
Abstract
Proteolysis targeting chimeras (PROTACs) technology has emerged as a novel therapeutic paradigm in recent years. PROTACs are heterobifunctional molecules that degrade target proteins by hijacking the ubiquitin–proteasome system. Currently, about 20–25% of all protein targets are being studied, and most works focus on their enzymatic functions. Unlike small molecules, PROTACs inhibit the whole biological function of the target protein by binding to the target protein and inducing subsequent proteasomal degradation. PROTACs compensate for limitations that transcription factors, nuclear proteins, and other scaffolding proteins are difficult to handle with traditional small-molecule inhibitors. Currently, PROTACs have successfully degraded diverse proteins, such as BTK, BRD4, AR, ER, STAT3, IRAK4, tau, etc. And ARV-110 and ARV-471 exhibited excellent efficacy in clinical II trials. However, what targets are appropriate for PROTAC technology to achieve better benefits than small-molecule inhibitors are not fully understood. And how to rationally design an efficient PROTACs and optimize it to be orally effective poses big challenges for researchers. In this review, we summarize the features of PROTAC technology, analyze the detail of general principles for designing efficient PROTACs, and discuss the typical application of PROTACs targeting different protein categories. In addition, we also introduce the progress of relevant clinical trial results of representative PROTACs and assess the challenges and limitations that PROTACs may face. Collectively, our studies provide references for further application of PROTACs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
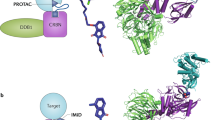
Proteolysis targeting chimeras (PROTACs) were first reported by Sakamoto et al. in 2001 [1]. PROTACs are heterobifunctional molecules that contain three components: the protein-of-interest (POI) binding moiety, a linker, and E3 ubiquitin ligase binding moiety (Fig. 1a) [2, 3]. PROTAC molecule can bind with E3 ligase and the target protein to form POI-PROTAC-E3 ligase ternary complex [4, 5]. Hijacking the ubiquitin-protease system (UPS) subsequently causes the target protein to be polyubiquitinated, which is then followed by the proteasomal degradation of protein. In eukaryotic cells, the UPS is the primary mechanism for maintaining protein homeostasis removing defective and damaged proteins [6, 7]. The UPS system degrades proteins by substrate-specific ubiquitination and recognition. Ubiquitination is a continuous three-step process that involves a cascade of three enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and substrate-specific ligases (E3) [8,9,10,11]. E1 activates the free ubiquitin (Ub) in an ATP-dependent process by forming a ubiquitin-E1 thioester bond, and then E1 subsequently transfers the activated Ub to E2 via trans-thioesterification [12]. Finally, the Ub-tagged E2 and target protein are recruited by E3 ligase to facilitate ubiquitin labeling on target proteins [13]. Such ubiquitination processes can be recycled to generate poly-ubiquitin chain tagged target protein, which directs the marked protein to 26S proteasome to undergo degradation [14]. PROTACs simultaneously recruit E3 ligase and POI, bringing POI and E3 Ligase in spatial proximity. PROTACs simulate specific recognition of substrate by E3 ligase and hijack the intracellular protein destruction mechanism to remove POIs from cells [15].

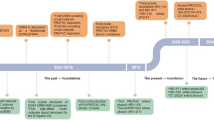
a The mechanism of PROTACs based on the UPS. UPS consists of specific enzymes (E1, E2 and E3) modifying proteins with ubiquitin and the proteasome degrading the ubiquitin-tagging proteins. PROTAC contains a POI ligand, an E3 ligand and a linker. The E3-PROTAC-POI ternary complex induces the polyubiquitination and proteasome-mediated degradation of POIs. b Milestones in the development of PROTAC technology
With the assistance of modern molecular biology methods and human genome information, target-based approaches have been applied to drug discovery [16]. Modern drug discovery focuses on finding small molecules with high binding affinity to target proteins, which modulate protein function by occupying the enzymatic activity site [17]. However, some proteins lack bindable sites or enzymatic activity sites, like transcription factors, RAS family proteins [18], scaffolding proteins and regulatory proteins [19], making them insensitive to traditional small molecule drugs. With the advent of PROTACs, it is possible to degrade “undruggable protein” without taking the presence of the active sites into account. PROTACs broaden the horizon for future drug discovery with unique advantages. A series of PROTACs drug using for degradation of androgen receptors (AR) [20] and estrogen receptors (ER) [21] have already entered in phase II clinical trials. By far, the PROTACs have proved effective in degrading a variety of proteins, such as representative AR, ER nuclear receptors, various kinases, transcription factors, and abnormal protein aggregates. In 2001, Craig Crews and his coworker reported the first heterobifunctional molecule, and it consisted of angiogenesis inhibitor ovalicin recruiting methionine aminopeptidase-2 (MetAP-2) and IкBα phosphopeptide recruiting E3 ubiquitin ligase β-TRCP [1]. The F-box protein β-transducin repeat-containing protein (β-TRCP) has been demonstrated to bind to IкBα, a negative regulator of NF-кB. Studies had shown that this PROTAC can effectively reduce MetAP-2 levels in vitro. This groundbreaking work persisted after extracellular studies showed promise. In 2003, PROTACs were applied to targeted the degradation of the ER and AR receptors [22]. IкBα phosphopeptide was connected to estradiol and dihydrotestosterone (DHT) respectively, making PROTACs are potent for degrading Erα and AR. The first cell-permeable PROTAC was developed by Schneekloth et al. in 2004 and contained the E3 ligase binding peptide ALAPYIP to recruit von Hippel-Lindau (VHL) which could induce the degradation of AR and FK506 binding protein 12 (FKBP12) [23, 24]. Additionally, a poly-D-arginine tag was incorporated into the carboxy terminus of the peptide sequence to confer cell permeability and resist nonspecific proteolysis [23]. These works are pioneering examples of PROTACs with in vivo validity. Although peptide based PROTACs have the advantages of high biocompatibility and low toxicity in vivo, it is impossible to ignore the limited cell permeability and synthetic problem caused by the large molecular weight. Along with the development of small molecule ligand for E3 ligase such as mouse double minute 2 (MDM2) [2], cell inhibitor of apoptosis protein (cIAP) [25], VHL, Cereblon (CRBN) [26], DCAF11 [27], DCAF15 [28], DCAF16 [29], KEAP1 [30], and RNF114 [31], PROTACs go into small molecularization rapidly [32]. The SARM-nutlin PROTAC was the first all-small molecule PROTAC that consisted of three parts: a specific substrate of AR, a ligand binding to E3 ligase MDM2, and a short soluble polyethylene glycol (PEG) linker joining these two moieties [2]. The in vitro results revealed that the SARM-nutlin PROTAC was capable of inducing AR proteasomal degradation and this is a significant improvement to the PROTACs.
Research on the CRBN E3 complex led to vital breakthroughs in 2010. CRBN, a substrate receptor subunit of CUL4-RBX1-DDB1-CRBN (CRL4CRBN) E3 ubiquitin ligase [33], has been identified as the direct target for thalidomide immunomodulatory drugs (IMiDs) [34]. These IMiDs bind CRBN ligase, which provided a binding site for multiple transcription factors with zinc finger domain such as IKZF1 and IKZF3, forming the basis of their anti-cancer effects [35,36,37,38]. Over the past few years, thalidomide and its derivatives have been successfully applied in PROTACs for target degradation of various protein classes for its efficient POI degradation and promising druggability [39]. ARV-825 (Fig. 2a) was the first PROTAC that incorporated pomalidomide and Bromodomain-containing protein 4 (BRD4) inhibitor OTX015 (Fig. 2b) [26]. This PROTAC could degrade BRD4 protein in Burkitt’s lymphoma (BL) cells effectively.

The representative PROTACs targeting diverse proteins
In 2012, Crews and Ciulli team identified the first small molecule ligand for VHL with micromolar dissociation constant [40]. To improve the affinity and lipophilicity of the first generation VHL ligand, Galdeano et al. discovered the second-generation ligands for VHL with the key hydroxyproline (Hyp) group as core recognition motif, VH032 was identified as the most potent ligand for VHL with nanomolar affinity. Identification of the novel VHL E3 ligands marked a milestone in PORTACs technology (Fig. 1b). These newly discovered VHL ligands opened up new opportunities to design VHL-recruiting PROTACs. The crystal structure of VH032 coupled with VHL was solved by Zengerle et al., providing the structural details for constructing the first VHL-recruiting PROTACs, MZ1 (Fig. 2b). Compound MZ1 potently and selectively removed BRD4 over BRD2 and BRD3. Gadd et al. later uncovered the crystal structure of the BRD4-MZ1-VHL complex, revealing MZ1 was “sandwiched” between BRD4 and E3 ligase and the new contact of BRD4-VHL is generated by MZ1-induced cooperative recognition, they found that PROTAC-induced electrostatic surface interactions between the target protein and E3 ligase are important for stabilizing the ternary complex [5]. The aforementioned groundbreaking research lay the foundation for later VHL-based PROTACs studies. To date, VHL-based PROTACs have successfully applied to the degradation of various disease-associated proteins, such as BCR-ABL [41], ALK (anaplastic lymphoma kinase) [42], and FAK (Focal adhesion kinase) [43].
PROTAC technology has been applied to the alternative treatment of various diseases. More and more targets have been confirmed to be “PROTACable” genome, and some PROTAC molecules have achieved clinical benefits. For instance, PROTAC targets that have entered the clinical trials include AR [44], ER [45], IRAK4, STAT3, BTK, BRD9, BCR-Xl [46], etc. There are at least 20 PROTACs in the clinical trials by the end of 2022 (Table 1), with more expected to follow. Among them, ARV-110 and ARV-471 from Arvinas are the most advanced PROTAC drugs in clinical research and have entered the clinical phase II study. Arvinas, C4 therapeutics, Kymera Therapeutics, and Captor Therapeutics are pioneering pharmaceutical companies in the field of PROTACs, which promote the clinical translation of PROTACs. Arvinas is dedicated to advancing its representative ARV-110 and ARV-471 into the market. Early clinical data of ARV-110 and ARV-471 demonstrated ideal safety, effective exposure and meaningful clinical efficacy for patients, proving the therapeutic feasibility of the approach. The research data indicate that ARV-110 is safe as an oral bioavailable degradation agent. Phase I trials have shown that ARV-110 reduced prostate-specific antigen (PSA) levels by more than 50% in 40% of patients with mCRPC in a population with a specific gene mutation. In addition, in the initial clinical study, the biopsy data of one patient showed a 70% ~ 90% decrease in AR. A Phase I clinical study of ER+ and HER2- breast cancer patients who had received an average of five continuous treatments showed that ARV-471 could significantly reduce the expression level of ER in tumor tissue of patients, reducing the ER level by 62% on average, up to 90% at most. In addition, the phase I clinical data of ARV-471 also showed that a high level of ER degradation (89%) was observed at all dose levels of 30-700 mg, and it was well tolerated. ARV-471 exhibited certain degradation effects on both wild type ER and ER mutants. ARV-471 is undergoing a phase II dose expansion clinical trial to evaluate the efficacy of ARV-471 in the treatment of ER + /HER2- patients with locally advanced or metastatic breast cancer.
The clinical trials results of the most advanced PROTAC drugs ARV-110 and ARV-471 were considered as weathervane for the development of PROTACs field. As a new drug paradigm, there are still considerable questions about the clinical transformation of PROTAC drugs. How PROTACs ensure therapeutic effects without meeting the Lipinski’s Rule-of-Five, and how to study, elucidate and minimize the complex off-target effects and side effects that might be caused by the heterobifunctional molecule form of PROTAC drug are unavoidable challenges in clinical trials. As the first two PROTAC drugs to enter phase II clinical trials, ARV-471 and ARV-110 have shown strong clinical performance in the early trials. If the results of the subsequent phase II and phase III clinical trials can achieve the expected goals, PROTAC may enter clinical use in the near future, which will bring a historic breakthrough in the research and development of targeted protein degradation drugs.
In this review, we list the targets that PROTACs have been applied for the first time in the last 5 years (Table 2). We briefly recount the advantages of PROTAC technology compared to other technologies. We then outline the recent progress of PROTACs for targeting diverse related proteins, especially those in clinical trials. The essential considerations for designing a new PROTAC molecule were also discussed and recommended. We try to provide a valuable reference for people in the related fields to design potent PROTACs.
The advantages of PROTACs
Diverse novel therapeutic strategies (e.g., small-molecule inhibitors, monoclonal antibodies and RNA interference (RNAi)), have become a well-established paradigm for drug discovery. The activity of small molecule-inhibitors usually depend on occupying the active pocket of the target, competing with endogenous ligands to inhibit the function of the target protein or enzyme. Long-term clinical application of small-molecule inhibitors faces the challenges of drug resistance and off-target effects [71]. Monoclonal antibody drugs regulate cellular responses by blocking extracellular protein–protein or protein–ligand interactions. The major advantages of monoclonal antibody drugs stem from their high affinity to the target protein, whereas, the deficiencies of the monoclonal antibody drugs involve their poor cell permeability, oral unavailability, and high cost. RNA interference is used to induce gene silencing by knocking down mRNA, due to the catalytic nature of RNAi, which is capable of degrading multiple equivalents of mRNA transcripts. However, the off-target effects, poor oral bioavailability, and unsatisfactory tissue penetration made drug delivery challenging to study [72]. As a promising therapeutic paradigm, PROTACs have unique advantages over small-molecule inhibitors, monoclonal antibodies and other therapeutic strategies (Table 3) [73]. When the target protein is degraded by the proteasome, PROTACs can disassociate from the complex and continue to exert the degradation effect (called “event-driven” mechanism), allowing low exposures to be efficacious. Additionally, PROTACs completely abolish the target’s functionalities, and even ligands with lower POI/E3 affinity can be employed for target degradation. In this section, we will briefly compare PROTACs with other therapeutic strategies.
