
Abstract
Background
The coronavirus disease 2019, also known as COVID-19, imposed various challenges to healthcare and became a pandemic accompanied by a high rate of mortality. This infection has many manifestations and affects nearly all body systems. The circulatory and coagulation systems also seem to be affected. Studies show elevated rates of thrombotic events within COVID-19 patients such as disseminated intravascular coagulation (DIC), deep vein thrombosis (DVT), and pulmonary embolism (PE). Incidences of such coagulopathies were correlated to poor patient prognosis and mortality. Given the importance, complication, and mortality caused by thrombotic events (TEs) in COVID-19 patients, the goals of this study are to collect and analyze data on coagulopathy in COVID-19 patients and the pathophysiology and molecular events behind it. We also aim to bring attention to the role of d-dimer in COVID-19 infection by presenting the most recent information available from research studies evaluating d-dimer as a potential biomarker for disease severity, as well as mortality in COVID-19 patients.
Main body
Various mechanisms are described for COVID-19 coagulopathies such as endothelial cell dysfunction, fibrinolysis inhibitor overexpression, immuno-thrombosis, and imbalance between pro- and anticoagulants, to name a few. d-dimer which is a degradation product of fibrin is a helpful diagnostic tool for the assessment of clots and thrombosis. Given the pro-thrombotic nature of COVID-19 infection, within the current narrative review, we studied the diagnostic value of d-dimer for PE prediction. Several studies utilized d-dimer as a predictive tool for detecting PE, and the results were varied. Different cutoff points are proposed ranging from 0.5 up to over 4 mg/L with varying sensitivity and specificity. Although CT pulmonary angiography (CTPA) is the standard model for the prediction of PE, radiation exposure, contrast nephropathy, higher cost, and lack of adequate access can shift our diagnosis into models based on d-dimer.
Short conclusion
In summary, various coagulopathies have been associated with COVID-19 infection, and a safe and early diagnosis is needed. d-dimer showed various successes in PE prediction and can be a good candidate for further research and diagnostic model and algorithm development.
Similar content being viewed by others


Introduction
Acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which is a part of the coronavirus family and is known as the cause of the coronavirus disease 2019 (COVID-19) infection, characterized by a highly contagious respiratory infection rapidly spread across countries and in March 2020, was announced as a worldwide viral pandemic by the World Health Organization (WHO) [1,2,3,4]. COVID-19 patients presented with a host of symptoms including fatigue, dyspnea, anosmia, cough, arthralgia, fever, chest pain, sore throats, and gastrointestinal symptoms [5, 6]. Respiratory failure, acute respiratory distress syndrome (ARDS), super-infection, acute hepatic, renal and cardiac damage, shock, fatal multi-organ dysfunction, and hypoxic encephalopathy are among the complications of severe COVID-19 infection [7,8,9].
Given the hypoxemic, ischemic, and inflammatory nature of COVID-19, many different systems can be affected by the infection such as the nervous [10], endocrine [11], gastrointestinal [12], immune [13], renal [14], and cardiovascular systems [15]. Cardiovascular damages including myocarditis, cardiac arrhythmias, endothelial cell damage, myocardial interstitial fibrosis, and thrombotic events (TE) are observed and reported within some of the COVID-19 patients [16]. The coagulation and the hemostatic pathways—as a part of the cardiovascular system—are not immune to COVID-19 damage, with many reports of coagulopathy being described in COVID-19 patients [17]. Increased levels of d-dimer and factor VIII in COVID-19 patients are described in studies as a sign of abnormal thrombotic parameters and display hemostatic system dysfunction [17].
Thrombotic events have been reported to be common in COVID-19 patients, especially in critically ill patients admitted to the intensive care unit (ICU), despite receiving prophylactic anticoagulants. Acute pulmonary embolism (PE), ischemic stroke, and deep vein thrombosis (DVT) are among common coagulopathies in COVID-19 patients, affecting about one-third of the ICU patients, and are attributed to poor patient prognosis [18,19,20,21,22,23,24]. Systematic studies report a 74% increase in mortality in patients experiencing thrombotic event (TE) in the forms of DVT, PE, and arterial TE during the active COVID-19 infection [25].
Given the importance, complication, and mortality caused by TEs in COVID-19 patients, the goals of this study are to collect and analyze data on coagulopathy in COVID-19 patients and the pathophysiology and molecular events behind it. We also aim to bring attention to the role of d-dimer in COVID-19 infection by presenting the most recent information available from research studies evaluating d-dimer as a potential biomarker for disease severity, as well as mortality in COVID-19 patients.
