemi-luminescence detection system (Bio-Rad). After developing, the membrane was stripped and re-probed using an antibody against β-actin (# 4970, Cell Signaling Technology, Danvers, MA) at 1:2500 dilution to confirm equal loading.
ccK18 Quantification by ELISA
The apoptotic cleavage of K18 exposes a neoepitope M30 (CK18-Asp396), and the caspase-generated cleavage fragments of K18 (ccK18) can be detected by the M30 antibody. The soluble ccK18 was detected in mouse plasma using the M30-Apoptosense ELISA (PEVIVA AB, Bromma, Sweden). Briefly, the M30 conjugate was diluted, and wash tablet was dissolved with distilled water. After adding 25 µL of standards, control, or samples per well and 75 µL of diluted M30 conjugate solution per well, the plate was incubated for 4 h on plate shaker at 600 rpm. The plate was then washed manually with 5 × 250 µL with wash buffer. After adding TMB substrate (200 µL/well) and incubation in darkness for 20 ± 1 min, 50 µL of stop solution per well was added and shaken for 5–10 s. The absorbance was read at 450 nm after 5–30 min using Biotek® Synergy™ 2 Multi Mode Microplate Reader (Vermont).
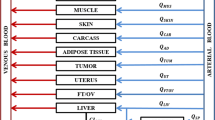
PBPK/PD Modeling
In our previous study (20), we developed a whole-body PBPK model (Fig. 2) to describe the time profiles of SHetA2 plasma and tissue concentrations in orthotopic SKOV3 xenograft tumor-bearing mice, after administration of a single IV dose (10 mg/kg) and oral dose (60 mg/kg). The PBPK model accounted for extensive distribution in 13 different tissues by perfusion-limited model and drug elimination through plasma by decomposition (CLPL) and metabolism in liver (CLLIV) and gut (CLGI). The oral absorption kinetics was described by two first-order absorption rate constants, kA and kA1, with each process associated with the fraction of the absorbed dose FAGG and (1-FAGG), respectively. In the current study, we extended this model by incorporating a PD model (Fig. 2) that characterizes the effect of SHetA2 on cyclin D1 in tumors, along with the time profiles of SHetA2 plasma and tissue concentrations, following multiple once-daily doses of 60 mg/kg. SHetA2 promotes the cyclin D1 reduction through proteasomal degradation (8, 16), and to capture this effect, we modeled the cyclin D1 turnover using the zero-order production rate constant (kin) and the first-order degradation rate constant (kout), with the drug effect (Smax, SC50, γ) as the stimulation of cyclin D1 loss. Smax represents the maximum fold change in cyclin D1 expression from the baseline, SC50 is the SHetA2 concentration causing 50% Smax, and γ is the slope factor. Additionally, we accounted for the time delay between changes in drug concentration and cyclin D1 reduction by adding transit compartments (TC1, TC2, and TC3). These compartments take into consideration events, such as phosphorylation and ubiquitination of cyclin D1 by SHetA2, which precede proteasomal degradation. We assumed that cyclin D1 levels remain stable during tumor progression, as supported by our in vivo data, which showed that cyclin D1 levels recovered to nearly their baseline levels 24 h after each dose, including both the first dose and repeated dosing. The value of the endogenous degradation rate constant for cyclin D1 (kout) in cancer cells was calculated from the data obtained from literature (24) and fixed to be 1.38 h−1. The change of cyclin D1 was described by the following equations:

A PBPK/PD model for SHetA2 in tumor-bearing mice. The PBPK model includes 14 perfusion-limited tissue compartments, elimination of SHetA2 from liver (CLliv), intestine (CLGI) and plasma (CLPL), and oral absorption kinetics (ka, ka1, FAGG). The PD model is based on the SHetA2 concentration in the tumor and depicts the stimulation of cyclin D1 degradation through three transit compartments (TC1-3). Cyclin D1 synthesis and degradation are depicted by the zero-order constant kin and the first-order constant kout, respectively. The initial effect E induced by drug concentration (Cdrug) is described by the maximum effect (Smax), the drug concentration for 50% effect (SC50), and the slope factor (γ). The transit compartments account for events such as cyclin D1 phosphorylation, ubiquitination, and proteasomal degradation, with τ denoting the transit time among compartments
In Eq. 1, Cdrug refers to the concentration of SHetA2 in either the cell culture medium (\({C}_{med}\)) or the tumor (\({C}_{tumor}\)), depending on whether the data is from in vitro or in vivo experiments, respectively. As SHetA2 undergoes degradation over time in cell culture medium or plasma in vitro, we accounted for this degradation using a first-order degradation rate constant (kdeg = 0.0127 h−1), which was calculated based on an in vitro stability study (25).
