
Abstract
Mantle cell lymphoma (MCL) is a subtype of non-Hodgkin’s lymphoma characterized by poor prognosis. The complexity of MCL pathogenesis arises from aberrant activities of diverse signaling pathways, including BTK, PI3K–AKT–mTOR and MYC-BRD4. Here, we report that MCL-related signaling pathways can be altered by a single small molecule inhibitor, SRX3305. Binding and kinase activities along with resonance changes in NMR experiments reveal that SRX3305 targets both bromodomains of BRD4 and is highly potent in inhibition of the PI3K isoforms α, γ and δ, as well as BTK and the drug-resistant BTK mutant. Preclinical investigations herein reveal that SRX3305 perturbs the cell cycle, promotes apoptosis in MCL cell lines and shows dose dependent anti-proliferative activity in both MCL and drug-resistant MCL cells. Our findings underscore the effectiveness of novel multi-action small molecule inhibitors for potential treatment of MCL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mantle cell lymphoma (MCL) is a mature B cell malignancy with characteristic t(11;14)(q13;q32) translocation juxtaposing IGH and CCND1 gene loci [1]. This translocation leads to overexpression of cyclin D1 and dysregulation of the cell cycle at the G1/S phase transition. While treatment with combination chemotherapeutic regimens is initially effective, nearly all patients relapse with a median survival of only 2.5 – 5 years [2, 3]. Several therapeutic options have been pursued to improve the outcome for MCL, including combination chemotherapy, high-dose chemotherapy followed by autologous stem cell transplant (ASCT) and monoclonal antibody therapy, all of which have been met with minimal success. Outside of ASCT, a therapy for which most patients are ineligible, MCL remains incurable to date. In recent years, several small molecule inhibitors have emerged for B-cell malignancies to provide improved survival outcomes with diminished therapy-associated morbidities. The therapeutic options for MCL are constantly growing in the era of novel targeted therapies – Ibrutinib, Acalabrutinib and Venetoclax – and a shift to chemotherapy-free regimens is highly warranted [3].
Aberrant B-cell receptor (BCR) signaling is a key mediator of MCL disease pathogenesis and contributes to enhanced survival and proliferation of malignant B-cells [4, 5]. Agents targeting Bruton's tyrosine kinase (BTK), a core effector of the BCR signaling pathway (e.g., Ibrutinib) [6,7,8] and other BCR pathway members, such as phosphatidylinositol-3 kinase δ (PI3K-δ; e.g., Idelalisib) [9], have demonstrated high response rates in MCL [10]; however, these responses were not durable, suggesting a combinatory approach to enhance patient outcome. Despite the high overall response rate seen in relapsed/refractory (R/R) MCL patients treated with Ibrutinib (68%), only 21% of patients reached a complete response with a median progression free survival of ~14 months [6]. Resistance to Ibrutinib is common, and there are few approved therapies capable of improving MCL patient outcome to an extent, making the post-FDA approved R/R MCL treatment setting an unmet clinical need [11].
Dysregulation of MYC plays an essential role in the pathogenesis of B-cell lymphomas, including MCL, and is associated with poor prognosis and aggressive clinical behavior [12,13,14]. Pharmacological inhibition of the BET family proteins BRD2, BRD3, BRD4, and BRDt has emerged as a therapeutic approach to target MYC-dependent transcription [15, 16] because BRD4 regulates MYC and a number of other lymphoma-relevant oncogenes (BCL2, CDK4/6, and cyclin D1) [17,18,19,20,21]. BRD4 is also essential for the transcriptional activity of NF-κB downstream of BCR signaling [22, 23], which makes it a vital therapeutic target in MCL. Various small molecule BET inhibitors, such as JQ1, I-BET-151 and OTX-015, have demonstrated anti-tumor activity in preclinical models of B-cell malignancies [24, 25], including MCL [20, 26]. Notably, due to the reported synergy with Ibrutinib, BET inhibitors may serve as a therapeutic option for MCL in the setting of Ibrutinib resistance [20, 26]. Recent studies have demonstrated the effectiveness of targeting multiple disease-relevant pathways with a combination of inhibitors (PI3Ki and BETi/BTKi) or with a single multi-action agent (PI3K-BET and PI3K-BRD4-CDK4/6). These therapeutic approaches are becoming an attractive way to achieve a durable response and combat drug resistance [10, 21, 27,28,29]. We recently introduced thienopyranone scaffold-based chemotypes for the combinatorial inhibition of BTK, PI3K-AKT and BRD4-MYC [30]. Here, we report on SRX3305, a second generation of the highly potent BTK/PI3K/BRD4 triple action inhibitors and demonstrate the effectiveness of this approach for potential treatment of MCL.