Main text
COVID-19’s pathogenesis
The initial wave of COVID-19 studies emphasized the cellular and molecular basis of phenotypes and virus-body interactions using omics technologies. The results did provide us with valuable information on pathways and mechanisms focused on certain cell types. Studies then aimed to highlight the innate immunological responses and mechanisms which govern protection and disease in COVID-19 [26, 27].
In the healthy alveoli of the lung, epithelial cells which are divided into type I and II pneumocytes are parts of the blood-alveolar barrier where gas exchange takes place, and a substance called surfactant is secreted to reduce local surface tension. Innate immune responses in the lungs are provided by alveolar macrophages (AM), and they are responsible for the maintenance of immune surveillance and responses through phagocytosis and antigen presentation [28].
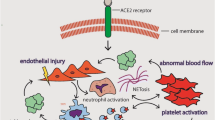
Following the attachment of the viral spike to the cellular membrane receptor-angiotensin-converting enzyme 2 (ACE2), cleavage and membrane fusion takes place. Viral mRNA is expressed, and following maturation, viruses are released from the cells [29]. Subsequent to pathogen infection, monocytes, neutrophils, and natural killer (NK) cells are recruited to provide an early innate immune reaction in response to inflammatory cytokines released by infected local epithelial and immune cells. Innate immune cells then phagocyte infected cells, release further inflammatory cytokine, and activate adaptive immune response via antigen processing [30].
COVID-19 has been shown to be a state of neutrophilia [31]. Neutrophils are recruited to interstitial spaces upon chemokine gradient. Alongside their phagocytic abilities and removal of infected cells, they appear to exert a particular process called NETosis, in which they produce extracellular traps within the inflammatory vessels of the lung and are believed to restrict further viral release. The NETosis process can also cause further endothelial damage and increased capillary permeability which can result in pulmonary edema [32, 33]. While AMs and inflammatory monocytes help viral clearance via Fc receptor cellular phagocytosis, M1 macrophages which are derived from monocyte during inflammation can also cause pulmonary damage by secreting increased levels of IL-6, nitric oxide synthase, tumor necrosis factor (TNF), and matrix metallopeptidases [21].
Infected epithelial cells were also shown to produce enhanced levels of IL-8 and Il-6 during active COVID-19 infection. Il-8 is a chemoattractant for both neutrophils and T cells. The activated cytotoxic T cells are important for fighting the infection, nonetheless can cause lung injury in the case of severe types of COVID-19 infection. In addition, T cells face exhaustion and have decreased functional diversity which is another contributing factor to the disease’s poor prognosis and pathophysiology [29]. Another characteristic of COVID-19 infection, particularly in those infected with severe forms, is the increased plasma concentrations of pro-inflammatory cytokines. The most involved and studied cytokines in COVID-19 pathogenesis include IL-6, IL-8, IL-17, IL-1β, and TNF. Excessive cytokine release could result in a hyper-inflammatory state called cytokine release syndrome (CRS). CRS can result in lethal pneumonia, multiple organ failure, and ARDS and is usually derived from viral damage or hyper-filtration and hyper-function of granulocyte within infected tissue [29, 34].
The inflammatory cytokines, especially IL-6, mediate signals that increase capillary permeability and leakage, which are responsible for lung failure and hypotension in ARDS patients. Also, it has been shown that pulmonary fibrosis is a pathology caused by cytokines such as IL-6 and transforming growth factor (TGF-β) and also M2 macrophages during pulmonary infections [35, 36]. Upregulation of IL‐6 and TNF‐α within SARS‐CoV‐2 infection can also activate nuclear factor‐κB (NF‐κB) through pattern‐recognition receptors (PRR). NF‐κB is capable of inducing IL‐6 amplifier which results in further pro-inflammatory cytokine and chemokine production, including IL-8, IL‐6, vascular endothelial growth factor (VEGF), and monocyte chemoattractant protein 1 (MCP‐1) [37]. STAT3 is also another transcription factor increased within COVID-19 via IL-6 and angiotensin II and can reduce lymphopoiesis, expression of E-cadherin, and antiviral responses which include interferon-mediated signaling, the activity of NK cells and helper T and cytotoxic CD8 cells. Thus, activation of STAT3 causes hyper-inflammation, viral persistence, vascular leak, and lung fibrosis [35].
In summary, COVID-19 pathophysiology is complex and mostly immune-based. Upon viral attachment and fusion via angiotensin receptors, the innate immune response is initiated via local and humoral granulocytes. Thereafter, various cytokines produced by infected and immune cells cause a hyper-inflammatory state, which then results in pulmonary edema, tissue injury, and finally lung fibrosis.