To adjust for the difference in medium concentration in vitro and tumor concentration in vivo, we applied a scaling factor \({\alpha }_{t}\) (e.g., \({\alpha }_{t}\bullet {C}_{tumor}\)) (26), which was estimated during fitting. The initial condition for all the differential equations was zero, except for CyclinD1, which was set to 100.
All the PK parameters were fixed during PD data fitting. The equations were solved simultaneously for both in vitro and in vivo data to estimate the PD model parameters using the maximum likelihood estimation implemented in ADAPT 5 (Biomedical Simulations Resource, CA) (27). The best model was selected based on the Akaike information criterion comparison, the goodness-of-fit, weighted residual plots, and reliability of parameter estimations. The variance model was: \({V}_{i}={\left({\sigma }_{1}+{\sigma }_{2}\cdot {Y}_{i}\right)}^{2}\), where Vi represents the variance of the ith data point, Yi is the ith model prediction, and σ1 and σ2 are variance model parameters. Figures were created using GraphPad Prism 8.42 (GraphPad Software, La Jolla, CA).
Sensitivity Analysis
A local sensitivity analysis was conducted for each parameter. In each scenario, an individual model parameter was adjusted while all other parameters were kept constant. Observations of the predicted changes in model output were recorded. The result is expressed as sensitivity coefficients which are calculated using Eq. (6). In this equation, p is the original parameter value, Δp is the change from the original parameter, r is the original output result, and Δr is the change in the model output resulting from altering the parameter.
Results
In this study, we investigated the plasma PK and tissue drug distribution as well as PD effects over the course of the treatment in tumor-bearing mice after multiple oral administrations of 60 mg/kg/day SHetA2 and characterized their relationships using a PBPK/PD model.
Characterization of Plasma and Tissue Distribution of SHetA2 Following Multiple Oral Doses
Fig. 3 displays the plasma and tumor concentration profiles of SHetA2 for a 7-day treatment with 60 mg/kg/day administered once daily. We utilized our previously developed PBPK model based on a single dose of SHetA2 (20) to predict the time-concentration profiles for repeated oral doses and compared the results with the observed data (Figs. 3–4). SHetA2 exhibited a multi-exponential plasma concentration decline and rapid and extensive tissue distribution in tumor-bearing mice model; we found that the model accurately predicted the observed data. The plasma and tumor pharmacokinetics of SHetA2 remained stable over the 7-day period of daily oral administration of 60 mg/kg SHetA2. No significant accumulation of the drug in plasma was observed, which was expected based on the estimated elimination half-life of 4.5 h from a single oral dose study (20). SHetA2 is distributed in the tumor tissue at tenfold higher concentrations compared to the blood. The mean trough concentrations of SHetA2 in the plasma were 18.8 ± 6.6 ng/mL. Fig. 4 shows the time-concentration profiles of SHetA2 in additional 12 non-tumor tissues. We observed no significant changes in SHetA2 concentration in these tissues throughout the treatment period, indicating that SHetA2 exhibits time-invariance PK of SHetA2 during oral absorption and drug disposition. Moreover, our PBPK model accurately predicted the drug concentrations for multiple dosing regimens, as evidenced by the consistent tissue concentrations observed after the 7th dose as well as the single dose (20).

Plasma and tumor drug concentration. The observed drug concentration (symbols) in plasma (a) and tumor (b) following a single (light blue circles) and 7-day (blue circles) oral dose of 60 mg/kg SHetA2 once daily in mice bearing orthotopic SKOV3 tumors. The observed data, presented as mean ± SD, were obtained from three mice at each time point. The lines represent the PBPK model-predicted drug concentration-time profiles. The light gray area represents the 95% prediction interval

Tissue drug concentration. The concentration-time profiles of SHetA2 in different tissues in SKOV3 tumor-bearing mice after a single (light blue circles) and multiple oral (blue circles) administrations of 60 mg/kg SHetA2 once daily. The observed data (symbols), presented as mean ± SD, were obtained from three mice at each time point. Note that the y-axis scale for stomach and GI panels is different from that of the other panels. The lines represent the PBPK model predicted drug concentration-time profiles
Changes in Cyclin D1 In Vitro and In Vivo
The effect of SHetA2 on cyclin D1 was investigated through in vitro and in vivo experiments, revealing a dose- and time-dependent relationship. In vitro cell experiments were conducted using different doses of SHetA2 to observe changes in cyclin D1 protein levels during 24-h drug exposure. Cyclin D1 was quantified using ELISA and confirmed by western blotting. Treatment with SHetA2 resulted in concentration-dependent decreases in cyclin D1 levels in two human ovarian cancer cell lines, with maximum inhibition at ~80% for A2780 cells and ~70% for SKOV3 cells, both at a half-maximum effective concentration of 4~5 µM (Fig. 5a, b). We next evaluated time-dependent changes in cyclin D1 over 24 h at 2.5 and 10 µM SHetA2 and the recovery of SHetA2-induced cyclin D1 reduction after the drug removal from the culture medium using SKOV3 cells. Time-dependent analysis showed that SHetA2 treatment induced a reduction in cyclin D1 levels as early as 4 h post-treatment, which continued for 24 h (Fig. 5c, d). When SHetA2 was removed from the culture medium, cyclin D1 levels began to recover at 4 h and restored to over 80% of their baseline at 24 h (Fig. 5e–g). As expected, SHetA2 had no effect on total mortalin levels.