Results
SRX3305 binds to BRD4 and inhibits BTK and PI3K
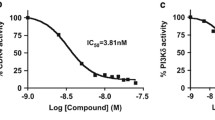
In effort to develop small molecule agents which act against MCL, we designed a series of thienopyranone (TP)-based compounds with the ability to concomitantly bind bromodomains (BDs) of BRD4, the catalytic domain of BTK, and the catalytic domain of PI3K. We have demonstrated that dual and triple action TP-scaffold compounds show promising results as anti-cancer agents [27,28,29], and the first generation of BTK/PI3K/BRD4 inhibitors, such as SRX3262, was cytotoxic to MCL cells [30]. To enhance efficacy of these compounds, a new set of chemotypes was generated [31] and screened in Alpha Screen and kinase assays. Among the new set of inhibitors, SRX3305 showed a substantial increase in potency for all three targets, BTK, PI3K and BRD4 compared to SRX3262 (Fig. 1a). SRX3305 displayed an IC50 of 6.5 nM, 15 nM, and 4 nM toward BTK, PI3Kɑ and PI3Kδ, respectively, and was a ~4-5-fold more potent inhibitor than SRX3262 towards these kinases (Fig. 1a-e). SRX3305 also retained its high inhibitory activity toward both bromodomains of BRD4, bromodomain 1 (BD1) and bromodomain 2 (BD2) and was ~2-fold more potent than SRX3262 (Fig. 1a, g, h). Importantly, SRX3305 showed enhanced binding to the drug-resistant C481S mutant of BTK compared to SRX3262 (IC50=9 μM and 25 μM, respectively), was more effective than the pan-kinase inhibitor Staurosporine for WT BTK (IC50=15 nM) and elicited an effect comparable to the PI3K inhibitor Idelalisib (IC50=2.5 nM) [32] on PI3Kδ (Fig. 1a, f).

SRX3305 is a potent triple activity BTK/PI3K/BRD4 inhibitor. a IC50 values (nM) of SRX3305 and other indicated inhibitors of BRD4, PI3K and BTK as measured by displacement binding and kinase assays. Values for SRX3262 are from [30] and values for Idelalisib are from [32]. b-h IC50 curves for indicated targets by indicated inhibitors. i, j Superimposed 1H,15N HSQC spectra of uniformly 15N-labeled BRD4 BD1 and BRD4 BD2, recorded while SRX3305 was titrated in. The spectra are color-coded according to the protein: inhibitor molar ratio (inset). k Structural overlay of the complexes: BRD4 BD1 (light blue) with the TP inhibitor SF2523 (pink) (PDB: 5U28) [27] and BRD4 BD1 (light green) with H4K5acK8ac peptide (yellow) (PDB: 3UVW) [33]. Water molecules are shown as red spheres
To gain insight into the mechanism of inhibition of BRD4, we produced 15N-labeled BRD4 BD1 and BRD4 BD2 and examined the binding of SRX3305 to each domain by NMR spectroscopy. Gradual addition of SRX3305 to either the BRD4 BD1 NMR sample or BRD4 BD2 NMR sample induced substantial chemical shift changes in 1H,15N heteronuclear single quantum coherence (HSQC) spectra of BD1 and BD2, indicating that both domains are targets of SRX3305 (Fig. 1i, j). These resonance changes were in slow exchange regime on the NMR timescale and suggested tight binding of SRX3305, confirming the nanomolar IC50 values, 77 nM for BD1 and 95 nM for BD2. Moreover, the pattern of these changes was similar to the pattern of changes observed in 1H,15N HSQC spectra of these domains due to the binding of other TP-scaffold compounds [27,28,29], including SF2523, as well as acetylated peptides [33], implying that bound SRX3305 occupies the acetyllysine-binding sites of BRD4 BD1 and BRD4 BD2 (Fig. 1k).
SRX3305 is cytotoxic to MCL cells
To establish the anti-cancer activity of SRX3305, we performed cytotoxicity experiments treating the MCL cell lines JeKo-1, Mino, Granta, JeKo-1 BTK C481S and Mino BTK C481S with increasing concentrations of SRX3305, SRX3306 or the BTK inhibitor Ibrutinib. The dose response curves generated from alamarBlue and CellTiter-Glo cell viability assays revealed strong anti-proliferative function of SRX3305 (Fig. 2a-d and Supplementary Fig. 1). SRX3305 showed an IC50 of 1 nM in Mino cells, 58 nM in JeKo-1 cells and 1.1 μM in Granta cells and had ~5-80-fold higher efficacy in these cell lines compared to SRX3306 and SRX3262. Moreover, SRX3305 was substantially more cytotoxic than Ibrutinib to these MCL cells due to the concurrent inhibition of not only BTK but also two additional targets, PI3K and BRD4. Cell viability assays using the JeKo-1 BTK C481S and Mino BTK C481S mutant cell lines further demonstrated that SRX3305 has higher efficacy (IC50=1 μM and 47 nM, respectively) compared to Ibrutinib (IC50=38 μM and 6 μM, respectively) (Fig. 2c, d). SRX3305 was minimally toxic to healthy donor peripheral blood mononuclear cells (PBMCs) (Fig. 2e) or bystander healthy stromal cells (Fig. 2f) and was less toxic to healthy donor B-cells when compared to the combination of single-target drugs (combo) required to effectively inhibit the same three targets (Fig. 2g). Furthermore, the anti-tumor effect of SRX3305 was confirmed in primary Eμ-Myc tumor samples (Fig. 2h).