Thrombosis and D-dimer
Rudolph Virchow was the first to explain the relationship between three critical components that lead to the development of thrombosis in the nineteenth century. These include endothelial injury, stasis of blood flow, and hypercoagulability. However, the pathophysiology is beyond the imbalance between platelets, coagulation factors, endogenous anticoagulants, and the fibrinolytic system. Thrombosis can be divided into venous and arterial types. Arterial thrombi can cause myocardial infarction, and venous thrombi may lead to pulmonary embolism and venous thromboembolism. In arterial thrombosis, platelet-rich thrombi are developed around atherosclerotic plaques which are ruptured and injured epithelium under intense pressure whereas venous thrombi are formed in areas with intact endothelium walls and lower pressure and flow. These clots are rich in fibrin and withhold activated platelets and erythrocytes [38].
d-dimer is a product of soluble fibrin that results from the decomposition and degradation of vascular clots through fibrinolysis. This molecule which is made up of two fragments of fibrin exhibits distinctive properties as a biomarker of hemostatic abnormalities as well as an indicator of intravascular thrombosis namely VTE, PE, DVT, and DIC [39, 40]. The d-dimer level is increased whenever a thrombosis or a DIC takes place due to activation of the coagulation system and is an indicator of destroyed fibrin being present in the bloodstream [40, 41].
Active malignancy causes a variable enhancement in d-dimer, which implies a higher risk of thrombosis. Following anticoagulation for a thrombotic episode, an elevated d-dimer implies a higher risk of recurrent thrombosis. There is also a notable increase of d-dimer within infections and sepsis which has been correlated with poor clinical outcomes. An elevated d-dimer associates with an escalated risk for incidental and recurrent VTE and thrombotic-related mortality. Although d-dimer levels can be increased in non-thrombotic pathological conditions, it still remains a sensitive yet specific marker of vascular thrombosis [42]. d-dimer also correlates significantly with the recurrence of VTE among both carriers and non-carriers of thrombophilia [43]. Taken together, thrombosis can lead to various complications and is divided into platelet-rich arterial and fibrin-rich venous thrombi. d-dimer which is a product resulting from fibrinolysis is elevated in TEs and can be a biomarker for a variety of disorders.
Molecular coagulopathy mechanism in COVID-19 patients
Viral infections have been demonstrated to induce pro-thrombotic states through modulating several coagulation proteins. SARS-CoV-1 and a bunch of other viral respiratory infections increase intravascular thrombi and fibrin accumulation in the lung [44]. Results of a retrospective study recognized VTE in 35 of 74 ICU patients (11 with PE) in spite of being on thrombosis prophylaxis [45].
In a retrospective cohort study of 119 H1N1 cases, nearly 6% of patients had vascular thrombotic events—either within arteries or veins—and the proposed mechanism can be explained through altered pro- and anticoagulant factor ratios, dysfunctional vascular endothelium, immobility stasis, and increased platelet activation [46]. d-dimer is produced within the blood as a result of the degradation and decomposition of stabilized fibrin polymer (fibrin cross-linked with factor XIII) by plasmin. That is, the thrombus or the clot is formed in the body through coagulation activation and is decomposed by fibrinolytic mechanisms. Even though many studies have indicated that elevated levels of d-dimer are attributed to the severity of the thrombosis, if a large amount of thrombi forms in the vessels but is not dissolved, the increase in d-dimer may be mild. Specifically, the increase in d-dimer that represented the suppressed fibrinolytic-type DIC, which is caused by sepsis, is relatively mild even in severe cases that result in death [47, 48].
DIC is another common complication of COVID-19 patients and increases mortality significantly [49]. In patients with viral infections like COVID-19 infection, sepsis-induced DIC is interrelated with organ failure. Microvascular thrombosis of extra-pulmonary organs has also been reported and may explain the acute onset of multi-organ failure [50]. Although DIC is usually characterized by intravascular coagulation and loss of localization, COVID-19–caused DICs seem to differ. Common findings of DIC are increased d-dimer, prolonged coagulation times namely PT, decreased platelet counts, and coagulation inhibitors concentration in the serum. Notwithstanding, COVID-19 patients go through a pro-thrombotic state without a consumption coagulopathy alongside increased venous thromboembolism [51]. The main characteristic of COVID-19 coagulopathy is a pronounced elevation of d-dimer without platelet reduction or prolongation of coagulation times, which suggests a process of generation of thrombin and a local rather than systemic fibrinolysis [52].