Degrading “undruggable” proteins
Although FDA has approved nearly 400 drugs targeting human proteins, there are about 3000 disease-related proteins which are far more than we can handle [74]. However, most of them do not have appropriate therapeutic drugs, because the lack of so-called druggable deep grooves and active pockets to occupy for small molecules [75], such as scaffolding proteins, transcriptional factors, and RAt Sarcoma (RAS) proteins, are deemed “undruggable” proteins for a long time [76]. Therefore, it is difficult to regulate these undruggable proteins by small molecules that can only rely on continuous occupancy of the binding pocket of the target protein to exert their pharmacological activity (called “occupancy-driven” mechanism) [77,78,79]. Fortunately, PROTAC-induced protein degradation has the potential to address these issues. Unlike traditional small-molecule inhibitors, PROTACs do not require high affinity for ligands and long lasting occupancy, and even low affinity ligands can induce efficient degradation of target proteins [80]. For these challenging undruggable targets, PROTACs can bind to targeted proteins without the existence of active pockets, thus leading to proteasome mediated degradation and complete inhibition of the biological functions of target proteins [81]. For example, RAS proteins are the most frequently mutated oncoproteins in the lung, colorectal, and pancreatic cancers [82]. RAS proteins comprise three isoforms, KRAS, NRAS and HRAS [83]. Among them, KRAS mutation is a deadly driver of cancers. Due to the lack of a well-defined binding pocket, KRAS has been viewed as an undruggable protein for many years. However, FDA fast-tracked the designation of two covalent inhibitors, AMGEN’s sotorasib (AMG 510) and Mirati Therapeutics’ adagrasib (MRTX849) (Fig. 2c), both of which demonstrated potent inhibition of KRASG12C in clinical trials. Unfortunately, long term and prolonged usage inevitably results a severe decrease in affinity and acquired drug resistance [84]. In 2020, Crews et al. designed and synthesized a series of KRASG12C PROTAC via tethering MRTX849 with VHL ligands. After a degradation activity screen, they identified one of the most potent PROTAC that rapidly induced the degradation of KRASG12C protein (DC50 = 0.59 μM, NCI-H2030 cells) and also exhibited degradation activity in other cells [85].
Improving selectivity and specificity
The main objective of medicinal chemistry researchers is the discovery of molecules with high selectivity to minimize adverse effects and toxicity brought on by off-target effects. However, it is difficult to achieve because of limited differences between proteins in the same family. Subtle differences in amino acid residues between the same family proteins are insufficient to provide adequate resolution for small molecular inhibitors. The unique mechanism of PROTAC endows it with the characteristics of dual selective substrate recognition. That is, in addition to the substrate selectivity of target protein ligands, the formation of stable POI-PROTAC-E3 ternary complexes before degradation also requires appropriate protein–protein interaction (PPI) between E3 ligase and target protein. Thus, selective recognition of target proteins from the whole protein level by E3 ligase improves the selectivity and specificity of PROTACs. A typical example of kinase isoform selectivity is targeting serum and glucocorticoid-induced protein kinase (SGK) and the SGK family contains three isoforms, SGK-1, SGK-2 and SGK-3. It was reported that mutant phosphoinositide 3-kinase (PI3K) can induce tumorigenesis through SGK3-dependent mechanism [86]. Some pieces of evidence suggest that various ATP-competitive inhibitors lack selectivity for all SGK isoforms, as they share similar affinity for different isoforms [87, 88]. However, the similar catalytic domain of the same family members prevented researchers from developing isoform-specific inhibitors. [89]. To address this problem, the highly specific SGK3-PROTAC1 (Fig. 2d) was developed. This PROTAC was designed by Tovell’s group based on the non-SGK3 selective inhibitor 308-R, to degrade SGK3 specifically [90]. At a low micromolar concentration of SGK3-PROTAC1, intracellular SGK3 levels can be significantly reduced without affecting SGK1 and SGK2. It could be assumed that the selectivity and specificity of SGK3-PROTAC1 derives from the selective recognition of SGK3 by VHL during the formation of ternary complexes induced by SGK3-PROTAC1.
Catalytic mode of action (MOA)
Traditional small-molecule inhibitors act in a dose-dependent manner, to achieve clinical effect by maximizing drug-receptor occupancy. Excessive drug concentrations lead to undesirable side effects and off-target effects [91]. PROTACs can initiate the degradation of target protein catalytic and escape from proteasome [92]. Theoretically, PROTACs can be delivered at lower doses, for longer dosing intervals, and with lower toxicity than small molecule inhibitors since their low concentration is sufficient to degrade proteins and is not constrained by equilibrium occupancy. Because of their catalytic nature, low doses of PROTACs may reduce the probability of off-target effects to occur [77].
Eliminate the accumulation of drug targets
The binding of small-molecule inhibitors to target proteins cansues increased protein accumulation even in a relatively short amount of time [93]. It can be attributed to two reasons: 1). drug binding to target proteins can stabilize the protein structure, thereby extending their half-life, and 2). long-term inhibition will cause upregulation of its compensatory expressio. In general, the accumulation of target protein can be detrimental to the efficacy of drugs. Therefore, for these proteins that are insensitive to inhibitors, it’s extremely suitable to take PROTAC-mediated protein degradation. For example, BRD4, as one of the important bromodomain and extraterminal domain (BET) family members [94]. Researchers demonstrated that targeting BRD4 is an effective means of suppressing MYC-driven cancers [95]. However, the small molecule BRD4 inhibitor, JQ1 (Fig. 2e) and OTX015 resulted in robust protein accumulation, and high concentration of inhibitor is required to suppress downstream c-MYC. In 2015, Lu et al. designed a potent BRD4 PROTAC (ARV-825) by hijacking CRBN E3 ligase, which induced a rapid and sustained degradation of BRD4 protein in all BL cell lines [26]. This highlights the advantages of PROTAC over small-molecule inhibitors.
Others
In addition to the points mentioned above, PROTACs also have other advantages. The occurrence of acquired drug resistance is often closely related to point mutations that can decrease the affinity of the inhibitor to the target protein. PROTACs are able to overcome drug resistance issues via the complete elimination of the target mechanism [96]. Besides, the event-driven model of PROTACs do not require high drug exposure to reduce the risk of off-target effects [97]. Unlike other DNA-level protein knockout techniques, PROTACs enable for the rapid degradation of target proteins in vivo at the post-translational level. In the field of targeted protein degradation (TPD), besides UPS based PROTACs, lysosome-targeting chimeras (LYTACs), autophagy-targeting chimeras (AUTACs), and antibody-based PROTACs (AbTACs) degrade target proteins through lysomal. PROTACs cannot degrade extracellular and membrane proteins. Therefore, lysosome induced protein degradation can compensate for the lack of PROTACs. LYTACs were first proposed by Banik et al. and consist of a ligand binds lysosome-targeting receptors (LTRs) and a ligand binds extracellular or membrane protein [98]. Currently, only poly-serine-O-mannose-6-phosphonate (M6Pn) and N-acetyl galactosamine (Tri-GalNAC) were LTRs ligands available [99]. The LYTACs have been used to successfully degrade apolipoprotein E4, epidermal growth factor receptor (EGFR), programmed death protein ligand 1 (PD-L1), and CD71 [99]. However, due to the large molecular weight, poor cell permeability, and the possible emergence of immune response in vivo, further studies are needed [100]. In 2019, Takahashi et al. developed AUTACs based on the autophagic process for the degradation of endogenous proteins [101]. AUTACs are a bifunctional molecule with a linker joints POI ligand and autophagic recruitment tag. However, currently published AUTACs are inefficient due to the lack of efficient autophagy pathway recruiters. The autophagic process is extremely complex and may have an impact on natural autophagy, the mechanism of action of AUTACs remain unclear, so it need to be studied in depth [100]. AbTACs utilize bispecific antibodies, with one arm targeting POIs and the other targeting RNF43 E3 ligases [102]. AbTACs can induce POIs internalization and subsequent lysosomal degradation, but the the exact degradation mechanism remains to be confirmed.
The typical application of PROTACs for targeting diverse proteins
In theory, PROTACs can degrade almost all intracellular proteins if there is an appropriate small molecule that specifically binds with those POI, but not all degraders outperform small-molecule inhibitors. Here, we summarize some typical PROTAC molecules that have demonstrated obvious inhibition activities, several of which have advanced to the clinical trial stage.
PROTACs for targeting protein kinases
The human genome encodes over 500 protein kinases [103], making it the largest protein family. Currently, traditional small-molecule inhibitors are the primary treatment options for protein kinases related diseases. A majority of kinase inhibitors focused on the inhibition of receptor tyrosine kinase (RTK) [104]. However, the emergence of drug resistance impaired the clinical benefit, so it is urgent to apply novel therapeutic strategy to overcome this challenge.
In 2013, Crews’s group reported the earliest kinase PROTACs, which was used to target PI3K to block the human epidermal growth factor receptor 3 (ErbB3)–PI3K-Akt (protein kinase B) signal pathway [105]. This PROTAC is composed of two heterospecific peptide sequences recruiting POI and E3 ligase. An ErbB3-derived sequence that can bind to PI3K after it has been phosphorylated. Another sequence derived from hypoxia-inducible factor-1α (HIF1α) can be identified by VHL [105]. The two moieties were conjugated by a PEG linker, and a cell-penetrating sequence was incorporated to improve cell permeability. However, this PROTAC only display moderate potency because of poor permeability and unstable linker [106].
FAK, a tyrosine kinase, regulates many aspects of tumor progression (e.g., invasion, metastasis, and angiogenesis). The leading FAK kinase inhibitor defactinib, failed in clinical trials to treat malignant pleural mesothelioma stem cancer for the lack of efficacy. FAK also has a scaffolding role other than kinase, but kinase inhibitors cannot inhibit kinase-independent function. Cromm et al. designed PROTAC-3 (Fig. 2f) which could effectively induce the degradation of FAK with the IC50 of 6.5 nM [43]. PROTAC-3 is a bifunctional molecule consisting of defactinib and VHL ligand. It effectively inhibits FAK kinase-independent signaling and kinase-dependent signaling by efficient induction of degradation.
Bruton’s tyrosine kinase (BTK) is a member of the non-receptor cytoplasmic tyrosine kinase of the TEC family and a key regulator of the B cell receptor (BCR) signaling pathway, which plays a critical role in the life activities of B-cells like proliferation, survival, and differentiation [107, 108]. BTK is widely expressed in B cell neoplasms, and the clinical interventions are generally performed by inhibiting the kinase activity of BTK [109]. In 2013, FDA approved the first-in-class covalent inhibitor ibrutinib for the treatment of several B-cell malignancies. Ibrutinib binds covalently to Cysteine481 (C481) of BTK with IC50 of 0.5 nM [110, 111]. However, it has been revealed that a cysteine to serine mutation at position 481 of BTK (C481S) is what causes acquired resistance to ibrutinib [112]. So, induction of BTK protein degradation using PROTAC technology has emerged as a promising alternative approach. To date, four BTK degraders have entered clinical trials. They are NX-2127 (NCT04830137) and, NX-5948 (NCT05131022) from Nurix Therapeutics, HSK-29116 (NCT04861779) and BGB-16673 (NCT05006716) small molecule drugs from Haisco and BeiGene respectively. NX-2127 is an oral dual-target small molecule that possesses the activity of BTK degrader and IMiD neosubstrates degrader. A phase I clinical trial of NX-2127 is currently underway for the treatment of relapsed or refractory B-cell malignancies. Preclinical data have demonstrated that NX-2127 could potently induce the degradation of both ibrutinib-sensitive BTKWT (wild type) and ibrutinib-resistant BTKC481S in multiple cancer cell lines and human peripheral blood mononuclear cells (PBMCs) with the DC50 < 5 nM. Additionally, NX-2127 inhibited cell proliferation of BTKC481S in TMD8 cells more effectively than ibrutinib. NX-2127 exhibits immunomodulatory activity through comprised of thalidomide IMiD [113]. Krönke et al. revealed that lenalidomide causes selective ubiquitination and degradation of CRBN neosubstrates Aiolos (IKZF3) and Ikaros (IKZF1) [35]. Lazarian et al. have shown that the overexpression of IKZF3 is a driver of BTK inhibitor resistance in chronic lymphocytic leukemia (CLL) [114]. Therefore, NX-2127 combines BTK degradation with IKZF degradation is expected to enhance its anti-tumor activity. NX-5948 is another BTK degrader designed by Nurix Therapeutics. Unlike NX-2127, NX-5948 lacks immunomodulatory activity and has the ability to cross the blood brain barrier (BBB) in animal models. NX-5948 displayed similar performance that preclinical data have shown that NX-5948 induced the degradation of BTK (50% degradation efficiency at < 1 n M) in lymphoma cell lines and PBMCs [115].