SRX3305 is cytotoxic to MCL cells. a-d Anti-proliferative activity of JeKo-1, Mino, JeKo-1 BTK C481S and Mino BTK C481S MCL cell lines treated with increasing concentrations of indicated inhibitors. Error bars represent mean ± SEM (n= independent 3 experiments). e PBMCs from healthy subjects (n = 4) were treated with increasing concentrations of SRX3305 or DMSO vehicle for 48 h. Viability was assessed via MTS assay and results are normalized to vehicle. f Human HS-5 bone marrow-derived stromal cell line was treated for 72 h with DMSO vehicle, indicated concentrations of SRX3305, 1 μM Ibrutinib, 1 μM Idelalisib or 1 μM JQ-1. Viability was assessed via MTS assay and results are normalized to vehicle. Results (mean ± SEM) are given from n = 4 independent experiments. *** P< 0.001 vs. vehicle. g B-cells were purified from healthy subject PBMCs (n = 3) using negative selection kit. Normal B-cells were then incubated with DMSO vehicle, 0.5 μM Ibrutinib, 0.5 μM Idelalisib, 0.5 μM JQ-1, a combination (combo) of the single inhibitors (Ibrutinib, Idelalisib, and JQ1 each at 0.5 μM), or 0.5 μM SRX3305. After 48 h, viability was assessed by MTS. Results (mean ± SEM) were normalized to vehicle. * P< 0.05 vs. vehicle and as indicted for combo vs. SRX3305. h Splenocytes isolated from morbid Eμ-Myc mice (n = 4) were stimulated ex vivo with PMA/Ionomycin and treated with DMSO vehicle or increasing inhibitor concentrations as indicated. After 48 h, proliferation was assessed by MTS and normalized to vehicle. Results are shown as (mean ± SD). Dose-dependent decrease in cell proliferation is observed with symbols denoting significance (* P< 0.05, *** P< 0.001)
SRX3305 shows improved efficacy in MCL and Ibrutinib-resistant MCL cells
The effect of SRX3305 on activation of BTK/PI3K signaling was examined in IgM stimulated JeKo-1 and Mino MCL cells using western blot analysis. Treatment of the cells with SRX3305 blocked activation of both BTK and AKT (a downstream effector of PI3K) as evidenced by the decrease of BTK phosphorylation at Tyr223 and AKT phosphorylation at Ser473 (Fig. 3a and Supplementary Fig. 2a). SRX3305 at a concentration of 0.5 μM inactivated the BTK/PI3K signaling to the same extent as the BTK inhibitor Ibrutinib at a concentration of 1 μM and SRX3262 at a concentration of 5 μM (Fig. 3b).

SRX3305 is efficacious in MCL and Ibrutinib-resistant MCL cells. a, b Western blot analysis of lysates from IgM-stimulated Mino cells treated with the indicated concentrations of Ibrutinib, SRX3305 or SRX3262 for 1 h. BTK and PI3K signaling was assessed by the levels of BTK, phosphorylated at Y223 BTK [pBTK(Y223)], AKT, and phosphorylated at S473 AKT [pAKT(S473)]. c, d Western blots analysis of lysates from IgM-stimulated Mino cells without and with washing out indicated inhibitors 24 h post treatment. e, f Western blot analysis of lysates from IgM-stimulated Granta cells treated with increasing concentrations of SRX3305, SRX3262 or Ibrutinib for 1 h
We next tested whether SRX3305 covalently binds to BTK. We treated Mino cells with SRX3305, SRX3262, Ibrutinib (irreversible covalent BTK inhibitor) and BMS-935177 (reversible BTK inhibitor) for 1 h and then performed inhibitor wash out experiments (Fig. 3c, d). As expected, cells treated with the reversible inhibitor BMS-935177 regained BTK phosphorylation after extensive washes with PBS buffer; however, cells treated with SRX3305, SRX3262 and Ibrutinib retained inhibition of BTK phosphorylation. SRX3305 and SRX3262 also retained inhibition of AKT phosphorylation in contrast to Ibrutinib in Mino cells (Fig. 3c) but not in the Mino BTK C481S mutant cells (Supplementary Fig. 2b, c). Furthermore, SRX3305 showed dose dependent anti-proliferative activity in Granta, an Ibrutinib-resistant cell line, and again it was as potent at a concentration of 0.5 μM as SRX3262 at a concentration of 5 μM (Fig. 3e, f). Collectively, these data demonstrate that SRX3305 is an irreversible inhibitor, which displays augmented efficacy in both MCL and Ibrutinib-resistant MCL cell lines.