COVID-19-associated coagulopathy (CAC) has emerged as a hallmark of disease severity in ICU patients [53]. Excessive enhancement of d-dimer and mild increase in PT time is commonly seen in COVID-19 patients. Thrombocytopenia is another hemostatic dysfunction in COVID-19 patients, but unlike other severe infections is not significantly related to mortality. Although coagulation factors are normal in COVID-19 patients, fibrinogen plasma concentration can significantly increase owing to acute phase response. Several studies indicated abnormal clotting in COVID-19 patients and enhanced viscoelastic parameters [51].
COVID-19—as discussed previously—is a hyper-inflammatory state with upregulation of several inflammatory cytokines including IL-1 and IL-6 and TNF [54]. Inflammation activates endothelial cells, platelets, monocytes, and tissue factors, altering fibrinolytic and endogenous anticoagulant pathways such as thrombomodulin, proteins C and S, and tissue factor pathway inhibitor (TFPI) [55]. Thrombin generation is usually a consequence of factor expression on macrophages and monocyte caused by IL-6 induction. The significant release of plasminogen activators caused by inflammation-driven endothelial cell disturbance can also explain COVID-19 coagulopathy [51]. Generally, an increase in d-dimer is believed to reflect the activation of both fibrinolysis and coagulation in vivo [47, 48]. Oppositely, one report has discussed the d-dimer origin of COVID-19. The paper suggests that d-dimer elevation reflects fibrin byproduct degradation accumulating within the lung parenchyma and the alveoli and thus causing lung damage [56].
Tissue hypoxia—often observed in severe COVID-19—increases thrombosis via inducing transcription factors, namely fibrinolysis inhibitors. For instance, thrombin-activatable fibrinolysis inhibitor (TAFI) and plasminogen activator inhibitor 1 (PAI-1) are among the stimulated transcription factors which can increase the risk of thrombi formation in COVID-19 patients [57].
Another mechanism of microvascular thrombosis in COVID-19 is endothelial activation which is caused by viral penetration of endothelial cells via ACE receptors [58]. microvascular thrombosis has been also reported following ARDS autopsies. Common findings include thickening of the vascular wall and lumen stenosis and also the formation of platelet-rich microthrombi, especially within the lungs which could be accompanied by hemorrhage or granulocyte accumulation [51].
Finally, immune thrombosis is also described in COVID-19 patients as coagulopathy caused by immune cells such as neutrophils [59]. As previously discussed, infections such as COVID-19 can induce neutrophils to produce NETs via inflammatory mediators, Toll-like receptors, or immunoglobulin production. NETs contain platelets, coagulation factors, and endothelial cells which are needed for coagulation. Also, two key components of NETs, namely citrullinated histone H3 and genomic DNA, are believed to induce coagulation and thrombin production. Histone and enzymes released by NETs can induce endothelial apoptosis and dysfunction which can contribute to thrombi formation [60].
In summary, various coagulopathies have been demonstrated within COVID-19 patents. Increased TEs are often characterized by increased d-dimer, thrombocytopenia, and increased PT time. Various mechanisms are involved including endothelial dysfunction, pro- and anticoagulant imbalance, enhanced platelet activation, inflammation-caused thrombosis, and immune thrombosis (Fig. 1).

A summary of the relationship between COVID-19 infection and clot formation
Pulmonary CT angiography for pulmonary thromboembolism
The standard criteria for COVID-19 confirmation are based on microbiological tests including reverse transcription polymerase chain reaction (RT-PCR) and sequencing. However, in urgent care, these tests may be unavailable [61]. Since one of the main clinical presentations and manifestations of COVID-19 is coagulopathy and PE, laboratory confirmation and imaging are needed for confirmation, immediate care, and anti-coagulant treatment initiation [62, 63].
There are several imaging techniques available to assess acute PE. Chest radiography, CTPA, CT venography (CTV), magnetic resonance pulmonary angiography (MRPA), nuclear medicine ventilation/perfusion scan, venous ultrasonography, echocardiography, and catheter pulmonary angiography are among the utilized imaging modalities [64,65,66].
The precise role of CT imaging within COVID-19 management is still debated; nonetheless, current guidelines recommend using non-contrast chest CT for diagnosis, assessment of severity, and monitorization of COVID-19 [67]. Chest CT scan plays an essential role in optimizing the management of COVID-19 patients, excluding alternative diagnoses and additional pathologies, especially by acute PE [68].