PROTACs for targeting nuclear receptors
Nuclear receptors (NRs) belong to the family of transcription factors. Unlike other traditional transcription factors, its main function is to convert external the signal to transcriptional output [21]. A typical NR includes three domains: two structural domains that bind DNA and ligand respectively, and an unstructured N-terminal regulatory domain that is highly variable in terms of both sequence and size [116]. Ligand agonist binding confers a conformational change that results in exposure of the nuclear localization signal (NLS), which allows NR to translocate to the nucleus and bind the response elements. Small-molecule inhibitors that bind to ligand binding domain have been designed to activate or block the signal transduction function of nuclear receptors. However, small-molecule inhibitors have several disadvantages. For instance, our understanding of the concept of pure inhibitors is not clear, as continual AR antagonists prove to be agonists when the AR gene is overexpressed or mutated [117, 118]. In addition, some ligands for orphan NRs have not yet been identified, thus making it more complicated to target NRs to treat diseases. The advent of PROTAC technology has made it possible to target a wider range of NRs. NRs such as AR and ER participate in various important physiological progress in the body, and are closely related to prostate cancer and breast cancer. Therefore, a series of PROTACs targeting ER or AR have been developed.
AR signaling is critical in the development and maintenance of the normal function of prostate. AR not only plays a key role in the maintenance of musculoskeletal and male sex-related functions but also in the progression of prostate cancer [119]. Inhibition of AR function with AR antagonists such as enzalutamide and apalutamide is a common strategy in the treatment of prostate cancer [120]. Unfortunately, castration-resistant eventually occurs in patients with antiandrogen therapy [121]. PROTACs emerged as an alternative potential therapeutic approach to compensate for the shortcomings of AR inhibitors. Salami et al. synthesized a potent AR PROTAC ARCC-4 (Fig. 2g), which comprised of enzalutamide derivative and E3 ligand recruiting VHL. Compared with its parent inhibitor enzalutamide, ARCC-4 can effectively degrade AR and AR mutants caused by long-term use of clinical inhibitors, without leading to the presence of drug resistance [118]. It is well-known that ARV-110 (Fig. 2g) is the first AR-targeting PROTAC in clinical trial. The latest clinical trial data indicated that ARV-110 has an acceptable safety profile. The maximum tolerated dose (MTD) has not been established and the determination of the recommended phase 2 dose (RP2D) continues. In addition, ARV-110 has demonstrated antitumor activity in patients with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamid and/or abiraterone administration [44]. Recently, Wang’s group reporteded two highly potent and orally bioavailable AR PROTACs, ARD-2128 and ARD-2585 (Fig. 2g). ARD-2128 features an optimized AR antagonist linked to thalidomide via a rigid linker, achieving 67% oral bioavailability and better antitumor activity than enzalutamide in mice [122]. ARD-2585 incorporates the same CRBN ligand as ARD-2128 and achieves DC50 values of ≤ 0.1 nM in the VCaP cell line and 51% of oral bioavailability in mice [123].
Breast cancer is a malignant tumor in which the breast tissue becomes cancerous and the patient is usually a female population. Breast cancer can be subdivided into three types based on the status of the tumor receptor: estrogen receptor-positive (ER +), human epidermal growth factor receptor 2 positive (HER2+), and triple-negative subtypes (ER-, PR-,HER2-) [124]. Among these, ER + breast cancer is most commonly diagnosed [125]. ER is a member of nuclear receptor family, and ERα and ERβ regulate the gene expression of estrogen. Nevertheless, ERα has been verified to be primarily responsible for converting the estrogen signaling in the female reproductive system and mammary tissue [126, 127]. Most selective estrogen receptor degraders (SERD) were designed to target ERα to treat ER + breast cancer [128]. SERD is a class of small molecules that bind with ERα and subsequently degraded by proteasome. Fulvestrant is the only SERD that has been approved and administered by monthly intramuscular injection for the treatment of postmenopausal women with breast cancer [129,130,131,132]. To address the shortcomings of poor oral bioavailability of fulvestrant [131], a series of SERD molecules have been developed. However, SERD molecules could not degrade ER completely, and long-term use can lead to drug resistance. PROTAC technology offers an alternative treatment option [133,134,135,136]. Arvinas developed an ER-targeting PROTAC, ARV-471 (Fig. 2h), which was approved by the FDA to enter clinical trial for the treatment of patients with locally advanced or metastatic ER-positive/HER2-negative breast cancer [137]. Results from the mid-stage trials revealed that ARV-471 markedly reduced the expression level of ER in tumor tissues by an average of 62% and up to 90%. In addition, ARV-471 may degrade wild-type and clinically relevant ERα mutants (Y537S and D538G) with DC50 values of about 2 nM in multiple ER-positive breast cancer cell lines [138].
PROTACs for targeting transcriptional factors
Transcriptional factors (TFs) are a class of proteins binding to DNA specific sequence to regulate gene transcription process [139]. TFs play a key role in multiple cell functions such as proliferation, differentiation and death. With the exception of nuclear receptors, direct targeting of transcription factors is particularly challenging for small-molecule inhibitors, thus rendering them considered as “undruggable protein” for decades [140,141,142]. Therefore, inducing protein degradation emerges as a potential modality for TFs [143]. Based on the specific structure of DNA-binding domains, TFs could be classified into tens of families [139]. It is worth noting that the C2H2 zincfinger, homeodomain and helix-loop-helix families account for over 80% of the total number of transcription factors [140].
Signal transducer and activator of transcription 3 (STAT3) is a key nuclear transcription factor that is phosphorylated on tyrosine 705 and integrates cytokine and growth factor signaling to regulate an array of cellular process [144, 145]. STAT family comprises seven proteins, among which STAT3 has been shown to be overexpressed in many types of cancer, especially breast cancer. Targeting STAT3 is a prevalent strategy for the treatment of various cancers, inflammatory and autoimmune disorders [146]. The phosphorylation of STAT3 at Tyr705 can trigger its dimerization and is closely related to the transcriptional regulation of target genes [147]. STAT3 dimerization relies on the interaction between the Src-homology 2(SH2) domain of two monomers. Based on this mechanism, researchers are keen to find small-molecule inhibitors that act on the SH2 domain to block STAT3 dimerization and transcriptional activity. However, several inhibitors acting on the STAT3 SH2 domain have demonstrated limited clinical value because of the existence of structural homology between STAT family members, making obstacles for specific STAT3 inhibitors development [148, 149]. Another problem stands out that the single STAT3 protein is still transcriptionally active [150], so developing inhibitors of the STAT3 SH2 domain is not a feasible approach to fully suppress the activity of STAT3. PROTACs have a promising prospect as a novel therapeutic for the degradation of targeted protein [151]. Here we introduced a specific and potent STAT3 PROTACs. Bai et al. reported the first STAT3 PROTAC SD-36 (Fig. 2i) that not only could effectively and specifically degraded STAT3 and has the antiproliferative activity of leukemia and lymphoma cell lines [147]. SD-36 consists of a selective STAT3 inhibitor SI-109 and lenalidomide and is a typical successful example of how PROTACs can be applied to target challenging proteins such as transcription factors.
Design and development of PROTACs
The degradation activity of PROTACs not only depends on the affinity of both ends to their respective target, but also relies on the formation of ternary complex that can form stable PPI. Currently, the construction of PROTACs largely relies on empirical analyses and structure–activity relationship (SAR) studies. However, synthetic difficulty presents significant limitations for rapid synthesis of a wealth of PROTAC compound libraries. By analyzing and summarizing published PROTACs structures, we will provide conventional strategies in PROTAC design to accelerate PROTACs discovery. In addition, we have listed some recently reported PROTACs that recruit traditional E3 ligases with corresponding degradation activity (Table 4).
E3 ligase and its ligand
Of the more than 600 ligases identified, only a few with small molecule ligands have been used for PROTAC targeting [163]. We list the commonly used E3 ligases and their ligands (Fig. 3). Cao et al. summarized and analyzed the structures of highly active PROTACs published over 20 years, and they found that CRBN, VHL, and cIAP ligands were used most frequently, of which CRBN accounted for 60.1%, VHL for 30.1%, and cIAP for 5.5% [164]. The main reason is that CRBN is widely expressed in tissues with high abundance and CRBN-based PROTACs have better degradation efficiency. In addition, CRBN ligands have better drug-like properties compared to the VHL ligand. PROTACs recruiting MDM2 and cIAP usually have high molecular weight and poor tissue permeability, indicating that the oral bioavailability may be a potential concern. Some other E3 ligases such as DCAF11 [27], DCAF15 [28], DCAF16 [29], KEAP1 [30], and RNF114 [31] etc., are less used for the following reasons: their ligands are derived from natural products with poor affinity, and are difficult to synthesize, and most of these E3 ligases are recruited by irreversible PROTACs, which have poor degradation activity and some potential toxicity. Of note, different recruited E3 ligases have been shown to induce different degrees of protein degradation [165]. The major reasons are as follow: different expression levels of E3 ligases in different cells may contribute to the different degradation efficiency. And some proteins have different degrees of selectivity for different E3 ligases. Therefore, in the process of designing the PROTACs, ligands targeting CRBN or VHL should be preferentially chosen, as these two E3 ligases have the widest range of applications. As an illustrative example, both ARV-110 and ARV-471 selected CRBN ligase as the E3 ligand. Here, we review the traditional E3 ligases and their ligands used in PROTAC design.

Representative small molecule ligands of E3 ligases used for PROTACs. Blue dots indicate the appropriate linker attachment site
Linker design strategies of PROTACs
Type of linkers
Maple’s group built a database containing more than 400 published PROTACs to find a general principle that has been applied in PROTAC [166]. A summary of the linker structures in the database (Table 5) reveals that the frequently used linkers in PROTACs design are PEG and (un)saturated alkane chains with varying lengths up to now [81]. Due to the facile chemical synthesis feature, alkyl linkers are often used for the synthesis of PROTAC molecules to identify the optimal linker length. However, introduction of alkyl linkers might reduce the cell permeability of PROTACs due to their high hydrophobicity. Alkyl chains containing heteroatoms (oxygen atoms or nitrogen atoms) have improved hydrophilicity over alkyl chains alone. In addition, incorporating PEG chain can enhance the solubility and uptake of PROTACs by cells. More than half of the published PROTACs structure contained alkyl and PEG motifs. Alkyl, PEG, and glycol chains are incorporated into the PROTACs to increase the flexibility. However, their introduction can affect the pharmacokinetics (PK) properties of PROTACs. In recent years, linear linkers are gradually replaced by rigid linkers, such as alkynes and saturated heterocycles (piperazine and piperidine). The incorporation of aromatic rings or alkyne chains imparts some rigidity and promotes stable ternary complex formation. It also facilitates the solubility and cell permeability of PROTAC [167]. Thus, making it orally bioavailable and clinically effective, such as ARV-110, ARV-471, and BTK PROTACs [168]. Click chemistry is commonly applied to construct PROTAC molecules in vivo, so the triazole group is chosen to link POI and E3 ligase ligand. However, it’s difficult to metabolize triazole in vivo, therefore, the introduction of triazole may help to enhance metabolic stability and prolong the durability of PROTACs [169]. The discovery process of ARV-110 is an example of great reference value in the development of PROTACs. In earlier study, AR antagonists and VH032 based ARCC-4 was discovered with efficient degradation activity. Given the lack of oral bioavailability of ARCC-4, the VHL ligand was replaced with a CRBN ligand and the linker was optimized accordingly to improve bioavailability. Then the warhead was further modified to obtain two PROTACs with superior in vivo and in vitro activity to ARCC-4, but both compounds had a high clearance rate. The activity and bioavailability were improved after switching to a rigid linker, and further optimization of the dose-escalation exposure finally led to the discovery of ARV-110 [170].
Length of linkers
The length of the linker also has a significant effect on the degradation activity of PROTAC [171]. Recently, Bemis et al. presented a model of linear linker length SARs studies which suggested that degraders with longer linkers are more likely to succeed in the preliminary design of PROTAC. Once the efficient PROTAC is identified, the length of the linker will be shortened step by step to identify the optimal linker length [172]. When the linker is too short, it is difficult for the 2 ligands to bind to their respective targets simultaneously because of steric hinderance effects, thus preventing the formation of ternary complex [171]. However, in case that the linker is excessively long, it will hinder the PPI, resulting in the failure of target protein ubiquitination [73]. Additionally, longer linkers have larger molecular weights, which make PROTACs less likely to cross cell membrane. Hence, the incorporation of rigid linkers, such as alkyne, piperazine and piperidine into linkers can effectively improve the pharmacokinetic profile and efficacy of PROTACs [123].
Choosing an appropriate linker attachment site
PROTACs have three necessary components, a warhead, an E3 ligase ligand, and a linker connecting them. Once the warhead and the E3 ligase ligands have been fixed, the selection and optimization of composition, length and attachment sites are essential factors to construct PROTACs. PROTAC molecules with suitable linkers have a significant impact on the activity and selectivity for POI degradation [54]. In general, its preferable to access the linker from the solvent-exposed position of the target protein, where it does not affect the binding affinity of its ligand. In most cases, researchers have identified the appropriate linkage position through co-crystal structure and SAR study. For warheads and E3 ligase ligands, the choice of linker attachment site requires to be considered without affecting the original affinity to its receptor. Most importantly, do not derive linkers from the critical active group of the ligand.