SRX3305 impacts gene expression, perturbs the cell cycle and promotes apoptosis in MCL
BRD4 is known to regulate expression of target genes, including MYC and other oncogenes [16, 34]. We treated JeKo-1 cells with SRX3305, the BRD4 inhibitor PLX51107, or control DMSO and analyzed expression of cMYC and HEXIM1, a pharmacodynamic marker for BRD4 inhibition, by qRT-PCR. As expected, treatment with SRX3305 led to a decrease in expression of cMYC and an increase in expression of HEXIM1 (Supplementary Fig. 3). Given the synergistic relationship of BRD4, PI3K and BTK, we assessed the apoptotic response induced by SRX3305. Treatment of JeKo-1 and Mino cells with increasing concentrations of SRX3305, up to 2 μM for 24 h, stimulated apoptosis, as measured by Annexin V and propidium incorporation in flow cytometry (Fig. 4 and Supplementary Figs. 4 and 5). SRX3305 at 2 μM induced apoptosis in JeKo-1 and Mino cells to the same extent as Staurosporine at 20 nM, whereas Ibrutinib showed no significant effect in JeKo-1 cells and a mild effect in Mino cells at a concentration of 5 μM. SRX3305 treatment of JeKo-1 cells led to alterations in the cell cycle phases with cell population in S phase increasing from 49 to 65% and cell population in G2 phase increasing from 8 to 18% (Supplementary Fig. 6). The effect was milder in Mino cells, where cell population in G2 phase increased from 12 to 19%. Together, these results indicate that SRX3262 inhibits MCL cell proliferation through inducing S/G2 phase cell cycle arrest and promoting apoptosis.

SRX3305 induces apoptosis and cell cycle arrest. a, b Flow cytometry analysis of apoptosis induced in the JeKo-1 (a) and Mino (b) MCL cell lines treated with control (DMSO) or the inhibitors Staurosporine, Ibrutinib, SRX3262 and SRX3305 and assessed after 24 h by evaluating the total percentage of Annexin V/ Propidium Iodide positively stained cells. Histograms show % apoptotic cells (Annexin V positive cells). Experiments were independently performed in duplicate (see also Supplementary Figs. 4 and 5)
Discussion
Given the absence of curative therapy for MCL, especially for patients with R/R disease, it is essential to explore new treatment options and strategies. Current anti-cancer treatments, such as the use of combinations of drugs to target multiple pathways, often suffer from additive toxicity, underscoring an urgent need for the development of potent anti-MCL agents with reduced drug-induced adverse effects. This could be achieved by developing highly specific inhibitors that concomitantly hit two or more major targets in MCL pathogenesis and thus provide a synergistically lethal effect.
In this study we have developed the most potent to date BRD4/PI3K/BTK inhibitor, SRX3305, that shows enhanced preclinical efficacy in MCL cells and overcomes Ibrutinib resistance by inducing cytotoxic mediated cell death in the acquired Ibrutinib resistance cells. SRX3305 is a single agent which simultaneously binds to the catalytic domains of BTK and PI3K and bromodomains of BRD4 and inhibits the critical signaling pathways known to be aberrantly activated in MCL, including BTK, PI3K–AKT–mTOR and MYC-BRD4.
Our findings demonstrate that SRX3305 perturbs the cell cycle, shows dose dependent anti-proliferative activity and promotes apoptosis in MCL cells. Moreover, ex vivo studies using primary Eμ-Myc tumor samples confirm the anti-tumor activity of SRX3305 in an aggressive model of B-cell lymphoma. Further studies will focus on testing efficacy and toxicity of SRX3305 across various B-cell malignancies, including in vivo models, and on the development of the next generation of triple action anti-MCL agents.