The preferred imaging modality for PE evaluation in suspected patients is CTPA, which is also a key component widely used in clinical diagnostic algorithms. The Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED) II trial indicated a high accuracy of CTPA, with a sensitivity of 83% and a specificity of 96%. When clinical data were included, the positive predictive value even increased to 96% [64]. In CTPA, acute PE is shown as low-density defects within vessel perfusion, which can be surrounded by opacified blood flow or a totally filling defect with unspecified blood flow. Other findings can include vascular remodeling, oligemia, plate-like atelectasis, pleural effusion, and parenchymal infarction [69]. The result of a retrospective observational study in Madrid indicated that pulmonary angiography with multi-detector computed tomography (MDCT), as well as iodine mapping, can demonstrate PTE and hypo-perfusion in COVID-19 patients. Several visual properties were described in the study to help PTE diagnoses such as crazy paving patterns, consolidation, septal thickening, or bronchiectasis [21]. A study by Silva et al. showed that different algorithms for PE detection can increase diagnosis specificity which then results in CTPA use reduction. When they employed the YEARS and PEFeD algorithms which are based on d-dimer levels and clinical variables, a 19% reduction in CPTA utilization was seen [70]. A meta-analysis showed a pooled sensitivity of 97% and specificity of 41% of d-dimer levels for PE diagnosis, whereas CTPA had a sensitivity of 94% and specificity of 98%. It is worth mentioning that such a comparison is made in non-COVID-19 patients, and the results in COVID-19 patients may vary [71]. The study of Ramadan et al. reported that 37% of CTPA were inconclusive for PTE rule out, and there might have been some missed clots. Even without the presence of thromboembolism visualized by CTPA, a large amount of microthrombi still could be present within the patient pulmonary vasculature [72].
Patients with considerably elevated d-dimer levels upon admission (2000–4000 g/mL) or notable d-dimer increases throughout the hospital stay are indicated for a CTPA. Dual-energy CT could help evaluate lung perfusion within COVID-19 patients both in the acute setting and to monitor lung sequelae in subsequent scans [73].
Due to immoderate radiation exposure, viable contrast reactions, and high overall costs, it would be better to prevent CTPA as possible [74]. Considering the mounted studies mentioned in the previous parts, plasma d-dimer levels may be used as an alternative for PE prediction or patient risk assessment prior to or for CTPA.
D-dimer as a biological marker for mortality and disease severity within COVID-19 patients
d-dimer, a degradation product of fibrin, is a small protein particle that is present in the blood after the thrombus is broken down by fibrinolysis. Measurement of circulating d-dimer levels is a sensitive tool in clinical settings for the diagnosis of PE and DIC [75]. In addition, underlying disorders such as cancer, stroke, diabetes, and pregnancy can cause elevated d-dimer levels in patients with COVID-19 [40]. Therefore, elevated d-dimer levels in COVID-19 patients rapidly identify patients with high disease severity, pulmonary complications, and risk of thromboembolism within veins as part of a pro-thrombotic condition. An original study in Spain reported that a higher threshold (2903 ng/mL) for d-dimer may predict the risk of PE in COVID-19 patients with a sensitivity of 81% [76]. Another retrospective study in Spain analyzed the value of d-dimer to assess CTPA for diagnosis of PE in COVID-19 patients. They concluded that d-dimer levels higher than 2.00 mg/L could be a sensitive cutoff point for ruling out PE within hospitalized COVID-19 patients. d-dimer increase of 4.00 mg/L since patient admission is useful for the detection of PE [77]. A meta-analysis study in 2021 evaluated the diagnostic value and accuracy of d-dimer within COVID-19. The results showed a pooled sensitivity of 77% of d-dimer for disease severity (95% CI: 73–80%), 75% for mortality (95% CI: 65–82%), and 90% for DVT prediction (95% CI: 90–90%). The specificity was found to be 71% (95% CI: 64–77%), 83% (95% CI: 77–87%), and 60% (95% CI: 60–60%), respectively [78].
Another Spanish study screened patients with d-dimer above 1 mg/L for asymptomatic DVT using Doppler ultrasound. Their results showed higher d-dimer levels within DVT patients (4.527 vs 2050 mg/L). They concluded a cutoff point of approximately 1.5 mg/L had a sensitivity and specificity of 95.7% and 29.3%, respectively. Positive predictive value (PPV) and negative predictive value (NPV) were reported to be 19% and 97.5%, respectively [79]. A study conducted by Cui et al. showed that when a d-dimer cutoff point of 1.5 mg/L is employed, VTE prediction is obtained with 85% sensitivity, 88.5% specificity, and 94.7 NPV [80].