Dose- and time-dependent effects of SHetA2 on cyclin D1 in vitro. Cyclin D1 protein levels were quantified in cell lysates using both ELISA (a, c, e, and f), which are presented as mean ± SD, and western blotting (b, d, and g). Two human ovarian cancer cell lines, A2780 and SKOV3, were treated with varying concentrations of SHetA2 (1, 2.5, 5, 7.5, 10, 15 μM) for 24 h (a and b). Time-dependent changes in cyclin D were monitored at different time points (0, 2, 4, 6, 8, 12, 24 h) after treating SKOV3 cells with 2.5 or 10 μM SHetA2 (c and d). The recovery of drug-induced cyclin D1 reduction upon drug removal was assessed at a dose of 10 μM SHetA2 (e, f, and g). After A2780 or SKOV3 cells were treated with control or SHetA2 for 24 h, the drug-containing culture medium was removed, cultures were washed, and the cells were incubated with fresh medium for an additional 24 h. The levels of cyclin D1 were monitored at 4, 8, and 24 h after drug removal. Representative western blots for mortalin and cyclin D1, along with GAPDH or cyclophilin G as the loading control (b, d, and g). Statistical analyses were performed in GraphPad Prism using one-way ANOVA tests
For in vivo experiments, the tumor samples were analyzed for cyclin D1 expression following treatment with a single dose or multiple doses. In vivo experiments demonstrated that SHetA2 induces a cyclic pattern of cyclin D1 expression over 7 days. Within the first 12 h of administration, cycling, D1 expression decreased but returned to near-baseline levels at 24 h post-dose for both single and multiple dose treatments (Fig. 6b). The PD profile of cyclin D1 mirrored the PK profile of SHetA2, with a delay. Once-daily treatment with 60 mg/kg of SHetA2 resulted in peak tumor concentrations around 4000 ng/mL (equivalent to ~10 µM) at 1 h, and a maximum reduction of ~70% in cyclin D1 observed at 12 h. Taken together, our in vitro and in vivo results indicate that changes in cyclin D1 expression in tumors are strongly correlated with SHetA2 tumor concentrations in a dose- and time-dependent manner, suggesting that cyclin D1 could serve as an effective proximal PD marker for SHetA2.

The time profiles of cyclin D1 after treatment with SHetA2 in vitro and in vivo. a Changes in cyclin D1 protein levels over time in SKOV3 treated with different SHetA2 concentrations (1–15 μM) during a 24-h incubation followed by drug removal (the data are from Fig. 5). Different time points (0, 2, 4, 6, 8, 12, 24 h) were monitored for the 2.5 and 10 μM groups, and only 24-h time point was measured for the other doses. The recovery of drug-induced cyclin D1 reduction upon drug removal was evaluated at 10 μM by washing the drug-containing culture medium and incubating with fresh medium for an additional 24 h. b The time profile of cyclin D1 levels in SKOV3 tumors in mice treated with a single (light blue circle) and 7-day (blue circles) oral dose of 60 mg/kg SHetA2 once daily. The symbols represent the observed cyclin D1 protein levels in cancer cells or tumors, quantified using the ELISA method, with data presented as mean ± SD (n = 3). Both in vitro and in vivo data were used to estimate the PD model parameters, and the lines represent the best-fit curves. c Sensitivity analysis for each parameter. Sensitivity coefficients are determined through the division of changes in the model’s output by variations in the parameters
Changes in ccK18 In Vivo
We investigated plasma levels of ccK18 as a non-invasive marker for SHetA2-induced apoptosis in tumor-bearing mice treated with either vehicle or SHetA2. ccK18 is a specific cleavage product of full-length cytokeratin 18 during apoptosis. This cleavage occurs at a specific site within the protein, resulting in the generation of a smaller fragment (28, 29). SHetA2 was administered via a single IV dose of 10 mg/kg or once-daily oral dose of 60 mg/kg for 7 days (Supplementary Fig. S1). The vehicle-treated animals had a mean value of approximately 700 U/L ccK18, with no significant change over the treatment period. Following treatment with a single IV dose of SHetA2, ccK18 levels slowly increased to a peak of 913 U/L over 2–4 h after dosing and returned to their baseline levels by 8–12 h postdosing (Fig. S1A). Once-daily oral SHetA2 treatment resulted in a significant increase in plasma levels of ccK18 (>1000 U/L) within first 4–6 h of administration, which also appeared to return levels close to those of control animals by 8 h postdosing (Fig. S1B-C). These results indicate that SHetA2 induces apoptosis. However, the measured ccK18 values were highly variable, and overall changes in ccK18 were not well correlated with the SHetA2 PK. These findings suggest that ccK18 may not be a suitable marker for SHetA2.