Photo-control linkers
Although the catalytic MOA offers fewer side effects for PROTACs over traditional small-molecule inhibitors. However, the unique catalytic feature of PROTACs also rendered inevitably adverse effects resulting from robust degradation in both normal and cancer cells. To overcome this problem, introduction of photocontrol linkers enabled the degradation of POI in a spatiotemporal manner [173, 174]. Incorporating photo-switchable azobenzene group into linker is a reversible way to control degradation activity with light. PROTAC will switch reversibly between “cis” and “trans” at different given wavelengths of irradiation, resulting in the conformation of corresponding inactive or active PROTAC. After irradiation at a certain wavelength, the inactive PROTAC will be converted to the active isoform. Only active PROTAC has the capability to form a stable ternary complex with target proteins, triggering subsequent ubiquitination and proteasome degradation. In addition, degradation also can be interrupted by switching back to the inactive form under certain wavelengths of light. Pfaff et al. have designed a bistable photoPROTACs using tetrafluoro azobenzene group to optically control the degradation of BET [175]. The photoPROTACs connected the VHL ligand and JQ1 together via a photoswitchable tetrafluoro azobenzene linker (Fig. 4a).Xue’s group developed a different method to optically control the degradation of target proteins through incorporation of a photocaging group to either warhead or E3 ligand to hinder the formation of a stable POI-PROTAC-E3 ligase complex [176]. Under the specified wavelength light irradiation, once the photocaging moiety is released, it can be reverted to the active conformation. In 2019, a photocaging strategy was first used in PROTAC. A photocaging moiety was conjugated to the warhead side in dBET1, creating pcPROTAC1 [176] (Fig. 4b). Under 365 nm wavelength irradiation, the 4,5-dimethoxy-2-nitrobenzyl (DMNB) was released, resulting in the formation of active dBET1. Additionally, photocaged PROTAC-3 introduced the photocaging groups into CRBN ligand side to hinder CRBN recruitment, Under 365 nm wavelength irradiation, the uncaged pcPROTAC-3 induced BTK degradation [177] (Fig. 4c). These studies proved the probability of introducing the photocaging groups to either side. However, conjugating to POI ligand would be better than E3 ligand, as it excludes the effect of the inhibitory activity that the target protein has in the absence of light exposure.

a photoPROTAC-1 comprising BRD-targeting JQ1 and a VHL ligand linked via a photoswitchable tetrafluoro azobenzene moiety. Light irradiation converts the inactive cis-photoPROTAC-1 into its active trans isomer, and vice versa, b irradiation of DMNB-protected PROTAC at 365 nm releases potent BET degrader dBET1, c irradiation of NVOC-protected PROTAC at 365 nm releases potent BTK degrader
Clickable linkers
Click reactions are valid bioorthogonal tools for the self-assembly of PROTACs in cells, and improve the poor permeability of PROTACs. Astex Pharmaceuticals has developed smaller precursor-based intracellular CLIPTACs (In-cell click-formed proteolysis targeting chimeras, CLIPTACs) system [178]. The small molecule precursors in this system have smaller molecular weights, such as the tetrazine (Tz)-tagged thalidomide derivative (~ 572 Da) and the trans-cyclo-octene (TCO)-tagged JQ-1 derivative (~ 609 Da). Heightman’s group developed two model CLIPTACs which can be synthesized intracellularly via click reaction of trans-cyclo-octene and tetrazine precursor molecules [178]. The results showed that CLIPTACs were capable of successfully inducing the degradation of BRD4 and ERK1/2 (extracellular regulated protein kinase) in three cell lines, including HeLa, A375 and HCT116 [178]. This pioneering strategy not only improves the cell permeability and solubility of PROTACs, but also eliminates the need for linker optimization, which is more flexible and convenient as only the protein ligand fraction needs to be changed for different target protein degradation.
Computer simulation accelerates PROTAC design
The rational design of PROTACs includes three components: warhead, E3 ligand and linker. Although the discovery process of warheads and E3 ligands is similar in nature to that of small molecules, but the design of linkers is somewhat challenging since POI and E3 ligases cannot interact in the absence of an effective PROTAC. As the importance of linker to the physicochemical properties and degradation activity of PROTACs are better understood, current research have focused on de novo PROTAC design. The length, composition, flexibility, and attachment sites of the linker all have a dramatic effect on the degradation efficiency. In addition, another design challenge arises from the fact that PROTAC molecules often have poor solubility, poor permeability, low bioavailability, and unpredictable hook effects, which hinder the clinical translation of PROTACs. Therefore, it’s urgent to discover new methods to improve the discovery efficiency of PROTACs. To accelerate the design progress of rational PROTACs, Zheng et al. created a novel depth-generating model (PROTAC-RL) [179]. A pair of E3 ligands and warheads are input into the model, and the designed linkers are output along with chemically feasible PROTACs having specific properties under the guidance of Reinforcement Learning (RL) [179]. Specifically, they first pre-trained a linker generation model (Proformer) based on transformer neural network. To overcome the challenge of low PROTAC training data, the model was first pre-trained through many quasi-PROTAC small molecules similar in size to PROTAC, and then the model was fine-tuned with real PROTACs set and augmented data. The Proformer was subsequently fed into a memory-based reinforcement learning framework, PROTAC-RL, and rewarded with experience to obtain PROTACs with ideal PK properties. To prove the validity, the research team identified BRD4 as POI and generated 5000 PROTACs. Relying on supercomputing capabilities, they further clustered and screened these virtual molecules. The researchers finally selected, synthesized and experimentally tested six PROTACs, three of which exhibited inhibitory activity, and one lead compound showed high anti-proliferative activity against tumor cell lines and good pharmacokinetics. Western blot assay results showed that compound 1–3 (Fig. 2j) decreased intracellular BRD4 at micromolar concentration. And all these compounds showed anti-proliferative activity against Molt4 cell line at micromolar concentrations. The entire research effort took only 49 days, indicating that the application of computer models can facilitate efficient rational PROTAC design and optimization.
Application of PROTACs in diseases
Over the last two decades, PROTACs have demonstrated unique advantages in addressing disease associated proteins. Currently, some representative PROTACs have reached clinical trials for the treatment of cancers. Except for cancer, PROTACs also offer great advantages in the treatment of other diseases, such as neurodegenerative diseases, immune system diseases or viral infection. Here, we summarize some PROTACs targets for these diseases (Table 6).
PROTACs targeting cancer-related targets
The indispensability of oncogenic proteins in the progression of cancer makes PROTAC particularly suitable for the treatment of cancer. Most of the current research on PROTACs focused on cancer-related targets. In the reported studies, researchers preferred kinases as degradation targets [103]. Statistically, kinases account for 45% of the total targets degraded by PROTAC [180,181,182,183,184], of which, more than half of PROTACs targeted RTK [185] and CMGC kinase group (CMGCs). BTK PROTACs have entered clinical trials and several compounds have shown good clinical benefits. PROTACs targeting kinases such as ALK, MEK and CDK have also been studied and investigated extensively in the literature. Besides the kinase-based PROTACs, there are still a large number of PROTACs focused on targeting nuclear receptors and epigenetic protein. So far, the most successful targets for PROTAC applications are AR and ER. Compared with kinase small-molecule inhibitors, the resistance of AR and ER is very complex and tricky. Due to the diversity of AR mutations, original inhibitor of AR, enzalutamide, may lose its inhibitory efficacy and even become a partial agonist. Therefore, PROTACs are particularly suitable for the treatment of AR related cancer, especially metastatic castrate-resistant prostate cancer.
PROTACs for treating neurodegenerative diseases
The most common neurodegenerative disorders include Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease [186]. They often occur in the elderly population and, are a class of diseases that cause cognitive impairment. Aggregation of misfolded proteins is one of the leading cause of neurodegenerative diseases, and the commonly misfolded proteins are β-amyloid, tau, alpha-synuclein, and polyglutamates [186].
Tau is an important microtubule-associated pathological protein of Alzheimer’s disease [187], which is difficult to regulate like many non-enzymatic proteins, because of the lack of active pockets. One of the most prominent features of AD and other neurodegenerative diseases is the accumulation of Tau [30]. Tau levels are higher in the brains of patients with AD than in healthy people. A high Tau level can promote its aggregation and also affect the toxicity of amyloid-β (Aβ). Thus, minimizing Tau aggregation is considered as a potential way to treat AD. Lu’s group designed and synthesized a peptide-based PROTAC bearing Keap1 E3 ligase ligand for the degradation of intracellular Tau, it showed high affinity with tau and keap1 in vitro and induced moderate degradation of Tau [30].
Huntington’s disease (HD) is caused by the variation of Huntington gene, and the abnormal mutant huntingtin (mHtt) produced by the variation that accumulates in the brain will affect neural and nerve cell function [188]. Consequently, inhibition or clearance of toxic mHtt aggregation is considered as a potential treatment modality [189]. Previous research mainly focused on the development of chemical small molecules that have inhibitory effects on mHtt aggregates. Unfortunately, because of the unclear machinery of chemical aggregation modulators, no applicable clinical results are available. Tomoshige et al. designed two small molecule PROTACs, conjugating probes for mHtt aggregates with a ligand for ubiquitin ligase cIAP1. Experimental data showed that the two compounds are capable of inducing the degradation of mHtt in living cells [190]. The effect is particularly pronounced in HD patients and mHtt with a much longer polyglutamine repeat sequence (145Q).
PROTACs for treating immune-related diseases
IRAK4
Interleukin-1 receptor-associated kinase 4 (IRAK4) plays an important role in toll-like receptors (TLRs) and interleukin1 receptors (1L-1R) signaling pathways [191]. IRAK4 belongs to a family of four kinases (IRAK4, IRAK1, IRAK2, and IRAK-M) [192]. IRAK4 receives signals from the upstream TLRs as well as the 1L-1R and activates its downstream NF-κB and JNK signaling pathways, which are closely related to human inflammatory responses and cancers. After TLRs or 1L-1R receptors are activated, IRAK4 binds with MyD88 and IRAK2 through the shared death domain (DD) to form a myddosome complex. The myddosome complex performs its phosphorylation function and activates the downstream IRAK1 and related factor 6 (TRAF6), thus activating the downstream NF-κB and JNK signaling pathways to transcribe genes associated with inflammation and cellular proliferation. Overactivation or dysfunction of IRAK4 can lead to different problems accordingly. In addition to its kinase activity, IRAK4 also has scaffolding signaling. Therefore, traditional small-molecule inhibitors are unable to block all the functions of IRAK4. As a promising technology, PROTACs can eliminate all the functions of protein. In 2020, Dai et al. reported several PROTACs which could selectively degrade IRAK4 [193]. Among all of the PROTACs, only one PROTAC induced the degradation of IRAK4.
HDAC3
The histone deacetylases (HDACs) family is a class of chromatin-modifying enzymes that silence transcription via the modification of histones [194]. HDACs family consists of eighteen isoenzymes that can be divided into four types [195]. Among them, HDAC1-3 and 8 belong to class I HDACs that play a key role in cell motility, immunoregulation, and proliferation [196]. However, the structure of HDAC3 contains a well-conserved catalytic structural domain that makes selective targeting of HDAC3 challenging. In 2020, Dekker et al. reported a novel HDAC3 PROTAC HD-TAC7 (Fig. 2k), which consists of CRBN ligand pomalidomide and selective class I HDAC inhibitors o-aminoanilide [197]. HD-TAC7 has a medium degradation potency but no effect on HDAC1 and HDAC 2. This year, Liao et al. unraveled VHL-based PROTAC XZ9002 (Fig. 2k) that could specifically degrade HDAC3 and inhibit tumor cell activity [198].
PROTACs targeting virus-related targets
It has been thought that PROTACs also can be applied in the antivirus field to reduce susceptibility to resistance mutations. With the drug resistance of conventional antiviral drugs, the effect of clinical treatment began to gradually deteriorate. Recent study leveraged PROTACs to develop a chemical knock-down antiviral to induce degradation of viral proteins. Wispelaere et al. designed a PROTAC which consists of a reversible-covalent inhibitor telaprevir that binds to the hepatitis C virus (HCV) protease active site and a ligand for CRBN ligase [199]. The compound DGY-08-097 (Fig. 2l), not only inhibits but also degrades the HCV NS3/4A protease, exhibiting efficiency in a cellular infection model [199].
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a serious threat to the lives and health of people around the world since its outbreak in 2019 [200]. Despite the fact that several vaccines have been designed worldwide against COVID-19, the high mutagenicity of the virus limits the effectiveness of vaccine. In 2021, Desantis et al. designed a series of indomethacin-based PROTACs pan-coronavirus antiviral agents [201]. Indomethacin (INM) has antiviral activity, but the mechanism behind it is not known. The antiviral activity of INM against SARS-CoV-2 probably came from its inhibitory activity to human prostaglandin E synthase type 2 (PGES-2). Previous study has reported that INM has inhibitory activity of PGES-2 in the nanomolar concentration [202, 203]. The PGES-2 has been shown to have an interaction with the NSP7 protein of SARS-CoV-2 [204, 205]. And the interaction of NSP7 with PGES-2 was also present in other coronaviruses [204, 206], suggesting that targeting PGES-2 may be a potential approach for INM-based antiviral PROTACs design. Desantis et al. designed four INM-based PROTACs, but the biological evaluation results showed that only two compounds were about 4.5-fold more potent than INM, as well as a wide-spectrum antiviral activity against the β-coronavirus HCoV-OC43 and α-coronavirus HCoV-229E [201].