Another retrospective study performed on 697 COVID-19 patients aimed to asses PTE, d-dimer, and CTPA. About a third of admitted patients experienced PTE and had significantly and notably higher d-dimer levels. Their results indicated a d-dimer cutoff of 0.5 mg/L has a specificity of 5.7% and sensitivity of 98.2% for the presence of PTE [81]. Artifoni et al. investigated the diagnostic value of d-dimer for VTE and PE within 79 COVID-19 patients. Lower limb duplex ultrasound and CTPA were used for confirmation of VTE and PE, respectively. They showed a significant increase in d-dimer within DVT patients. d-dimer cutoff point at 1 mg/L had an NPV of 90% and 98% for VTE and PE, respectively. The PPV for VTE was 44% which increased up to 67% if the cutoff is considered at 3 mg/L [82]. Leonard-Loranat et al. also investigated a study with the same purpose. The confirmation was done by a CT angiography. Their findings support that a d-dimer level above 2.6 mg/L is highly suggestive of PE with a sensitivity of 100% and specificity of 67% [83]. The cutoff point of 2.6 mg/L in another study had a sensitivity of 89.7% and a specificity of 59.5% [84]. Lastly, a study performed by Ventuora-Diaz et al. revealed that a cutoff of 2.903 was optimal for PE detection with sensitivity and specificity of 81% and 59%, respectively [76].
A few other studies on d-dimer and PE prediction are worth mentioning. Tuck et al.’s study showed a sensitivity of 81% and a specificity of 70% when a 1.5 mg/L cutoff is employed. When the cutoff was increased to 2 mg/L, sensitivity was reduced by 1% but specificity increased by 6%, which made authors recommend higher d-dimer thresholds for PE exclusion [85]. Another study on 3583 COVID-19 patients and a cohort of 13,091 patients using 2 mg/L cutoff revealed a sensitivity of 70.3% and 70.5%, specificity of 82.4% and 67.8%, and NPV of 98.5% and 99.5%, respectively. However, when the cutoff was reduced to 0.5 mg/L, sensitivity increased to 99.3% and 92% and NPV increased to 99.9% and 99.5%, but specificity dropped to 34.3% and 17%, respectively [86]. Ten percent of CTPAs could have been avoided as a result of two studies with d-dimer lower than 0.5 mg/L, with 98.3% and 98.2% sensitivity, 10.8% and 5.7% specificity, 98.7% and 87.1% NPV, and 8.4% and 33.3% PPV [81, 87]. Other studies showed a 100% and 72% sensitivity and 90.62% and 74% specificity with 2.494 and 2.247 mg/L cutoff, respectively [22, 88]. 2.5 mg/L cutoff had an 80% sensitivity and 51% specificity for PE in a Spanish retrospective study [89]. A complete list of mentioned studies along with a few others is presented in Table 1.
Apart from TE prediction, d-dimer was also shown to be a tool for survival and severity prediction in COVID-19. ICU patients are likely to have higher d-dimer levels than non-critically ill patients (mean plasma levels of 2.4 compared to 0.5 mg/L). d-dimer levels greater than 1 mg/L were observed in 81% of non-survivors, which was found to be 24% in survivors. In another study, it was stated that d-dimer levels greater than 3 mg/L were observed within non-survivor COVID-19 patients [95]. Gudot et al. proposed the use of d-dimer as an ICU referral guide. Their conclusion highlights using a 1 mg/L cutoff with sensitivity and specificity of 72.4% and 60.0%, respectively. PPV and NPV were reported to be 58.6% and 74.2%, respectively [96] (Table 1).
Thus, utilizing d-dimer evaluation can help with risk stratification and the earlier start of treatments that may help reduce COVID-19-related morbidity and mortality [75]. However, the d-dimer level (as a static value or as a trend over time), when viewed alone, limits the performance characteristics of COVID-19 as a prognostic test [97].
Conclusions
The COVID-19 pandemic imposed various challenges for treatment and complication controls. Studies revealed a hyper-inflammatory nature of the diseases which can along with other pathophysiological mechanisms such as endothelial dysfunction and hyper-coagulation cause increased risk of TE within patients. d-dimer—a thrombin protein fragment—is a useful test for coagulation and thrombosis screening. Various studies evaluated the use of d-dimer for COVID-19 TE evaluation, especially PE. Even though the results are varied, mostly suggest a significant increase of d-dimer in patients with VTE and adequate sensitivity with controversial specificity. Given the challenges of CTPA such as cost and contrast iatrogenic complications, d-dimer can be easily and frequently used for VTE risk assessment in COVID-19 patients.