PBPK/PD Modeling of SHetA2-Induced Cyclin D1 Reduction
Our in vitro and in vivo results demonstrated that the changes in cyclin D1 in tumors well correlated with the SHetA2 concentrations. To characterize the SHetA2-induced cyclin D1 degradation (8), we explored various PD models, including a simple indirect model with SHetA2’s kout stimulation, both with and without a delay function, and transduction processes either before or after an Emax model. Our final PD model (Fig. 2) provided the best fit for the effect of SHetA2 on cyclin D1. This model depicts that the activity of SHetA2 mediated through three transit compartments, the number of which we estimated through fitting, and then promoting the degradation of cyclin D1. We simultaneously fitted the PD model to both in vitro (dose- and time-dependent and drug removal studies; Fig. 6a) and in vivo data (Fig. 6b), and the estimated parameters are listed in Table I. The model effectively characterizes the in vitro dose-response relationship, yielding estimated values of Smax at 2.44 and SC50 at 4.11 µM. Notably, the model predicts that SHetA2 achieves its maximum inhibitory effect (reducing cyclin D1 expression level by over 70%) in SKOV3 cells at tumor concentrations higher than 10 µM, with an SC50 value (4.11 µM) that closely matches the initial value calculated from in vitro data.
In terms of tumor, the PD model adequately captures the cyclin D1 levels in both the first and seventh doses, achieving steady state quickly following the PK profile. With a once-daily (QD) dose of 60 mg/kg, the cyclin D1 profile fluctuates between 50 and 80%. The difference in drug concentration between in vivo tumors and tissue culture medium was accounted for using the scaling factor (αt), which was estimated to be 1.78. This indicates that the effective concentration of SHetA2 in the tumor is higher than the measured drug concentration.
The free form of cyclin D1 undergoes rapid degradation, but when it binds with mortalin, it is protected from degradation. When SHetA2 binds to mortalin, it releases cyclin D1, thereby promoting its degradation. To model the natural turnover of cyclin D1 both in the absence and presence of SHetA2 treatment, we have fixed kout at its experimentally determined value (1.38 h−1), which corresponds to the degradation rate of its bound form with mortalin. This allows us to approximate the degradation rate of cyclin D1 induced by SHetA2 (e.g., kout∙[1+S(t)]). This degradation process is relatively faster than other processes involved in PD model, making kout less sensitive to the profile. Our sensitivity analysis of the parameters (Fig. 6c) revealed that kout had the lowest sensitivity score, while Smax, SC50, and αt exhibited considerable sensitivity within the PD model.
Using the developed PBPK/PD model, we conducted simulations of various dosing regimens to explore the behavior of cyclin D1 expression at different doses (Fig. 7). Our findings indicate that twice-daily (BID)/three times daily (TID) regimen may achieve a more stable inhibition effect on cyclin D1 expression compared to QD dosing. Additionally, the simulations suggest that dosing higher than 90 mg/kg BID/60 mg/kg TID will likely result in inhibition levels close to the maximum (~70%). These insights offer valuable information for optimizing dosing regimens in future clinical trials.

The simulated time profiles of SHetA2 concentration (a) and cyclin D1 (b) in tumors in mice treated with different dosage regimens of SHetA2
Discussion
Epithelial ovarian cancer remains a difficult-to-treat gynecological malignancy, highlighting the need for novel treatments that offer better efficacy and safety profiles for patients with recurrent ovarian cancer. SHetA2 is a novel agent with demonstrated anticancer activity in various cancer types and has entered a phase I clinical trial for patients with recurrent cervical, ovarian, or endometrial cancer. In this study, we investigated PD effects of SHetA2 in human ovarian cancer cell lines and in vivo models and their relationship with the plasma and tumor drug concentration using a PBPK/PD modeling approach.