Other PROTACs
In 2020, Rao et al. reported the first PROTAC of HMG-CoA reductase (HMGCR), which is the rate-limiting enzyme in the cholesterol biosynthetic pathway [207, 208]. They synthesized a series of PROTACs by tethering Atorvastatin and CRBN ligands. After optimization and screening, they ultimately found the most potent degrader P22A (Fig. 2m) with DC50 of 0.1 μM [209]. This PROTAC stressed the potential application for the treatment of hypercholesterolemia and cardiovascular disease. In addition, PROTACs are a promising therapeutic approach in other non-oncoproteins. Li et al. reported the first PROTAC that induced degradation of α1A-adrenergic receptor (α1A-AR) and is also the first PROTAC for G protein-coupled receptors (GPCRs) [210]. They connected α1A-AR inhibitor prazosin with pomalidomide by different linkers and finally found the potent compound 9c (Fig. 2m). 9c could inhibit the proliferation of PC-3 cells and cause tumor growth slowdown, which provided a new strategy for the treatment of prostate cancer. Hu et al. presented the first PROTAC of indoleamine 2,3-dioxygenase 1 (IDO1) [65]. IDO1 has been extensively reported as key immune checkpoint, which overexpressed in multiple cancers [211]. Hu et al. discovered the first PROTAC 2c (Fig. 2m) which induced the pronounced and sustained degradation of IDO1. Si et al. showed that PROTAC of hematopoietic progenitor kinase1 (HPK1) helped to improve CAR-T cell-based immunotherapy [212]. PROTAC technology is so widespread in the field of disease treatment, making it a powerful tool for drug discovery.
Disadvantages and future challenges of PROTAC
As an emerging technology, PROTAC has attracted great attention from academia and the pharmaceutical industry. The development of any new technology comes with various opportunities and challenges, and PROTAC is no exception. The prospect of potential opportunities and challenges for PROTAC will contribute to the research and development of targeted protein-degrading drugs. Although PROTAC has unique advantages over other drug discovery paradigm, it also has some disadvantages, which bring nonnegligible issues and challenges:
-
Pharmaceutical property: PROTAC molecule is more complex than traditional small-molecule drugs and has more potential metabolic sites, which affects the metabolic stability of PROTAC molecules. At the same time, traditional small-molecule inhibitors generally follow the “Rule of Five”, but most of the reported PROTACs tend to have a molecular weight greater than 700, resulting in poor permeability, low solubility and unsatisfactory oral bioavailability [213]. Therefore, how to improve physicochemical properties of PROTAC molecule will be the key to its successful drug formation if “the Rule of Five” are not satisfied.
-
Resistance: First, PROTACs can cause drug resistance through the change in the genome of the core component of the E3 ligase complex. Significantly reduced expression of CRBN gene or CUL2 gene can also cause resistance to PROTACs [214, 215]. Studies have shown that deletion of the CRBN genome is the main reason for myeloma cells to develop resistance to IMiDs. Secondly, the action of PROTAC depends on specific E3 ligase subtype, and the expression of specific E3 ligase limits the application of PROTAC in different cell types. Although the human genome encodes hundreds of E3 ubiquitin ligases, only a few E3 ligases and small molecule ligands have been used for PROTACs. Therefore, finding more kinds of E3 ligases for the research and development of PROTAC drugs might be the way to solve drug resistance [216].
-
“Hook effect” and “Off target”: How to avoid Hook effect and off-target effect is also a major challenge for PROTAC drugs development. The higher the concentration of drugs, the better degradation effect is not necessarily for PROTACs, which is often referred to as the “Hook effect”. In the research of PROTACs, it has been found that significantly higher concentration than DC50 will result in self-inhibition effect to compensate degradation efficiency, called “Hook effect” [217, 218]. In addition, the mechanism of off-target effects of PROTACs molecules have not been fully understood [219]. PROTACs can completely degrade target protein, thus inhibit all functions of target protein. However, in this process, normal protein may be accidentally injured, off-target effect and toxicity are also one of the biggest challenges. For example, studies have shown that thalidomide derivatives can cause degradation of transcription factors such as IKZF1, IKZF3 and GSTP1 [214]. Further studies found that the degradation of thalidomide derivatives on transcription factors such as GSPT1 was due to their “molecular glue” effect.
-
Target selection: To date, what targets are appropriate for PROTAC technology to achieve better benefits than small-molecule inhibitors are not fully understood and most of the target proteins of the PROTACs are part of the “druggable” protein. In fact, one of the greatest advantages of PROTAC technology is its potential to handle “undruggable” target. Because PROTAC technology only needs temporarily mediate the formation of ternary complexes, low affinity POI ligands can be incorporated into PROTAC molecules. Unfortunately, there are only few PROTAC molecules targeting “undruggable” proteins to date. Therefore, another challenge for PROTACs is the need to develop more molecules that target “undruggable” proteins and thus embody the advantages of PROTAC technology.
Discussion and conclusion
As an emerging paradigm for drug discovery, PROTACs have attracted great attention from academia and industry. Although PROTAC technology has many advantages in drug development, there are still many obstacles and challenges in the process of discovery and clinical application, such as off-target, cell permeability, stability, and large molecular weight, etc. In addition, the issues of oral bioavailability and drug integrity are also ongoing challenges for PROTAC drug development. It is worth noting that PROTAC still has many advantages in clinical application compared with other traditional small-molecule inhibitors. First, PROTAC plays a role by inducing the degradation of pathogenic proteins, so it can promote the degradation of multiple rounds of target proteins, assisting to eliminate off-target effects and accumulation of drug targets. PROTAC can also degrade some proteins that are considered “undruggable”, such as transcription factors. Secondly, PROTAC has the advantages of improving selectivity and specificity, overcoming drug resistance. In short, the current status of PROTAC drug development is the coexistence of both advantages and disadvantages, but how to solve these problems will be the key to the success of PROTAC drug development.
The discovery of efficient PROTAC molecules is a time-consuming and challenging process, such as the optimization of linker length and structure. It is urgent to summarize a general method for designing efficient PROTAC molecules. At present, the design and optimization of PROTAC mainly focus on the structure–activity relationships research of POI ligands and linker. Among them, linker is not only critical to the degradation activity of PROTACs, but also greatly affects the membrane permeability, metabolic stability and drug availability. Therefore, how to effectively design and link POI and E3 ligands is the key to the molecular design of PROTACs. Up to now, the principles guiding the design of linker, including length and composition, have not been fully understood. On the other hand, photo-PROTAC designed based on “photo control linkers” also has some advantages over traditional drugs, which is also introduced in this article. It is expected that the newly emerging photo-PRTOAC can become a leading way among PROTAC drugs. In this review, we summarized the general principles in the design of PROTAC, providing a systematic understanding for the research and design of PROTACs. In addition, E3 ligase is also crucial in the composition of the ternary complex. However, among the hundreds of E3 ligases encoded by the human genome, only a few E3 ligases are used in PROTACs, and the progress in discovering new E3 ligases and their ligands is far behind the research of PROTACs. So far, the majority of PROTACs induce target protein degradation by recruiting E3 ligases CRBN, VHL, MDM2 and IAP, and the research on PROTACs by only these E3 ligases is still far from enough. Therefore, it is necessary to explore more novel E3 ligases to accelerate the development of PROTACs. However, it can be predicted that the number of E3 ligands may increase significantly in the future, which will provide more options for the design of PROTACs.
PROTAC technology has been developed for nearly 20 years, and some molecules have entered clinical trials, which reveals the huge therapeutic potential of PROTACs in tumor, immune disease, neurodegenerative disease, cardiovascular disease and viral infection. There are also studies around the world using this technology to treat COVID-19. So far, two PROTAC drugs ARV-110 and ARV-471 have entered the phase II clinical trial, which are used to treat prostate cancer and breast cancer respectively. Although more than ten drugs are in clinical trials, clinical research data are still insufficient, and more clinical studies are needed to prove the prospects of PROTAC technology. With the deepening of research, these obstacles will be basically solved in the near future. Once more drugs enter the clinical application, it will open a new era of drug research and development.
Although there are still many obstacles and challenges to be overcame, PROTACs have great therapeutic potential with its unique advantages. It is believed that in the future, with the development of technology and in-depth research, the design and synthesis of PROTACs will be gradually optimized, which will eventually open up a broad road for the treatment of various diseases, and is expected to provide clinical therapeutic benefits in the near future. In a word, PROTAC technology not only provides a powerful tool for the research in the field of pharmaceutical chemistry, but also brings great hope for the development of clinical drugs in the future.
Availability of data and materials
Not applicable.
Abbreviations
- PROTACs:
-
Proteolysis targeting chimeras
- POI:
-
Protein of interest
- UPS:
-
Ubiquitin-protease system
- MetAP-2:
-
Methionine aminopeptidase-2
- β-TRCP:
-
β-Transducin repeat-containing protein
- ER:
-
Estrogen
- AR:
-
Androgen
- DHT:
-
Dihydrotestosterone
- VHL:
-
Von Hippel-Lindau
- FKBP12:
-
FK506 binding protein 12
- MDM2:
-
Mouse double minute 2
- cIAP:
-
Cell inhibitor of apoptosis protein
- CRBN:
-
Cereblon
- IMiDs:
-
Immunomodulatory drugs
- BL:
-
Burkitt’s lymphoma
- Hyp:
-
Hydroxyproline
- ALK:
-
Anaplastic lymphoma kinase
- FAK:
-
Focal adhesion kinase
- RNAi:
-
RNA interference
- SGK:
-
Serum and glucocorticoid-induced protein kinase
- PI3K:
-
Phosphoinositide 3-kinase
- MOA:
-
Mode of action
- RTK:
-
Receptor tyrosine kinase
- HIF1α:
-
Hypoxia-inducible factor-1α
- PEG:
-
Polyethylene glycol
- BTK:
-
Bruton’s tyrosine kinase
- BCR:
-
B cell receptor
- C481:
-
Cysteine481
- WT:
-
Wild type
- IKZF3:
-
Ikaros family zinc finger 3
- PBMCs:
-
Peripheral blood mononuclear cells
- CLL:
-
Chronic lymphocytic leukemia
- NRs:
-
Nuclear receptors
- NLS:
-
Nuclear localization signal
- MTD:
-
Maximum tolerated dose
- RP2D:
-
Recommended phase 2 dose
- mCRPC:
-
Metastatic castrate-resistant prostate cancer
- PSA:
-
Prostate specific antigen
- ER+:
-
Estrogen receptor-positive
- HER2+:
-
Human epidermal growth factor receptor 2 positive
- SERD:
-
Selective estrogen receptor degraders
- TFs:
-
Transcriptional factors
- STAT3:
-
Signal transducer and activator of transcription 3
- SH2:
-
Src-homology 2
- PPI:
-
Protein–protein interaction
- SAR:
-
Structure–activity relationship
- DMNB:
-
4,5-Dimethoxy-2-nitrobenzyl
- CLIPTACs:
-
In-cell click-formed proteolysis targeting chimeras
- Tz:
-
Tetrazine
- TCO:
-
Trans-cyclo-octene
- ERK1/2:
-
Extracellular regulated protein kinase
- CMGCs:
-
CMGC kinase group
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-β
- HD:
-
Huntington’s disease
- mHtt:
-
Mutant huntingtin
- IRAK4:
-
Interleukin-1 receptor-associated kinase 4
- TLRs:
-
Toll-like receptors
- 1L-1R:
-
Interleukin1 receptors
- DD:
-
Death domain
- HDACs:
-
Histone deacetylases
- HCV:
-
Hepatitis C virus
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- INM:
-
Indomethacin
- PGES-2:
-
Human prostaglandin E synthase type 2
- HMGCR:
-
HMG-CoA reductase
- RL:
-
Reinforcement Learning
- MWs:
-
Molecular weights
- BRD4:
-
Bromodomain-containing protein 4
- α1A-AR:
-
α1A-adrenergic receptor
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- HPK1:
-
Hematopoietic progenitor kinase1
- GPCRs:
-
G protein-coupled receptors
- EGFR:
-
Epidermal growth factor receptor
- TPD:
-
Targeted protein degradation
- RAS:
-
RAt Sarcoma
- LYTACs:
-
Lysosome-targeting chimeras
- AUTACs:
-
Autophagy-targeting chimeras
- AbTACs:
-
Antibody-based PROTACs
- LTRs:
-
Lysosome-targeting receptors
- M6Pn:
-
Poly-serine-O-mannose-6-phosphonate
- GalNAC:
-
N-acetyl galactosamine
- PD-L1:
-
Programmed death protein ligand 1
References
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci. 2001;98(15):8554–9. https://doi.org/10.1073/pnas.141230798.
Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18(22):5904–8. https://doi.org/10.1016/j.bmcl.2008.07.114.
Yao T, Xiao H, Wang H, Xu X. Recent advances in PROTACs for drug targeted protein research. Int J Mol Sci. 2022;23(18):10328. https://doi.org/10.3390/ijms231810328.
Buckley DL, Buckley DL, Crews CM. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem. 2014;53(9):2312–30. https://doi.org/10.1002/anie.201307761.
Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514–21. https://doi.org/10.1038/nchembio.2329.
Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. https://doi.org/10.1146/annurev.biochem.78.081507.101607.
Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421–35. https://doi.org/10.1038/s41580-019-0101-y.
Kliza K, Husnjak K. Resolving the complexity of ubiquitin networks. Front Mol Biosci. 2020;7:21. https://doi.org/10.3389/fmolb.2020.00021.
Konstantinidou M, Li J, Zhang B, Wang Z, Shaabani S, Brake FT, et al. PROTACs– a game-changing technology. Expert Opin Drug Discov. 2019;14(12):1255–68. https://doi.org/10.1080/17460441.2019.1659242.
Cromm PM, Crews CM. Targeted protein degradation: from chemical biology to drug discovery. Chem Biol. 2017;24(9):1181–90. https://doi.org/10.1016/j.chembiol.2017.05.024.
Crews CM, Hu Z. Recent developments in PROTAC-mediated protein degradation: from bench to clinic. ChemBioChem. 2021. https://doi.org/10.1002/cbic.202100270.
Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. https://doi.org/10.1146/annurev-biochem-060310-170328.
Chen Y, Jin J. The application of ubiquitin ligases in the PROTAC drug design. Acta Biochim Biophys Sin. 2020;52(7):776–90. https://doi.org/10.1093/abbs/gmaa053.
Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181(1):102–14. https://doi.org/10.1016/j.cell.2019.11.031.
Salami J, Crews CM. Waste disposal-an attractive strategy for cancer therapy. Science. 2017;355(6330):1163–7. https://doi.org/10.1126/science.aam7340.
Eder J, Herrling P. Trends in modern drug discovery. Handb Exp Pharmacol. 2015;232:3–22. https://doi.org/10.1007/164_2015_20.
Valeur E, Jimonet P. New modalities, technologies, and partnerships in probe and lead generation: enabling a mode-of-action centric paradigm. J Med Chem. 2018;61(20):9004–29. https://doi.org/10.1021/acs.jmedchem.8b00378.
Kargbo RB. PROTAC-mediated degradation of KRAS protein for anticancer therapeutics. ACS Med Chem Lett. 2020;11(1):5–6. https://doi.org/10.1021/acsmedchemlett.9b00584.
Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30. https://doi.org/10.1038/nrd892.
Neklesa TK, Snyder L, Willard RR, Vitale N, Pizzano J, Gordon DA, et al. ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol. 2019;37:259–259. https://doi.org/10.1200/jco.2019.37.7_suppl.259.
Flanagan JJ, Neklesa TK. Targeting nuclear receptors with PROTAC degraders. Mol Cell Endocrinol. 2019;493:110452. https://doi.org/10.1016/j.mce.2019.110452.
Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2(12):1350–8. https://doi.org/10.1074/mcp.T300009-MCP200.
Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126(12):3748–54. https://doi.org/10.1021/ja039025z.
Hon W-C, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, et al. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature. 2002;417(6892):975–8. https://doi.org/10.1038/nature00767.
Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin−ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132(16):5820–6. https://doi.org/10.1021/ja100691p.
Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22(6):755–63. https://doi.org/10.1016/j.chembiol.2015.05.009.
Zhang X, Luukkonen LM, Eissler CL, Crowley VM, Yamashita Y, Schafroth MA, et al. DCAF11 supports targeted protein degradation by electrophilic proteolysis-targeting chimeras. J Am Chem Soc. 2021;143(13):5141–9. https://doi.org/10.1021/jacs.1c00990.
Li L, Mi D, Pei H, Duan Q, Wang X, Zhou W, et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct Target Ther. 2020;5(1):129. https://doi.org/10.1038/s41392-020-00245-0.
Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol. 2019;15(7):737–46. https://doi.org/10.1038/s41589-019-0279-5.
Lu M-C, Liu T, Jiao Q, Ji J-A, Tao M, Liu Y, et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251–9. https://doi.org/10.1016/j.ejmech.2018.01.063.
Yang Y, Zhou C, Wang Y, Liu W, Liu C, Wang L, et al. The E3 ubiquitin ligase RNF114 and TAB1 degradation are required for maternal-to-zygotic transition. EMBO Rep. 2017;18(2):205–16. https://doi.org/10.15252/embr.201642573.
Ishida T, Ciulli A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS Discov Adv Life Sci R D. 2020;26(4):484–502. https://doi.org/10.1177/2472555220965528.
Nguyen TV. USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. 2021;118(40):e2111391118. https://doi.org/10.1073/pnas.2111391118.
Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi A, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326–35. https://doi.org/10.1038/leu.2012.119.
Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–5. https://doi.org/10.1126/science.1244851.
Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53. https://doi.org/10.1038/nature13527.
Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, et al. Structure of the human Cereblon–DDB1–lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21(9):803–9. https://doi.org/10.1038/nsmb.2874.
Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9. https://doi.org/10.1126/science.1244917.
Lee J, Lee Y, Jung YM, Park JH, Yoo HS, Park J. Discovery of E3 ligase ligands for target protein degradation. Molecules. 2022;27(19):6515. https://doi.org/10.3390/molecules27196515.
Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew Chem Int Ed Engl. 2012;51(46):11463–7. https://doi.org/10.1002/anie.201206231.
Zhao Q, Ren C, Liu L, Chen J, et al. Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase. J Med Chem. 2019;62(20):9281–98. https://doi.org/10.1021/acs.jmedchem.9b01264.
Kang CH, Lee DH, Lee CO, Du Ha J, Park CH, Hwang JY. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem Biophys Res Commun. 2018;505(2):542–7. https://doi.org/10.1016/j.bbrc.2018.09.169.
Cromm PM, Samarasinghe KT, Hines J, Crews CM. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J Am Chem Soc. 2018;140(49):17019–26. https://doi.org/10.1021/jacs.8b08008.
Petrylak DP, Gao X, Vogelzang NJ, Garfield MH, Taylor IW, Taylor I, et al. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J Clin Oncol. 2020;38:3500–3500. https://doi.org/10.1200/jco.2020.38.15_suppl.3500.
Xie H, Liu J, Alem Glison DM, Fleming JB. The clinical advances of proteolysis targeting chimeras in oncology. Explor Target Anti-Tumor Ther. 2021;2(6):511–21. https://doi.org/10.37349/etat.2021.00061.
He Y, Koch R, Budamagunta V, Zhang P, Zhang X, Khan S, et al. DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J Hematol OncolJ Hematol Oncol. 2020;13(1):95. https://doi.org/10.1186/s13045-020-00928-9.
You I, Erickson EC, Donovan KA, Eleuteri NA, Fischer ES, Gray NS, et al. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem Biol. 2020;27(1):66-73.e7. https://doi.org/10.1016/j.chembiol.2019.11.014.
Kargbo RB. PROTAC compounds targeting α-synuclein protein for treating neurogenerative disorders: Alzheimer’s and Parkinson’s diseases. ACS Med Chem Lett. 2020;11(6):1086–7. https://doi.org/10.1021/acsmedchemlett.0c00192.
Gasic I, Groendyke BJ, Nowak RP, Yuan JC, Kalabathula J, Fischer ES, et al. Tubulin resists degradation by cereblon-recruiting PROTACs. Cells. 2020;9(5):1083. https://doi.org/10.3390/cells9051083.
Shi W, Feng Z, Chi F, Zhou J, Qiu Q, Jiang Y, et al. Structure-based discovery of receptor tyrosine kinase AXL degraders with excellent anti-tumor activity by selectively degrading AXL and inducing methuosis. Eur J Med Chem. 2022;234:114253. https://doi.org/10.1016/j.ejmech.2022.114253.
Wang Z, He N, Guo Z, Niu C, Song T, Guo Y, et al. Proteolysis targeting chimeras for the selective degradation of Mcl-1/Bcl-2 derived from nonselective target binding ligands. J Med Chem. 2019;62(17):8152–63. https://doi.org/10.1021/acs.jmedchem.9b00919.
Zhang X, Thummuri D, He Y, Liu X, Zhang P, Zhou D, et al. Utilizing PROTAC technology to address the on-target platelet toxicity associated with inhibition of BCL-X L. Chem Commun. 2019;55(98):14765–8. https://doi.org/10.1039/C9CC07217A.
Xue G, Chen J, Liu L, Zhou D, Zuo Y, Fu T, et al. Protein degradation through covalent inhibitor-based PROTACs. Chem Commun. 2020;56(10):1521–4. https://doi.org/10.1039/C9CC08238G.
Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14(7):706–14. https://doi.org/10.1038/s41589-018-0055-y.
Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57(26):3564–75. https://doi.org/10.1021/acs.biochem.8b00391.
Chi JJ, Li H, Zhou Z, Izquierdo-Ferrer J, Xue Y, Wavelet CM, et al. A novel strategy to block mitotic progression for targeted therapy. EBioMedicine. 2019;49:40–54. https://doi.org/10.1016/j.ebiom.2019.10.013.
Zhou F, Chen L, Cao C, Yu J, Luo X, Zhou P, et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem. 2020;187:111952. https://doi.org/10.1016/j.ejmech.2019.111952.
Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun. 2019;55(18):2704–7. https://doi.org/10.1039/C9CC00163H.
Steinebach C, Lindner S, Udeshi ND, Mani DC, Kehm H, Köpff S, et al. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem Biol. 2018;13(9):2771–82. https://doi.org/10.1021/acschembio.8b00693.
Zhou L, Chen W, Cao C, Shi Y, Ye W, Hu J, et al. Design and synthesis of α-naphthoflavone chimera derivatives able to eliminate cytochrome P450 (CYP)1B1-mediated drug resistance via targeted CYP1B1 degradation. Eur J Med Chem. 2020;189:112028. https://doi.org/10.1016/j.ejmech.2019.112028.
Potjewyd F, Turner A-MW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem Biol. 2020;27(1):47-56.e15. https://doi.org/10.1016/j.chembiol.2019.11.006.
Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Chem Biol. 2017;25(1):67. https://doi.org/10.1016/j.chembiol.2017.09.009.
Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A, et al. Chemically induced degradation of anaplastic lymphoma kinase (ALK). J Med Chem. 2018;61(9):4249–55. https://doi.org/10.1021/acs.jmedchem.7b01655.
Smalley JP, Adams GE, Millard CJ, Song Y, Norris JKS, Schwabe JWR, et al. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem Commun. 2020;56(32):4476–9. https://doi.org/10.1039/D0CC01485K.
Hu M, Zhou W, Wang Y, Yao D, Ye T, Yao Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B. 2020;10(10):1943–53. https://doi.org/10.1016/j.apsb.2020.02.010.
Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem. 2019;62(2):448–66. https://doi.org/10.1021/acs.jmedchem.8b00909.
Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D, et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife. 2019;8:e45457. https://doi.org/10.7554/elife.45457.
Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S, et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun. 2017;8(1):830. https://doi.org/10.1038/s41467-017-00954-1.
Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, et al. Development and characterization of a Wee1 kinase degrader. Cell Chem Biol. 2020;27(1):57-65.e9. https://doi.org/10.1016/j.chembiol.2019.10.013.
Hu M, Li Y, Li J, Zhou H, Liu C, Liu Z, et al. Discovery of potent and selective HER2 PROTAC degrader based Tucatinib with improved efficacy against HER2 positive cancers. Eur J Med Chem. 2022;244:114775. https://doi.org/10.1016/j.ejmech.2022.114775.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26. https://doi.org/10.1038/nrc3599.
Zou Y, Ma D, Wang Y. The PROTAC technology in drug development: the PROTAC technology in drug development. Cell Biochem Funct. 2019;37(1):21–30. https://doi.org/10.1002/cbf.3369.
Martín-Acosta P, Xiao X. PROTACs to address the challenges facing small molecule inhibitors. Eur J Med Chem. 2021;210: 112993. https://doi.org/10.1016/j.ejmech.2020.112993.
Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10(2):207–38. https://doi.org/10.1016/j.apsb.2019.08.001.
Bond MJ, Crews CM. Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation. RSC Chem Biol. 2021;2(3):725–42. https://doi.org/10.1039/D1CB00011J.
Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. https://doi.org/10.1146/annurev-pharmtox-010715-103440.
Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138–44. https://doi.org/10.1016/j.pharmthera.2017.02.027.
Moon S, Lee B-H. Chemically induced cellular proteolysis: an emerging therapeutic strategy for undruggable targets. Mol Cells. 2018;41(11):933–42. https://doi.org/10.14348/molcells.2018.0372.
Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14(4):248–62. https://doi.org/10.1038/nrc3690.
Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Chem Biol. 2017;25(1):78. https://doi.org/10.1016/j.chembiol.2017.09.010.
Troup RI, Fallan C, Baud MGJ. Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Ther. 2020;1(5):273–312. https://doi.org/10.37349/etat.2020.00018.
Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–52. https://doi.org/10.1038/s41573-020-0068-6.
Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, et al. Exploring targeted degradation strategy for oncogenic KRASG12C. Chem Biol. 2020;27(1):19. https://doi.org/10.1016/j.chembiol.2019.12.006.
Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11(8):1913–22. https://doi.org/10.1158/2159-8290.CD-21-0365.
Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent Sci. 2020;6(8):1367–75. https://doi.org/10.1021/acscentsci.0c00411.
Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth Factors. 2010;28(6):394–408. https://doi.org/10.3109/08977194.2010.518616.