Availability of data and materials
Not applicable.
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- DIC:
-
Disseminated intravascular coagulation
- DVT:
-
Deep vein thrombosis
- PE:
-
Pulmonary embolism
- CTPA:
-
CT pulmonary angiography
- SARS-CoV-2:
-
Acute respiratory syndrome coronavirus 2
- WHO:
-
World Health Organization
- ARDS:
-
Acute respiratory distress syndrome
- ICU:
-
Intensive care unit
- TE:
-
Thrombotic event
- AM:
-
Alveolar macrophages
- ACE2:
-
Angiotensin-converting enzyme 2
- NK:
-
Natural killer
- IL:
-
Interleukin
- TNF:
-
Tumor necrosis factor
- CRS:
-
Cytokine release syndrome
- TGF:
-
Transforming growth factor
- NF‐κB:
-
Nuclear factor‐κB
- PRR:
-
Pattern‐recognition receptors
- VEGF:
-
Vascular endothelial growth factor
- MCP-1:
-
Monocyte chemoattractant protein-1
- CAC:
-
COVID-19-associated coagulopathy
- TFPI:
-
Tissue factor pathway inhibitor
- TAFI:
-
Thrombin-activatable fibrinolysis inhibitor
- PAI-1:
-
Plasminogen activator inhibitor-1
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- CT:
-
Computed tomography
- CTV:
-
CT venography
- MRPA:
-
Magnetic resonance pulmonary angiography
- PIOPED:
-
Prospective investigation of pulmonary embolism diagnosis
- MDCT:
-
Multi-detector computed tomography
- PPV:
-
Positive predictive value
- NPV:
-
Negative predictive value
References
Lone SA, Ahmad A (2020) COVID-19 pandemic–an African perspective. Emerg Microbes Infect 9(1):1300–8
Hasöksüz M, Kilic S, Sarac F (2020) Coronaviruses and SARS-CoV-2. Turkish J Med Sci 50(9):549–56
Eslamzadeh M, Bordbar MRF, Ghalibaf AM, Modaresi F, Emadzadeh M, Farhoudi F (2022) The role of personality traits in following quarantine orders during the COVID-19 pandemic. Int Clin Psychopharmacol 37(4):173
Dadgarmoghaddam M, Najafi MN, Ebrahimi A, Talaei A, Najafnajafi N, Ghalibaf AM. Social mental health during COVID-19 pandemic in Iran: a cross-sectional study among the general population of Razavi Khorasan Province, Iran. Iran Red Crescent Med J. 2021;23(11). https://ircmj.com/index.php/IRCMJ/article/view/1010
Fernández-de-las-Peñas C, Palacios-Ceña D, Gómez-Mayordomo V, Florencio LL, Cuadrado ML, Plaza-Manzano G et al (2021) Prevalence of post-COVID-19 symptoms in hospitalized and non-hospitalized COVID-19 survivors: a systematic review and meta-analysis. Eur J Intern Med 92:55–70
Shakeri M, Ghalibaf AA, Ghodsi-Moghadam M, Ghorbannezhad G, Ahmadi SP, Mashahiri S, et al. Clinical manifestations and associated mortality factors of COVID-19: a large population-based study in the northeast of Iran during 2020-2021. Epidemiol Health Syst J. 2023;10(1):31-8
SeyedAlinaghi S, Afsahi AM, MohsseniPour M, Behnezhad F, Salehi MA, Barzegary A, et al. Late complications of COVID-19; a systematic review of current evidence. Archives of academic emergency medicine. 2021;9(1). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7927752/
AkbariRad M, Allahyari A, Ravanshad S, Mahali SN, Ebrahimzadeh F, Mozdourian M et al (2022) Clinical characteristics of 200 COVID-19 patients in Mashhad, Iran, February and March 2020. J Fam Med Prim Care 11(5):1949
Saeedi N, Gohari NSF, Ghalibaf AAM, et al (2023) COVID-19 infection: a possible induction factor for development of autoimmune diseases? Immunol Res 71:547–553. https://doi.org/10.1007/s12026-023-09371-7
Emmi A, Boura I, Raeder V, Mathew D, Sulzer D, Goldman JE et al (2022) COVID-19, nervous system pathology, and Parkinson’s disease: bench to bedside. Int Rev Neurobiol 165:17–34
Pal R, Banerjee M (2020) COVID-19 and the endocrine system: exploring the unexplored. J Endocrinol Investig 43(7):1027–31
Gulen M, Satar S (2020) Uncommon presentation of COVID-19: gastrointestinal bleeding. Clin Res Hepatol Gastroenterol 44(4):e72–e6
Tufan A, Güler AA, Matucci-Cerinic M (2020) COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turkish J Med Sci 50(9):620–32