Materials and methods
Cell lines, primary donor samples and reagents
Human Mantle Cell Lymphoma (MCL) cell lines Mino, JeKo-1 and Granta-519 (Granta) were received from Thomas Kipps and David Weinstock labs. BTK C481S mutant was cloned into a pMSCV-ires-tdTomato expression vector using the Gateway vector conversion system (Invitrogen). Cells were transfected and selected for homogeneity. Parental Mino and JeKo-1 cells served as WT BTK. Mino and JeKo-1 cells were grown in RPMI-1640 with 20% fetal bovine serum (FBS) and Granta cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS. All cells were supplemented with 1% penicillin/streptomycin and incubated at 37 °C with 5% CO2. Cell lines were tested for mycoplasma and checked for authenticity against the ICLAC (International Cell Line Authentication Committee; http://iclac.org/databases/cross-contaminations/) list. Healthy donor peripheral blood mononuclear cells (PBMCs) were obtained from the Elutriation Core Facility per approved IRB protocol at the University of Nebraska Medical Center. For a subset of experiments, B-cells were purified from healthy donor PBMCs using B cell negative selection kit from StemCell Technologies (Vancouver, Canada) following manufacture’s protocol. Healthy donor cells were cultured in RPMI-1640 supplemented with 100 μg/mL streptomycin (P/S), 100 U/mL penicillin, 2 μM L-glutamine and 10% heat-inactivated fetal bovine serum (hi-FBS). Human HS-5 stromal cell line was obtained from the ATCC and cultured in DMEM supplemented with P/S and 10% hi-FBS. All antibodies were purchased from Cell Signaling Technology. Ibrutinib, Staurosporine, Idelalisib (CAL101) and JQ1 were purchased from Selleck Chemicals. SRX3305, SRX3306, SRX3302 and SRX3262 were synthesized and provided by SignalRx Pharmaceuticals, Inc. (Cumming, GA).
Binding and kinase assays
Adapta kinase activity assays were carried out by ThermoFisher Scientific (Waltham, MA) to measure IC50 of the indicated inhibitors for PI3K isoforms. Kinase activity assays were carried out by Reaction Biology (Malvern, PA) to measure IC50 of the indicated inhibitors for BTK WT and BTK C481S (Ibrutinib-resistant mutant). Alpha Screen assays were carried out by Reaction Biology (Malvern, PA) to measure IC50 of the indicated inhibitors for His-tagged BRD4 BD1 and BD2 bromodomains.
Protein expression and purification
The human pGEX6P-BRD4 BD1 (aa 43-180) and pGEX4T-BRD4 BD2 (aa 342-460) constructs were generated as previously described [28]. Briefly, BRD4 BD1 and BD2 bromodomain constructs were expressed in E. coli BL21 (DE3) RIL in M19 minimal media supplemented with 15NH4Cl and purified as GST fusions. Cells were induced with IPTG at a final concentration of 1 mM and grown overnight at 16 °C. The cells were harvested by centrifugation, resuspended in 10 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, and lysed by sonication. The proteins were purified using glutathione Sepharose 4B beads, and the GST tag was cleaved with PreScission protease. The proteins were purified using a S100 column (GE Healthcare), equilibrated in 10 mM HEPES pH 7.5, 1 mM TCEP and 150 mM NaCl. The fractions of BRD4 BD1 and BD2 were assessed for purity by SDS-PAGE, buffer exchanged into PBS pH 6.8 and concentrated to ~10-20 mg/mL.
NMR spectroscopy
NMR spectroscopy experiments were performed on a Varian INOVA 600 MHz spectrometer. Experiments were carried out on uniformly 15N-labeled BRD4 BD1 (0.15 mM) and BRD4 BD2 (0.25 mM) in PBS buffer pH 6.8, containing 10% D2O. 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the BRD4 BD1 and BD2 were collected while SRX3305 was titrated in the NMR samples.
Cell viability assay
JeKo-1 (parental/WT BTK or BTK C481S mutant), Mino (parental/WT or BTK C481S mutant), and Granta cells were plated in 96 well plates at a density of 2x104 in 100 μL media and incubated overnight. Cells were treated with DMSO (0.1% final) or increasing concentrations of the indicated inhibitors for 48 h. 100 μL of Alamar blue (ThermoFisher Scientific) and Cell titer Glow assay reagent (Promega Cat # G9241) was added to each well, and the cells were incubated for 10 min at room temperature. Luminescence signal was measured in a VarioScan multimode reader. For Alamar Blue, fluorescence signals were read (emission at 590 nm after excitation at 560 nm). Averaged values were normalized as a percentage of DMSO (control) and analyzed by nonlinear regression then plotted for dose response using GraphPad Prism (GraphPad Software, Inc.). Experiments were performed in triplicate.