SHetA2 is a nontoxic anticancer drug that induces G1 cell cycle arrest and causes apoptosis in human ovarian cancer cell lines, regardless of their histology and inhibits growth of ovarian xenografts tumors (1, 3, 8, 9). SHetA2 degrades cyclin D1 protein by inducing phosphorylation of cyclin D1 on T286, targeting it for ubiquitination and proteasomal degradation (8). Additionally, SHetA2 selectively induces apoptosis in cancer cells by acting on mortalin; releasing its client proteins Bcl-2, p53, and p66shc; and degrading Bcl-2 protein, which in turn promotes intrinsic apoptosis and prevents intraperitoneal ovarian cancer establishment (21, 30). Despite the direct target of SHetA2, mortalin cannot be used as a proximal PD marker for SHetA2 as the drug does not affect the cellular levels of mortalin protein (21). Thus, we proposed cyclin D1 protein and/or cellular byproducts from caspase activity upon apoptosis, such as ccK18, as proximal PD markers for monitoring the effects of SHetA2 on the targets.
Our study found that SHetA2 reduces cyclin D1 protein levels in cancer cells in a dose-dependent manner, with up to 70% inhibition observed and an IC50 of 4~5 µM. In vitro time-course analysis revealed that cyclin D1 expression decreased within 4 h of SHetA2 exposure and began to recover upon drug removal. Furthermore, our in vivo studies showed that cyclin D1 tumor expression exhibited a cyclic pattern in response to SHetA2 tumor profiles, suggesting its potential use as a measure of SHetA2’s activity at the target site. Our investigation of circulating ccK18 levels as a non-invasive marker for SHetA2-induced apoptotic activity showed that SHetA2 treatment resulted in an increase in ccK18 levels, but with wide fluctuations around the baseline and high variability, without a clear trend observed with changes in SHetA2 concentration, which may be due to down-regulation or loss of CK18 expression in cancers arising from CK18 positive tissues linking to tumor dedifferentiation (31). While ccK18 is a sensitive biomarker for other diseases (28, 29), such as nonalcoholic fatty liver disease (32), it may not be a reliable marker for SHetA2.
Given Rb’s crucial role as a growth checkpoint in the G1 phase, inhibiting cyclin D1 and CDK4/6 represents an important target for anticancer therapeutics. Palbociclib, ribociclib, and abemaciclib are three FDA-approved CDK4/6 inhibitors. These inhibitors directly bind to CDK4/6 in its APT packet when bound to cyclin D1, leading the deactivation of the cyclin D1-CDK4/6 complex, reducing Rb phosphorylation, maintaining the G1 phase checkpoint, and inhibiting cancer cell growth. Cyclin D1 overexpression is associated with the development and progression of cancer, and deregulation of cyclin D1 degradation is thought to be responsible for increased cyclin D1 levels in several cancers (9). In addition to its role in cell cyclin regulation, cyclin D1 performs other functions, such as inhibiting mitochondrial metabolism and enhancing cytosolic glycolysis, that help tumors survive in stressful environments (33, 34). Therefore, complementary targeting of Rb phosphorylation by reducing cyclin D1 using SHetA2, and CDK4/6 inhibition would be an effective combination strategy, as confirmed by the SHeA2-palbociclib combination in preclinical models of cervical cancer (14).
Another aim of this study was to assist in the design of a phase 1 study to determine the required dosing regimens to achieve sufficient drug concentrations for tumor target modulation. The relatively fast recovery of cyclin D1, likely due to its short half-life (~ 30 min) (35, 36), and the duration of cyclin D1 reducion, which largely depends on SHetA2 exposure time, indicates the need to maintain drug concentraion above the effective concentration. In tumor-bearing mice, a 60-mg/kg QD achieved a tumor concentration above the IC50 value for the first 2.5 h post-dose, and cyclin D1 levels remained between 50 and 80%. Our simulations showed that the same total daily dose of BID or TID can achive similar PD effect on cyclin D1, in which 60 mg/kg BID or 40 mg/kg TID extended the time above 4 μM tumor concentration and kept stable cyclin D1 reduction. Increasing the dose to 90 or 120 mg/kg BID or 60 or 80 mg/kg TID prolonged the period above 4 μM to 4 h and 6 h, respectively. Consequently, cyclin D1 expression was maximally reduced to around 30–35% of normal levels with very little variation. Considering convenience and patient compliance, these findings support the design of our ongoing phase 1 trial, in which the drug is administered orally twice a day. Tumor biopsies from these patients will provide an opportunity to confirm drug concentrations exceeding 4 µM and cyclin D1 reduction.