Halland N, Schmidt F, Weiss T, Saas J, Li Z, Czech J, et al. Discovery of N-[4-(1H-Pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as highly active and selective SGK1 inhibitors. ACS Med Chem Lett. 2015;6(1):73–8. https://doi.org/10.1021/ml5003376.
Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68(18):7475–83. https://doi.org/10.1158/0008-5472.CAN-08-1047.
Gong G, Wang K, Dai X, Zhou Y, Basnet R, Chen Y, et al. Identification, structure modification, and characterization of potential small-molecule SGK3 inhibitors with novel scaffolds. Acta Pharmacol Sin. 2018;39:1902–12. https://doi.org/10.1038/s41401-018-0087-6.
Tovell H, Testa A, Zhou H, Shpiro N, Crafter C, Ciulli A, et al. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem Biol. 2019;14(9):2024–34. https://doi.org/10.1021/acschembio.9b00505.
Adjei AA. What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24(25):4054–5. https://doi.org/10.1200/JCO.2006.07.4658.
Bondeson DP, Mares A, Smith IED, Eunhwa K, Campos SA, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11(8):611–7. https://doi.org/10.1038/nchembio.1858.
Martinez Molina D, Nordlund P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu Rev Pharmacol Toxicol. 2016;56(1):141–61. https://doi.org/10.1146/annurev-pharmtox-010715-103715.
Wu S, Jiang Y, Hong Y, Chu X, Zhang Z, Tao Y, et al. BRD4 PROTAC degrader ARV-825 inhibits T-cell acute lymphoblastic leukemia by targeting “Undruggable” Myc-pathway genes. Cancer Cell Int. 2021;21(1):230. https://doi.org/10.1186/s12935-021-01908-w.
Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4(6):a014241. https://doi.org/10.1101/cshperspect.a014241.
Burke MR, Smith AR, Zheng G. Overcoming cancer drug resistance utilizing PROTAC technology. Front Cell Dev Biol. 2022; 10. https://doi.org/10.3389/fcell.2022.872729.
Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - past, present and future. Drug Discov Today Technol. 2019;31:15–27. https://doi.org/10.1016/j.ddtec.2019.01.002.
Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584(7820):291–7. https://doi.org/10.1038/s41586-020-2545-9.
Pei J, Wang G, Feng L, Zhang J, Jiang T, Sun Q, et al. Targeting lysosomal degradation pathways: new strategies and techniques for drug discovery. J Med Chem. 2021;64(7):3493–507. https://doi.org/10.1021/acs.jmedchem.0c01689.
Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020;41(7):464–74. https://doi.org/10.1016/j.tips.2020.04.005.
Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019;76(5):797-810.e10. https://doi.org/10.1016/j.molcel.2019.09.009.
Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143(2):593–8. https://doi.org/10.1021/jacs.0c10008.
Tan L, Gray NS. When kinases meet PROTACs: when kinases meet PROTACs †. Chin J Chem. 2018;36(10):971–7. https://doi.org/10.1002/cjoc.201800293.
Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353–77. https://doi.org/10.1038/nrd.2018.21.
Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A. 2013;110(22):8942–7. https://doi.org/10.1073/pnas.1217206110.
Henning RK, Varghese JO, Das S, Nag A, Tang G, Tang K, et al. Degradation of Akt using protein-catalyzed capture agents: DEGRADATION OF AKT USING PCC AGENTS. J Pept Sci. 2016;22(4):196–200. https://doi.org/10.1002/psc.2858.
Sun Y, Zhao X, Ding N, Gao H, Gao H, Wu Y, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28(7):779–81. https://doi.org/10.1038/s41422-018-0055-1.
del Mar Noblejas-López M, Nieto-Jiménez C, Burgos M, Gómez-Juárez M, Montero JC, Esparís-Ogando A, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38(1):383–383. https://doi.org/10.1186/s13046-019-1387-5.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–32. https://doi.org/10.1038/nrc3702.
Pan Z, Scheerens H, Li S-J, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1):58–61. https://doi.org/10.1002/cmdc.200600221.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–80. https://doi.org/10.1073/pnas.1004594107.
Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94. https://doi.org/10.1056/NEJMoa1400029.
Haertle L, Barrio S, Munawar U, Han S, Zhou X, Vogt C, et al. Cereblon enhancer methylation and IMiD resistance in multiple myeloma. Blood. 2021;138(18):1721–6. https://doi.org/10.1182/blood.2020010452.
Lazarian G, Yin S, ten Hacken E, Sewastianik T, Uduman M, Font-Tello A, et al. A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell. 2021;39(3):380-393.e8. https://doi.org/10.1016/j.ccell.2021.02.003.
Robbins DW, Noviski M, Rountree R, Tan M, Brathaban N, Ingallinera T, et al. Nx-5948, a selective degrader of BTK with activity in preclinical models of hematologic and brain malignancies. Blood. 2021;138(Supplement 1):2251. https://doi.org/10.1182/blood-2021-147473.
Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–72. https://doi.org/10.1146/annurev-physiol-021909-135917.
Pollock JA, Wardell SE, Parent AA, Parent AA, Stagg DB, Ellison SJ, et al. Inhibiting androgen receptor nuclear entry in castration-resistant prostate cancer. Nat Chem Biol. 2016;12(10):795–801. https://doi.org/10.1038/nchembio.2131.
Salami J, Alabi SB, Willard RR, Vitale NJ, Wang J, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1(1):100–100. https://doi.org/10.1038/s42003-018-0105-8.
Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308. https://doi.org/10.1210/er.2002-0032.
Sanford M. Enzalutamide: a review of its use in metastatic, castration-resistant prostate cancer. Drugs. 2013;73(15):1723–32. https://doi.org/10.1007/s40265-013-0129-9.
Palmbos PL, Hussain M. Non-castrate metastatic prostate cancer: have the treatment options changed? Semin Oncol. 2013;40(3):337–46. https://doi.org/10.1053/j.seminoncol.2013.04.007.
Han X, Zhao L, Xiang W, Qin C, Miao B, McEachern D, et al. Strategies toward discovery of potent and orally bioavailable proteolysis targeting chimera degraders of androgen receptor for the treatment of prostate cancer. J Med Chem. 2021;64(17):12831–54. https://doi.org/10.1021/acs.jmedchem.1c00882.
Xiang W, Zhao L, Han X, Qin C, et al. Discovery of ARD-2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J Med Chem. 2021;64(18):13487–509. https://doi.org/10.1021/acs.jmedchem.1c00900.
Tong CWS, Wu M, Cho WCS, To KKW. Recent advances in the treatment of breast cancer. Front Oncol. 2018;8:227. https://doi.org/10.3389/fonc.2018.00227.
Anderson WF, Katki HA, Rosenberg PS. Incidence of breast cancer in the United States: current and future trends. J Natl Cancer Inst. 2011;103(18):1397–402. https://doi.org/10.1093/jnci/djr257.
Nilsson S, Koehler KF, Gustafsson J-Å. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov. 2011;10(10):778–92. https://doi.org/10.1038/nrd3551.
Jia M, Dahlman-Wright K, Gustafsson J-Å. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab. 2015;29(4):557–68. https://doi.org/10.1016/j.beem.2015.04.008.
Wang Y, Tang S-C. The race to develop oral SERDs and other novel estrogen receptor inhibitors: recent clinical trial results and impact on treatment options. Cancer Metastasis Rev. 2022. https://doi.org/10.1007/s10555-022-10066-y.
Howell A, Sapunar F. Fulvestrant revisited: efficacy and safety of the 500-mg dose. Clin Breast Cancer. 2011;11(4):204–10. https://doi.org/10.1016/j.clbc.2011.02.002.
Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90(Suppl 1):S2-6. https://doi.org/10.1038/sj.bjc.6601629.
Robertson JFR, Harrison M. Fulvestrant: pharmacokinetics and pharmacology. Br J Cancer. 2004;90(Suppl 1):S7-10. https://doi.org/10.1038/sj.bjc.6601630.
Robertson JFR, Lindemann J, Garnett S, Anderson E, Nicholson RI, Kuter I, et al. A good drug made better: the fulvestrant dose-response story. Clin Breast Cancer. 2014;14(6):381–9. https://doi.org/10.1016/j.clbc.2014.06.005.
Mottamal M, Kang B, Peng X, Wang G. From pure antagonists to pure degraders of the estrogen receptor: evolving strategies for the same target. ACS Omega. 2021;6(14):9334–43. https://doi.org/10.1021/acsomega.0c06362.
Cyrus K, Wehenkel M, Choi E-Y, Lee H, Swanson HI, Kim KB. Jostling for position: optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem. 2010;5(7):979–85. https://doi.org/10.1002/cmdc.201000146.
Kargbo RB. PROTAC-mediated degradation of estrogen receptor in the treatment of cancer. ACS Med Chem Lett. 2019;10(10):1367–9. https://doi.org/10.1021/acsmedchemlett.9b00397.
Tecalco-Cruz AC, Zepeda-Cervantes J, Ramírez-Jarquín JO, Rojas-Ochoa A. Proteolysis-targeting chimeras and their implications in breast cancer. Explor Target Anti-Tumor Ther. 2021;2(6):496–510. https://doi.org/10.37349/etat.2021.00060.
Qin H, Zhang Y, Lou Y, Pan Z, Song F, et al. Overview of PROTACs targeting the estrogen receptor: achievements for biological and drug discovery. Curr Med Chem. 2021; 28. https://doi.org/10.2174/0929867328666211110101018.
Qi S-M, Dong J, Xu Z-Y, Cheng X-D, Zhang W, Zhang W-D, et al. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol. 2021;12:692574. https://doi.org/10.3389/fphar.2021.692574.
Liu J, Chen H, Kaniskan HÜ, Xie L, Chen X, Jin J, et al. TF-PROTACs enable targeted degradation of transcription factors. J Am Chem Soc. 2021;143(23):8902–10. https://doi.org/10.1021/jacs.1c03852.
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10(4):252–63. https://doi.org/10.1038/nrg2538.
Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Targeting CREB for cancer therapy: friend or foe. Curr Cancer Drug Targets. 2010;10(4):384–91. https://doi.org/10.2174/156800910791208535.
Darnell JE. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–9. https://doi.org/10.1038/nrc906.
Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer. 2019;19(11):611–24. https://doi.org/10.1038/s41568-019-0196-7.
Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, et al. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem. 2013;288(7):4723–32. https://doi.org/10.1074/jbc.m112.378984.
Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138(11):2570–8. https://doi.org/10.1002/ijc.29923.
Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–48. https://doi.org/10.1038/nrclinonc.2018.8.
Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(5):498-511.e17. https://doi.org/10.1016/j.ccell.2019.10.002.
Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem. 2012;55(15):6645–68. https://doi.org/10.1021/jm300207s.
Pallandre J-R, Borg C, Rognan D, Boibessot T, Luzet V, Yesylevskyy S, et al. Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors. Eur J Med Chem. 2015;103:163–74. https://doi.org/10.1016/j.ejmech.2015.08.054.
Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18(4):443–51. https://doi.org/10.1038/cr.2008.41.
Heppler LN, Frank DA. Inhibit versus destroy: are PROTAC degraders the solution to targeting STAT3? Cancer Cell. 2019;36(5):459–61. https://doi.org/10.1016/j.ccell.2019.10.010.
Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2019;62(2):699–726. https://doi.org/10.1021/acs.jmedchem.8b01413.
Alabi S, Jaime-Figueroa S, Yao Z, Gao Y, Hines J, Samarasinghe KTG, et al. Mutant-selective degradation by BRAF-targeting PROTACs. Nat Commun. 2021;12(1):920. https://doi.org/10.1038/s41467-021-21159-7.
Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J Med Chem. 2020;63(3):1216–32. https://doi.org/10.1021/acs.jmedchem.9b01566.
Wang M, Lu J, Wang M, Yang C-Y, Wang S. Discovery of SHP2-D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J Med Chem. 2020;63(14):7510–28. https://doi.org/10.1021/acs.jmedchem.0c00471.
He Y, Zhang X, Chang J, Kim H-N, Zhang P, Wang Y, et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat Commun. 2020;11(1):1996. https://doi.org/10.1038/s41467-020-15838-0.
Yang X, Wang Z, Pei Y, Song N, Xu L, Feng B, et al. Discovery of thalidomide-based PROTAC small molecules as the highly efficient SHP2 degraders. Eur J Med Chem. 2021;218:113341. https://doi.org/10.1016/j.ejmech.2021.113341.
Donoghue C, Cubillos-Rojas M, Gutierrez-Prat N, Sanchez-Zarzalejo C, Verdaguer X, Riera A, et al. Optimal linker length for small molecule PROTACs that selectively target p38α and p38β for degradation. Eur J Med Chem. 2020;201:112451. https://doi.org/10.1016/j.ejmech.2020.112451.
Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79(1):251–62. https://doi.org/10.1158/0008-5472.CAN-18-2918.
Schiemer J, Horst R, Meng Y, Montgomery JI, Xu Y, Feng X, et al. Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat Chem Biol. 2021;17(2):152–60. https://doi.org/10.1038/s41589-020-00686-2.
Zhang X, He Y, Zhang P, Budamagunta V, et al. Discovery of IAP-recruiting BCL-XL PROTACs as potent degraders across multiple cancer cell lines. Eur J Med Chem. 2020;199:112397. https://doi.org/10.1016/j.ejmech.2020.112397.