Cytotoxicity assays using non-tumor cells
MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assays were used to determine inhibitor-induced cytotoxicity. Briefly, healthy donor primary cells (~0.7e6/well) or HS-5 stromal cells (~25,000/well) were treated with vehicle (DMSO) or increasing inhibitor concentrations for up to 72 h in 96-well plates and then the CellTiter 96® AQueous assay (Promega, Madison, WI) was preformed according to manufacturer's instruction to determine cell proliferation. Absorbance signal from each well was acquired at 490 nm on a Tecan Infinite® M1000 Pro microplate reader (Männedorf, Switzerland).
Ex vivo proliferation assay using primary Eμ-Myc tumor samples
Spleens were harvested from terminally diseased Eμ-Myc mice (Jackson Laboratory) housed at the Animal Research Facility (UNMC), adhering to institutional animal care guidelines. Murine splenocytes (~0.7e6/well) were stimulated with 1X PMA/ionomycin (BioLegend) and treated with vehicle (DMSO), ibrutinib, idelalisib, JQ-1, or SRX3305 for 48 h in 96-well plates. The CellTiter 96® AQueous assay (Promega, Madison, WI) was then preformed according to manufacturer's instructions to determine cell proliferation. Absorbance signal from each well was acquired at 490 nm on a Tecan Infinite® M1000 Pro microplate reader (Männedorf, Switzerland).
Western blot
JeKo-1, Mino (parental/WT or BTK C481S mutant) and Granta cells were plated in 10 cm tissue culture dishes at a cell density of 2 x 106 and were incubated overnight. The cells were serum starved for 6 h and then treated in the presence of increasing concentration of SRX3305 or Ibrutinib for 1 h, followed by stimulation with 10 μg/mL of goat F(ab) 2 anti-human antibody (Southern Biotech, Birmingham, Cat # 2022-01) for 15 min at 37 °C. Whole cell lysates of the treated and untreated JeKo-1 and Mino cells were prepared using RIPA buffer supplemented with protease, phosphatase/protease inhibitor cocktails (Thermo Scientific) and 0.1% NP40. Protein concentration in cell lysates was determined using a bicinchoninic acid (BCA) assay kit (Thermo Fisher). Equal amounts of lysate were resolved on a 4-12% SDS-PAGE, transferred to nitrocellulose membranes, and probed with one or more of the following antibodies: anti-pBTK-Y223 (Cat # 5082S), anti-pAKT-S473 (Cat # 9271S), total anti-BTK (Cat # 8547S), total anti-AKT (Cat # 4691), and anti-β-actin (Cat # SC69879). Secondary antibodies were chosen according to the species of origin of the primary antibody. Protein bands were detected using the Pierce enhanced chemiluminescence (ECL) substrate (Thermo Fisher) and imaging system. Uncropped western blots images are shown in Supplementary Fig. 7.
FITC Annexin V apoptosis assay
Jeko-1 and Mino cells were seeded at a density of 2x106 cells and incubated overnight. Cells were treated with the specified concentrations of Ibrutinib, SRX3305, SRX3262 or controls DMSO and Staurosporine and incubated for 24 h. Cells were harvested and washed twice with cold PBS, and then labeled with Annexin V-FITC and PI (BD Pharmingen, Cat # 556419) or Annexin APC (Thermo Fisher, Cat # A35110) according to the manufacturer’s protocol (BD Pharmingen). The labeled cells were analyzed using BD Accuri C6 Flow cytometer. Experiments were performed in duplicate.
RNA isolation and qRT-PCR
5x106 Jeko-1 cells were seeded in 10 cm plate and incubated overnight. The cells were treated with 1 μM PLX51107 (BRD4 Inhibitor), 0.5 μM SRX3305 or DMSO for 24 hr. RNA was extracted post 24 hr by RNeasy mini kit (Qiagen). 1 μg of total RNA was converted to cDNA using iScript cDNA synthesis kit (BIO-RAD). cDNA amplification was performed with 1×SYBR green supermix on a QuantStudio™ 3 Real-Time PCR System. cDNAs were amplified using specific cMYC and HEXIM1 primers, and the data were normalized to GAPDH.
Cell cycle analysis
JeKo-1 and Mino cells were treated with the specified concentrations of SRX3305, SRX3262, or Ibrutinib for 24 h. Cells were harvested, washed once with PBS, and then fixed with 70% ethanol and stored at -20 °C overnight. The fixed cells were collected by centrifugation, washed twice with PBS, resuspended with assay buffer (PI-100 μL/mL, Triton-X-50 μL/mL and RNase A-100 μg/mL), and incubated in the dark for 30 min at RT. The samples were analyzed using BD Accuri C6 Flow cytometer. Experiments were performed in duplicate.