Although cyclin D1 provides a direct insight into SHetA2 activity at the target site, a limitation of this study was the lack of quantification of tumor burden, which hindered our ability to correlate cyclin D1 reduction with tumor growth inhibition. For the intraperitoneal orthotopic xenograft model, chosen to closely mimic the ovarian cancer tumor microenvironment and drug distribution, the initial tumor progression is slow, followed by a rapid progression over a two-month period. Therefore, a longer duration of treatment is required to observe significant tumor reduction. However, previous studies have demonstrated that SHetA2 exhibits antitumor activity of approximately 40–60% at a 60 mg/kg QD dose in xenograft tumor models (1,2,3, 7, 14, 17, 37). Benbrook et al. (5) demonstrated that QD doses of 30 or 60 mg/kg SHetA2 reduced incidence of colorectal tumors by over 50% in the APCmin/+ mouse model of colorectal cancer in association with 50 or 90% reduction of cyclin D1 in the tumors. Furthermore, considering the limitations of our study in terms of dosing levels, it is imperative to expand our investigations to a broader spectrum of dosing regimens. Future research should involve exploring multiple dose levels and dosing schedules to better capture the dose-response relationship. Additionally, the use of a single cell line in our in vivo study underscores the need for further investigations employing diverse cell lines and patient-derived xenograft models. This is essential for corroborating our findings, especially considering the heterogeneous molecular and pathohistological characteristics of ovarian cancer, which includes p53 mutations. Lastly, we acknowldege that we did not measure protein binding or cell uptake of SHetA2 in our in vitro experiments. Therefore, we used the scaling factor (αt) for in vitro-to-in vivo correlation, which has been shown to be useful in studies where direct measuments are not available (26). This study provides quantitative insights into the mechanism underlying the biological activity of SHetA2 in tumor and highlights the need for further research to develop an accesible and translatable PD markers for SHetA2 in the first in-human study.
Conclusion
Our study has demonstrated that SHetA2 exhibits a consistent PK/PD profile during a 7-day dosing regimen, and we have characterized the drug concentration-cyclin D1 expression relationship using a PBPK/PD model. Our results have identified dose regimens for achieving optimal drug concentration and target modulation. These insignts, along with the PBPK/PD model, provide valuable information for the development of dosing schedules and study designs for future clinical trials of SHetA2.
Data Availability
The data presented in this study are available on request from the corresponding author.
Abbreviations
- BID:
-
Two times a day
- TID:
-
Three times a day
- CDKs:
-
Cyclin-dependent kinases
- CHOP:
-
CAAT/enhancer binding protein homologous protein
- ELISA:
-
Enzyme-linked immunoassay
- Flex-Hets:
-
Flexible heteroarotinoids
- HPLC/UV:
-
High-pressure liquid chromatography/ultraviolet
- NSCLC:
-
Non-small-cell lung cancer
- QD:
-
Once a day
- Rb:
-
Retinoblastoma protein
- PBPK:
-
Physiologically based pharmacokinetics
- PD:
-
Pharmacodynamics
- PVDF:
-
Polyvinylidene difluoride
References
Benbrook DM, Kamelle SA, Guruswamy SB, Lightfoot SA, Rutledge TL, Gould NS, et al. Flexible heteroarotinoids (Flex-Hets) exhibit improved therapeutic ratios as anti-cancer agents over retinoic acid receptor agonists. Investig New Drugs. 2005;23:417–28.
Liu T, Masamha CP, Chengedza S, Berlin KD, Lightfoot S, He F, et al. Development of flexible-heteroarotinoids for kidney cancer. Mol Cancer Ther. 2009;8(5):1227–38. https://doi.org/10.1158/1535-7163.Mct-08-1069.
Naylor MF, Thompson DM, Lightfoot SM, Thompson DM, Lightfoot S, Benbrook BM. Anti-cancer activities and interaction of imiquimod and Flex-Het, SHetA2, in melanoma and ovarian cancer. J Cancer Ther. 2013;4:7–19.
Benbrook DM, Lightfoot S, Ranger-Moore J, Liu T, Chengedza S, Berry WL, et al. Gene expression analysis of biological systems driving an organotypic model of endometrial carcinogenesis and chemoprevention. Gene Regul Syst Bio. 2008;2:21–42. https://doi.org/10.4137/grsb.s344.
Benbrook DM, Guruswamy S, Wang YH, Sun ZJ, Mohammed A, Zhang YT, et al. Chemoprevention of colon and small intestinal tumorigenesis in APCmin/+ mice by SHetA2 (NSC721689) without toxicity. Cancer Prev Res. 2013;6(9):908–16. https://doi.org/10.1158/1940-6207.Capr-13-0171.