Meng F, Xu C, Park K-S, Kaniskan HÜ, Wang GG, Jin J. Discovery of a first-in-class degrader for nuclear receptor binding SET domain protein 2 (NSD2) and Ikaros/Aiolos. J Med Chem. 2022;65(15):10611–25. https://doi.org/10.1021/acs.jmedchem.2c00807.
Bricelj A, Steinebach C, Kuchta R, Gütschow M, Sosič I. E3 ligase ligands in successful PROTACs: an overview of syntheses and linker attachment points. Front Chem. 2021;9:707317. https://doi.org/10.3389/fchem.2021.707317.
Cao C, He M, Wang L, He Y, Rao Y. Chemistries of bifunctional PROTAC degraders. Chem Soc Rev. 2022;51(16):7066–114. https://doi.org/10.1039/D2CS00220E.
Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Eunhwa K, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem. 2016;55(2):807–10. https://doi.org/10.1002/anie.201507634.
Maple HJ, Clayden N, Baron A, Stacey C, Felix R. Developing degraders: principles and perspectives on design and chemical space. MedChemComm. 2019;10(10):1755–64. https://doi.org/10.1039/C9MD00272C.
Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang C-Y, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. 2019;62(2):941–64. https://doi.org/10.1021/acs.jmedchem.8b01631.
Kargbo RB. PROTAC-mediated degradation of Bruton’s tyrosine kinase as a therapeutic strategy for cancer. ACS Med Chem Lett. 2021;12(5):688–9. https://doi.org/10.1021/acsmedchemlett.1c00178.
He S, Dong G, Cheng J, Wu Y, Sheng C. Strategies for designing proteolysis targeting chimaeras (PROTACs). Med Res Rev. 2022;42(3):1280–342. https://doi.org/10.1002/med.21877.
Han X, Sun Y. Strategies for the discovery of oral PROTAC degraders aimed at cancer therapy. Cell Rep Phys Sci. 2022;3(10):101062. https://doi.org/10.1016/j.xcrp.2022.101062.
Cyrus K, Wehenkel M, Choi E-Y, Han H-J, Lee H, Swanson H, et al. Impact of linker length on the activity of PROTACs. Mol Biosyst. 2011;7(2):359–64. https://doi.org/10.1039/c0mb00074d.
Bemis TA, La Clair JJ, Burkart MD. Unraveling the role of linker design in proteolysis targeting chimeras. J Med Chem. 2021;64(12):8042–52. https://doi.org/10.1021/acs.jmedchem.1c00482.
Naro Y, Darrah K, Deiters A. Optical control of small molecule-induced protein degradation. J Am Chem Soc. 2020;142(5):2193–7. https://doi.org/10.1021/jacs.9b12718.
Liu J, Peng Y, Wei W. Light-controllable PROTACs for temporospatial control of protein degradation. Front Cell Dev Biol. 2021;9:678077. https://doi.org/10.3389/fcell.2021.678077.
Pfaff P, Samarasinghe KTG, Crews CM, Carreira EM. Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent Sci. 2019;5(10):1682–90. https://doi.org/10.1021/acscentsci.9b00713.
Xue G, Wang K, Zhou D, Zhong H, Pan Z. Light-induced protein degradation with photocaged PROTACs. J Am Chem Soc. 2019;141(46):18370–4. https://doi.org/10.1021/jacs.9b06422.
Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, et al. Light-induced control of protein destruction by opto-PROTAC. Sci Adv. 2020;6(8):eaay5154. https://doi.org/10.1126/sciadv.aay5154.
Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci. 2016;2(12):927–34. https://doi.org/10.1021/acscentsci.6b00280.
Zheng S, Tan Y, Wang Z, Li C, Zhang Z, Sang X, et al. Accelerated rational PROTAC design via deep learning and molecular simulations. Nat Mach Intell. 2022;4(9):739–48. https://doi.org/10.1038/s42256-022-00527-y.
Riching KM, Schwinn MK, Vasta JD, Robers MB, Machleidt T, Urh M, et al. CDK family PROTAC profiling reveals distinct kinetic responses and cell cycle-dependent degradation of CDK2. SLAS Discov Adv Life Sci R D. 2021;26(4):560–9. https://doi.org/10.1177/2472555220973602.
Kargbo RB. PROTAC compounds targeting TRK for use in cancer therapeutics. ACS Med Chem Lett. 2020;11(6):1090–1. https://doi.org/10.1021/acsmedchemlett.0c00235.
Burslem GM, Schultz AR, Bondeson DP, Eide CA, Savage Stevens SL, Druker BJ, et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 2019;79(18):4744–53. https://doi.org/10.1158/0008-5472.CAN-19-1236.
Adhikari B, Bozilovic J, Diebold M, Schwarz JD, Hofstetter J, Schröder M, et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat Chem Biol. 2020;16(11):1179–88. https://doi.org/10.1038/s41589-020-00652-y.
Pang X-J, Liu X-J, Liu Y, Liu W-B, Li Y-R, Yu G-X, et al. Drug discovery targeting focal adhesion kinase (FAK) as a promising cancer therapy. Molecules. 2021;26(14):4250. https://doi.org/10.3390/molecules26144250.
Burslem GM, Song J, Chen X, Chen X, Chen X, Hines J, et al. Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J Am Chem Soc. 2018;140(48):16428–32. https://doi.org/10.1021/jacs.8b10320.
Hyun S, Shin D. Chemical-mediated targeted protein degradation in neurodegenerative diseases. Life. 2021;11(7):607. https://doi.org/10.3390/life11070607.
Chu T-T, Gao N, Li Q-Q, Chen P-G, Yang X-F, Chen Y-X, et al. Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Chem Biol. 2016;23(4):453–61. https://doi.org/10.1016/j.chembiol.2016.02.016.
Fiorillo A, Morea V, Colotti G, Ilari A. Huntingtin ubiquitination mechanisms and novel possible therapies to decrease the toxic effects of mutated Huntingtin. J Pers Med. 2021;11(12):1309. https://doi.org/10.3390/jpm11121309.
Harding R, Tong Y, Tong Y. Proteostasis in Huntington’s disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol Sin. 2018;39(5):754–69. https://doi.org/10.1038/aps.2018.11.
Tomoshige S, Nomura S, Ohgane K, Hashimoto Y, Ishikawa M. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angew Chem Int Ed. 2017;56(38):11530–33. https://doi.org/10.1002/anie.201706529.
Chaudhary D, Robinson S, Romero DL. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J Med Chem. 2015;58(1):96–110. https://doi.org/10.1021/jm5016044.
Nunes JP, McGonagle GA, Eden J, Kiritharan G, et al. Targeting IRAK4 for degradation with PROTACs. ACS Med Chem Lett. 2019;10(7):1081–5. https://doi.org/10.1021/acsmedchemlett.9b00219.
Zhang J, Fu L, Shen B, Liu Y, Wang W, Cai X, et al. Assessing IRAK4 functions in ABC DLBCL by IRAK4 kinase inhibition and protein degradation. Cell Chem Biol. 2020;27(12):1500-1509.e13. https://doi.org/10.1016/j.chembiol.2020.08.010.
Nguyen HCB, Adlanmerini M, Hauck AK, Lazar MA. Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature. 2020;584(7820):286–90. https://doi.org/10.1038/s41586-020-2576-2.
Fischer F, Alves Avelar LA, Murray L, Kurz T. Designing HDAC-PROTACs: lessons learned so far. Future Med Chem. 2022;14(3):143–66. https://doi.org/10.4155/fmc-2021-0206.
Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res MCR. 2007;5(10):981–9. https://doi.org/10.1158/1541-7786.MCR-07-0324.
Cao F, de Weerd S, Chen D, Zwinderman MRH, van der Wouden PE, Dekker FJ. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur J Med Chem. 2020;208:112800. https://doi.org/10.1016/j.ejmech.2020.112800.
Xiao Y, Wang J, Zhao LY, Chen X, Zheng G, Zhang X, et al. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem Commun Camb Engl. 2020;56(68):9866–9. https://doi.org/10.1039/d0cc03243c.
de Wispelaere M, Du G, Donovan KA, Zhang T, Eleuteri NA, Yuan JC, et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat Commun. 2019;10(1):3468. https://doi.org/10.1038/s41467-019-11429-w.
Martinez-Ortiz W, Zhou M-M. Could PROTACs protect us from COVID-19? Drug Discov Today. 2020;25(11):1894–6. https://doi.org/10.1016/j.drudis.2020.08.007.
Desantis J, Mercorelli B, Celegato M, Croci F, Bazzacco A, Baroni M, et al. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur J Med Chem. 2021;226:113814. https://doi.org/10.1016/j.ejmech.2021.113814.
Al-Horani RA, Kar S. Potential anti-SARS-CoV-2 therapeutics that target the post-entry stages of the viral life cycle: a comprehensive review. Viruses. 2020;12(10):E1092. https://doi.org/10.3390/v12101092.
Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231(25):232–5. https://doi.org/10.1038/newbio231232a0.
Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science. 2020;370(6521):eabe9403. https://doi.org/10.1126/science.abe9403.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68. https://doi.org/10.1038/s41586-020-2286-9.
Terracciano R, Preianò M, Fregola A, Pelaia C, Montalcini T, Savino R. Mapping the SARS-CoV-2-host protein-protein interactome by affinity purification mass spectrometry and proximity-dependent biotin labeling: a rational and straightforward route to discover host-directed anti-SARS-CoV-2 therapeutics. Int J Mol Sci. 2021;22(2):E532. https://doi.org/10.3390/ijms22020532.
Lu X-Y, Shi X-J, Hu A, Wang J-Q, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature. 2020;588(7838):479–84. https://doi.org/10.1038/s41586-020-2928-y.
Luo G, Li Z, Lin X, Li X, Chen Y, Xi K, et al. Discovery of an orally active VHL-recruiting PROTAC that achieves robust HMGCR degradation and potent hypolipidemic activity in vivo. Acta Pharm Sin B. 2021;11(5):1300–14. https://doi.org/10.1016/j.apsb.2020.11.001.
Li M-X, Yang Y, Zhao Q, Wu Y, Song L, Yang H, et al. Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-Hydroxy-3-methylglutaryl coenzyme A reductase. J Med Chem. 2020;63(9):4908–28. https://doi.org/10.1021/acs.jmedchem.0c00339.
Li Z, Lin Y, Song H, Qin X, Yu Z, Zhang Z, et al. First small-molecule PROTACs for G protein-coupled receptors: inducing 1A-adrenergic receptor degradation. Acta Pharm Sin B. 2020;10(9):1669–79. https://doi.org/10.1016/j.apsb.2020.01.014.
Zamanakou M, Germenis AE, Karanikas V. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol Lett. 2007;111(2):69–75. https://doi.org/10.1016/j.imlet.2007.06.001.
Si J, Shi X, Sun S, Zou B, Li Y, An D, et al. Hematopoietic progenitor kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T cell-based immunotherapies. Cancer Cell. 2020;38(4):551-566.e11. https://doi.org/10.1016/j.ccell.2020.08.001.
Cantrill C, Chaturvedi P, Rynn C, Petrig Schaffland J, Walter I, Wittwer MB. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov Today. 2020;25(6):969–82. https://doi.org/10.1016/j.drudis.2020.03.012.
Zhang L, Riley-Gillis B, Vijay P, Shen Y. Acquired resistance to BET-PROTACs (proteolysis-targeting chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol Cancer Ther. 2019;18(7):1302–11. https://doi.org/10.1158/1535-7163.MCT-18-1129.
Ottis P, Palladino C, Thienger P, Britschgi A, Heichinger C, Berrera M, et al. Cellular resistance mechanisms to targeted protein degradation converge toward impairment of the engaged ubiquitin transfer pathway. ACS Chem Biol. 2019;14(10):2215–23. https://doi.org/10.1021/acschembio.9b00525.
Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18(12):949–63. https://doi.org/10.1038/s41573-019-0047-y.
An S, Fu L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–62. https://doi.org/10.1016/j.ebiom.2018.09.005.
Miles LE. Properties, variants, and applications of the immunoradiometric assay method. Ric Clin Lab. 1975;5(1):59–72. https://doi.org/10.1007/BF02910016.
Gao H, Sun X, Rao Y. PROTAC technology: opportunities and challenges. ACS Med Chem Lett. 2020;11(3):237–40. https://doi.org/10.1021/acsmedchemlett.9b00597.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22277086, 22007070), the Science and Technology Department of Sichuan Province (2022ZDZX0028, 2022NSFSC1409), 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC21075) and the Post-Doctor Research Project, West China Hospital, Sichuan University (2021HXBH006).
Code availability
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to this work. Zi Liu wrote and prepared the original draft. Mingxing Hu, Yu Yang, Haoxuan Zhou, Chengyali Liu and Hongqun Ma wrote, reviewed, and edited the manuscript. Chenghao Du, Yuanwei Chen, Lei Fan, Youling Gong and Yongmei Xie supervised and revised the study. All authors approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This article does not involve animal and human experiments.
Consent for publication
Not applicable.
Competing interests
Author Yuanwei Chen, Lei Fan, Hongqun Ma are employees in Hinova Pharmaceuticals Inc., but has no potential relevant financial or non-financial interests to disclose. The other authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Z., Hu, M., Yang, Y. et al. An overview of PROTACs: a promising drug discovery paradigm. Mol Biomed 3, 46 (2022). https://doi.org/10.1186/s43556-022-00112-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-022-00112-0