BCR Pathway stimulation
JeKo-1, Mino (parental/WT or BTK C481S mutant) and Granta cells (1x 106) were serum starved for 6 h and then incubated with or without SRX3305, SRX3262, Ibrutinib or controls for 1 h. Treated cells were then resuspended in 1 mL of RPMI-1640, supplemented with 20% FBS and stimulated with 10 μg/mL of goat F(ab’)2 anti-human IgM antibody at 37 °C for 10 min. Stimulated cells were harvested and lysed with RIPA buffer followed by western blot analysis for pBTK and pAKT.
Inhibitor washout assay
Cells were incubated with SRX3305, SRX3262, Ibrutinib or controls for 1 h, washed three times with PBS, and incubated in RPMI-1640 supplemented with 20% FBS and penicillin/streptomycin for 24 h. The BCR pathways in the washed cells were stimulated with 10 μg/mL of goat F(ab’)2 anti-human IgM antibody, and the cells were harvested and lysed. The resulting lysates were analyzed by western blot.
Statistical analysis
All data were quantified and plotted using GraphPad Prism 8.0 and are presented as the means ± SEM of two or more experiments unless indicated otherwise. In cytotoxicity assays utilizing non-tumor cells and primary tumor differences in cell viability between conditions of interest were assessed using ANOVA models followed by Dunnett’s post-hoc test. Log-transformations of the data were applied when necessary before modeling to stabilize variances. P values < 0.05 were considered statistically significant.
Availability of data and materials
The structures of the described compounds used in this study are proprietary information of SignalRx Pharmaceuticals, Inc. (Patent number WO2020023340A1, 2020), and are not publicly available. All other data are available from the authors upon reasonable request.
Abbreviations
- MCL:
-
Mantle cell lymphoma
- BTK:
-
Bruton’s tyrosine kinase
- BCR:
-
B-cell receptor
- PI3K:
-
Phosphatidylinositol-3 kinase
- R/R:
-
Relapsed/refractory
References
Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750–62. https://doi.org/10.1038/nrc2230.
Avivi I, Goy A. Refining the mantle cell lymphoma paradigm: impact of novel therapies on current practice. Clin Cancer Res. 2015;21(17):3853–61. https://doi.org/10.1158/1078-0432.CCR-15-0488.
Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94(6):710–25. https://doi.org/10.1002/ajh.25487.
Saba NS, Liu D, Herman SE, Underbayev C, Tian X, Behrend D, et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood. 2016;128(1):82–92. https://doi.org/10.1182/blood-2015-11-681460.
Merolle MI, Ahmed M, Nomie K, Wang ML. The B cell receptor signaling pathway in mantle cell lymphoma. Oncotarget. 2018;9(38):25332–41. https://doi.org/10.18632/oncotarget.25011.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16. https://doi.org/10.1056/NEJMoa1306220.
Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, et al. Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood. 2015;126(6):739–45. https://doi.org/10.1182/blood-2015-03-635326.
Ma J, Lu P, Guo A, Cheng S, Zong H, Martin P, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166(6):849–61. https://doi.org/10.1111/bjh.12974.
Kahl BS, Spurgeon SE, Furman RR, Flinn IW, Coutre SE, Brown JR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–405. https://doi.org/10.1182/blood-2013-11-537555.
de Rooij MF, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood. 2015;125(14):2306–9. https://doi.org/10.1182/blood-2014-12-619163.
Martin P, Maddocks K, Leonard JP, Ruan J, Goy A, Wagner-Johnston N, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood. 2016;127(12):1559–63. https://doi.org/10.1182/blood-2015-10-673145.
Hernandez L, Hernandez S, Bea S, Pinyol M, Ferrer A, Bosch F, et al. c-myc mRNA expression and genomic alterations in mantle cell lymphomas and other nodal non-Hodgkin’s lymphomas. Leukemia. 1999;13(12):2087–93. https://doi.org/10.1038/sj.leu.2401599.
Choe JY, Yun JY, Na HY, Huh J, Shin SJ, Kim HJ, et al. MYC overexpression correlates with MYC amplification or translocation, and is associated with poor prognosis in mantle cell lymphoma. Histopathology. 2016;68(3):442–9. https://doi.org/10.1111/his.12760.
Nguyen L, Papenhausen P, Shao H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes (Basel). 2017;8(4). https://doi.org/10.3390/genes8040116.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Research support, N.I.H., extramural research support, Non-U.S. Gov't. Nature. 2010;468(7327):1067–73. https://doi.org/10.1038/nature09504.
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. https://doi.org/10.1016/j.cell.2011.08.017.
Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. https://doi.org/10.1016/j.cell.2013.09.053.
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. https://doi.org/10.1016/j.cell.2013.03.036.
Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24(6):777–90. https://doi.org/10.1016/j.ccr.2013.11.003.