Doppalapudi RS, Riccio ES, Davis Z, Menda S, Wang A, Du N, et al. Genotoxicity of the cancer chemopreventive drug candidates CP-31398, SHetA2, and phospho-ibuprofen. Mutat Res. 2012;746(1):78–88. https://doi.org/10.1016/j.mrgentox.2012.03.009.
Kabirov KK, Kapetanovic IM, Benbrook DM, Dinger N, Mankovskaya I, Zakharov A, et al. Oral toxicity and pharmacokinetic studies of SHetA2, a new chemopreventive agent, in rats and dogs. Drug Chem Toxicol. 2013;36(3):284–95. https://doi.org/10.3109/01480545.2012.710632.
Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009;69(16):6565–72. https://doi.org/10.1158/0008-5472.CAN-09-0913.
Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. https://doi.org/10.1186/1476-4598-6-24.
Gillett C, Smith P, Gregory W, Richards M, Millis R, Peters G, et al. Cyclin D1 and prognosis in human breast cancer. Int J Cancer. 1996;69(2):92–9. https://doi.org/10.1002/(sici)1097-0215(19960422)69:2%3c92::Aid-ijc4%3e3.0.Co;2-q.
Liu J, Liao Q, Zhang Y, Sun S, Zhong C, Liu X. Cyclin D1 G870A polymorphism and lung cancer risk: a meta-analysis. Tumour Biol. 2012;33(5):1467–76. https://doi.org/10.1007/s13277-012-0397-0.
Kang S, Kim JW, Park NH, Song YS, Kang SB, Lee HP. Cyclin D1 polymorphism and the risk of endometrial cancer. Gynecol Oncol. 2005;97(2):431–5. https://doi.org/10.1016/j.ygyno.2005.01.023.
Quan LL, Liu JY, Qu LX, La H, Wang HL, Chen XX, et al. Expression of Cyclin D1 gene in ovarian cancer and effect of silencing its expression on ovarian cancer cells based on the Oncomine database. Bioengineered. 2021;12(2):9290–300. https://doi.org/10.1080/21655979.2021.2000225.
Kennedy AL, Rai R, Isingizwe ZR, Zhao YD, Lightfoot SA, Benbrook DM. Complementary targeting of Rb phosphorylation and growth in cervical cancer cell cultures and a xenograft mouse model by SHetA2 and palbociclib. Cancers (Basel). 2020;12(5). https://doi.org/10.3390/cancers12051269.
Benbrook DM, Guruswamy S, Wang Y, Sun Z, Mohammed A, Zhang Y, et al. Chemoprevention of colon and small intestinal tumorigenesis in APC(min/+) mice by SHetA2 (NSC721689) without toxicity. Cancer Prev Res (Phila). 2013;6(9):908–16. https://doi.org/10.1158/1940-6207.CAPR-13-0171.
Benbrook DM. SHetA2 attack on mortalin and colleagues in cancer therapy and prevention. Front Cell Dev Biol. 2022;10:848682. https://doi.org/10.3389/fcell.2022.848682.
Rai R, Chandra V, Kennedy AL, Zuna RE, Benbrook DM. Distinct mechanism of cervical cancer cell death caused by the investigational new drug SHetA2. Front Oncol. 2022;12:958536. https://doi.org/10.3389/fonc.2022.958536.
Moxley KM, Chengedza S, Benbrook DM. Induction of death receptor ligand-mediated apoptosis in epithelial ovarian carcinoma: the search for sensitizing agents. Gynecol Oncol. 2009;115(3):438–42. https://doi.org/10.1016/j.ygyno.2009.09.007.
Sharma A, Benbrook DM, Woo S. Pharmacokinetics and interspecies scaling of a novel, orally-bioavailable anti-cancer drug, SHetA2. PLoS ONE. 2018;13(4):e0194046. https://doi.org/10.1371/journal.pone.0194046.
Sharma A, Li M, Thavathiru E, Ibrahim M, Garcia-Contreras L, Benbrook DM, et al. Physiologically based pharmacokinetic modeling and tissue distribution characteristics of SHetA2 in tumor-bearing mice. AAPS J. 2020;22(2):51. https://doi.org/10.1208/s12248-020-0421-z.
Benbrook DM, Nammalwar B, Long A, Matsumoto H, Singh A, Bunce RA, et al. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Investig New Drugs. 2014;32(3):412–23. https://doi.org/10.1007/s10637-013-0041-x.
Kaplan-Lefko PJ, Graves JD, Zoog SJ, Pan Y, Wall J, Branstetter DG, et al. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biol Ther. 2014;9(8):618–31. https://doi.org/10.4161/cbt.9.8.11264.
Sharma A, Thavathiru E, Benbrook DM, Woo S. Bioanalytical method development and validation of HPLCUV assay for the quantification of SHetA2 in mouse and human plasma: application to pharmacokinetics study. J Pharm Technol Drug Res. 2017;6(1):2. https://doi.org/10.7243/2050-120X-6-2.
Shi Q, Li Y, Li S, Jin L, Lai H, Wu Y, et al. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat Commun. 2020;11(1):5513. https://doi.org/10.1038/s41467-020-19349-w.
Zhang Y, Hua Y, Benbrook DM, Covey JM, Dai G, Liu Z, et al. High performance liquid chromatographic analysis and preclinical pharmacokinetics of the heteroarotinoid antitumor agent, SHetA2. Cancer Chemother Pharmacol. 2006;58(5):561–9. https://doi.org/10.1007/s00280-006-0211-z.
Diegmiller R, Salphati L, Alicke B, Wilson TR, Stout TJ, Hafner M. Growth-rate model predicts in vivo tumor response from in vitro data. CPT Pharmacometrics Syst Pharmacol. 2022;11(9):1183–93. https://doi.org/10.1002/psp4.12836.
D’Argenio DZ, Schumitzky A, X W. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource. 2009.
Cha SI, Ryerson CJ, Lee JS, Kukreja J, Barry SS, Jones KD, et al. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir Res. 2012;13(1):105. https://doi.org/10.1186/1465-9921-13-105.
Greystoke A, Dean E, Saunders MP, Cummings J, Hughes A, Ranson M, et al. Multi-level evidence that circulating CK18 is a biomarker of tumour burden in colorectal cancer. Br J Cancer. 2012;107(9):1518–24. https://doi.org/10.1038/bjc.2012.416.
Ramraj SK, Elayapillai SP, Pelikan RC, Zhao YD, Isingizwe ZR, Kennedy AL, et al. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int J Cancer. 2020;147(4):1086–97. https://doi.org/10.1002/ijc.32830.
Menz A, Weitbrecht T, Gorbokon N, Buscheck F, Luebke AM, Kluth M, et al. Diagnostic and prognostic impact of cytokeratin 18 expression in human tumors: a tissue microarray study on 11,952 tumors. Mol Med. 2021;27(1):16. https://doi.org/10.1186/s10020-021-00274-7.
Hempel F, Roderfeld M, Muntnich LJ, Albrecht J, Oruc Z, Arneth B, et al. Caspase-cleaved keratin 18 measurements identified ongoing liver injury after bariatric surgery. J Clin Med. 2021;10(6). https://doi.org/10.3390/jcm10061233.
Tchakarska G, Roussel M, Troussard X, Sola B. Cyclin D1 inhibits mitochondrial activity in B cells. Cancer Res. 2011;71(5):1690–9. https://doi.org/10.1158/0008-5472.CAN-10-2564.
Caillot M, Bourgeais J, Dakik H, Coste E, Mazure NM, Lelievre E, et al. Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis. 2020;9(7):68. https://doi.org/10.1038/s41389-020-00253-3.
Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–511. https://doi.org/10.1101/gad.12.22.3499.
Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(8):957–72. https://doi.org/10.1101/gad.11.8.957.
Chandra V, Rai R, Benbrook DM. Utility and mechanism of SHetA2 and paclitaxel for treatment of endometrial cancer. Cancers (Basel). 2021;13(10). https://doi.org/10.3390/cancers13102322.
Acknowledgements
The authors thank Drs. Lucila Garcia-Contreras and Mariam Ibrahim for their help with SHetA2 oral formulation.
Funding
This work was made possible in part by the NCI RAPID Program and NIH R01 (R01CA196200, R01CA200126, R01CA258546, and R01CA258546).
Author information
Authors and Affiliations
Contributions
Participated in research design: Woo, Sharma, and Benbrook.
Conducted experiments: Sharma, Woo, Benbrook, Chandra, and Rai.
Contributed new reagents or analytical tools: Sharma, Woo, and Benbrook.
Performed data analysis: Sharma, Liu, and Woo.
Wrote or contributed to the writing of the manuscript: Sharma, Liu, Benbrook, Chandra, Rai, and Woo.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, A., Liu, X., Chandra, V. et al. Pharmacodynamics of Cyclin D1 Degradation in Ovarian Cancer Xenografts with Repeated Oral SHetA2 Dosing. AAPS J 26, 5 (2024). https://doi.org/10.1208/s12248-023-00874-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-023-00874-7