Tarantelli C, Bernasconi E, Gaudio E, Cascione L, Restelli V, Arribas AJ, et al. BET bromodomain inhibitor birabresib in mantle cell lymphoma: in vivo activity and identification of novel combinations to overcome adaptive resistance. ESMO Open. 2018;3(6):e000387. https://doi.org/10.1136/esmoopen-2018-000387.
Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, et al. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell. 2015;27(6):837–51. https://doi.org/10.1016/j.ccell.2015.05.006.
Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, et al. Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287(34):28840–51. https://doi.org/10.1074/jbc.M112.359505.
Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene. 2014;33(18):2395–404. https://doi.org/10.1038/onc.2013.179.
Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol. 2015;6(3):128–41. https://doi.org/10.1177/2040620715576662.
Spriano F, Stathis A, Bertoni F. Targeting BET bromodomain proteins in cancer: The example of lymphomas. Pharmacol Ther. 2020;215:107631. https://doi.org/10.1016/j.pharmthera.2020.107631.
Sun B, Shah B, Fiskus W, Qi J, Rajapakshe K, Coarfa C, et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015;126(13):1565–74. https://doi.org/10.1182/blood-2015-04-639542.
Andrews FH, Singh AR, Joshi S, Smith CA, Morales GA, Garlich JR, et al. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc Natl Acad Sci U S A. 2017;114(7):E1072–80. https://doi.org/10.1073/pnas.1613091114.
Vann KR, Pal D, Morales GA, Burgoyne AM, Durden DL, Kutateladze TG. Design of thienopyranone-based BET inhibitors that bind multiple synthetic lethality targets. Sci Rep. 2020;10(1):12027. https://doi.org/10.1038/s41598-020-68964-6.
Burgoyne AM, Vann KR, Joshi S, Morales GA, Vega FM, Singh A, et al. A triple action CDK4/6-PI3K-BET inhibitor with augmented cancer cell cytotoxicity. Cell Discov. 2020;6:49. https://doi.org/10.1038/s41421-020-0181-z.
Pal D, Vann KR, Joshi S, Sahar NE, Morales GA, El-Gamal D, et al. The BTK/PI3K/BRD4 axis inhibitor SRX3262 overcomes Ibrutinib resistance in mantle cell lymphoma. iScience. 2021;24(9):102931. https://doi.org/10.1016/j.isci.2021.102931.
Morales GA, Garlich JR, Durden DL. Single molecule compounds providing multi-target inhibition of btk and other proteins and methods of use thereof. Patent number WO2020023340A1. 2020.
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–4. https://doi.org/10.1182/blood-2010-03-275305.
Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Research Support, Non-U.S. Gov’t. Cell. 2012;149(1):214–31. https://doi.org/10.1016/j.cell.2012.02.013.
Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–74. https://doi.org/10.1073/pnas.1108190108.
Acknowledgements
We thank Muamera Zulcic for help with experiments, Donald Durden for providing the compounds and discussion, and Guillermo Morales for discussion.
Code availability
Not applicable.
Funding
This work was supported in part by grants from NIH HL151334 and CA252707 to T.G.K. and CA215651 supporting D.P., and institutional funds to D.E.
Author information
Authors and Affiliations
Contributions
K.R.V., D.P., A.L.S., N.S., and M.K. performed experiments and together with D.E. and T.G.K. analyzed the data. K.R.V., D.E. and T.G.K. wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Human samples were used following a protocol approved by the institutional review board at University of Nebraska Medical Center (UNMC). Primary mouse samples were obtained through an approved institutional animal care and use committee protocol at UNMC.
Consent for publication
Not applicable.
Competing interests
Corresponding author, Tatiana G. Kutateladze, is a member of the Editorial Board for Molecular Biomedicine and was not involved in the review or decisions related to this manuscript. Other authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure 1.
Anti-proliferative activity of Granta cells treated with increasing concentrations of SRX3305 or Ibrutinib. Supplementary Figure 2. SRX3305 inhibits BTK/PI3K signaling. Supplementary Figure 3. qRT-PCR analysis of cMYC and HEXIM1 expression levels in JeKo-1 cells treated with DMSO, 1 μM PLX51107 (BRD4 inhibitor) or 0.5 μM SRX3305. Supplementary Figure 4. SRX3305 induces apoptosis in JeKo-1 cells. Supplementary Figure 5. SRX3305 induces apoptosis in Mino cells. Supplementary Figure 6. Cell cycle arrest in Jeko-1 and Mino cells. Supplementary Figure 7. Uncropped western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vann, K.R., Pal, D., Smith, A.L. et al. Combinatorial inhibition of BTK, PI3K-AKT and BRD4-MYC as a strategy for treatment of mantle cell lymphoma. Mol Biomed 3, 2 (2022). https://doi.org/10.1186/s43556-021-00066-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-021-00066-9