Kaye AD, Okeagu CN, Tortorich G, Pham AD, Ly EI, Brondeel KC et al (2021) COVID-19 impact on the renal system: pathophysiology and clinical outcomes. Best Pract Res Clin Anaesthesiol 35(3):449–59
Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol 17(5):259–60
Farshidfar F, Koleini N, Ardehali H. Cardiovascular complications of COVID-19. JCI Insight. 2021;6(13). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8410051/
Martín-Rojas RM, Pérez-Rus G, Delgado-Pinos VE, Domingo-González A, Regalado-Artamendi I, Alba-Urdiales N et al (2020) COVID-19 coagulopathy: an in-depth analysis of the coagulation system. Eur J Haematol 105(6):741–50
Klok FA, Kruip MJH, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145–7
Hanff TC, Mohareb AM, Giri J, Cohen JB, Chirinos JA (2020) Thrombosis in COVID-19. Am J Hematol 95(12):1578–89
Vlachou M, Drebes A, Candilio L, Weeraman D, Mir N, Murch N et al (2021) Pulmonary thrombosis in COVID-19: before, during and after hospital admission. J Thromb Thrombolysis 51(4):978–84
Dueñas P, Krauel A, Rojas A, MT RP, Izquierdo D, de la Guardia L, Sánchez T (2020) Blue lungs in Covid-19 patients: a step beyond the diagnosis of pulmonary thromboembolism using MDCT with iodine mapping. Archivos de bronconeumologia 57:35–46
Ooi M, Rajai A, Patel R, Gerova N, Godhamgaonkar V, Liong S (2020) Pulmonary thromboembolic disease in COVID-19 patients on CT pulmonary angiography–prevalence, pattern of disease and relationship to D-dimer. Eur J Radiol 132:109336
Suarez Castillejo C, Toledo-Pons N, Calvo N, Ramon-Clar L, Martínez J, Hermoso de Mendoza S et al (2022) A prospective study evaluating cumulative incidence and a specific prediction rule in pulmonary embolism in COVID-19. Front Med 9:936816
Tirumani SH, Rahnemai-Azar AA, Pierce JD, Parikh KD, Martin SS, Gilkeson R et al (2021) Are asymptomatic gastrointestinal findings on imaging more common in COVID-19 infection? Study to determine frequency of abdominal findings of COVID-19 infection in patients with and without abdominal symptoms and in patients with chest-only CT scans. Abdom Radiol 46(6):2407–14
Malas MB, Naazie IN, Elsayed N, Mathlouthi A, Marmor R, Clary B (2020) Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: a systematic review and meta-analysis. EClinicalMedicine 29–30:100639
Sette A, Crotty S (2021) Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184(4):861–80
Miri M, Badriahmadi S, Shamshirian A, Ghalibaf AM, Mozdourian M. Electrolyte imbalance and COVID-19 severity in hospitalized patients. Nephro-Urol Monthly. 2022(In Press). https://brieflands.com/articles/num-128085.html
Paludan SR, Mogensen TH (2022) Innate immunological pathways in COVID-19 pathogenesis. Sci Immunol 7(67):eabm5505
Yuki K, Fujiogi M, Koutsogiannaki S (2020) COVID-19 pathophysiology: a review. Clin Immunol 215:108427
Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW et al (2017) Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol 10(6):1529–41
McKenna E, Wubben R, Isaza-Correa JM, Melo AM, Mhaonaigh AU, Conlon N et al (2022) Neutrophils in COVID-19: not innocent bystanders. Front Immunol 13:864387
Reusch N, De Domenico E, Bonaguro L, Schulte-Schrepping J, Baßler K, Schultze JL, et al. Neutrophils in COVID-19. Front Immunol. 2021;12. https://www.frontiersin.org/articles/10.3389/fimmu.2021.652470/full
Alon R, Sportiello M, Kozlovski S, Kumar A, Reilly EC, Zarbock A et al (2021) Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat Rev Immunol 21(1):49–64
Darif D, Hammi I, Kihel A, El Idrissi Saik I, Guessous F, Akarid K (2021) The pro-inflammatory cytokines in COVID-19 pathogenesis: what goes wrong? Microb Pathog 153:104799
Jafarzadeh A, Nemati M, Jafarzadeh S (2021) Contribution of STAT3 to the pathogenesis of COVID-19. Microb Pathog 154:104836
Karizi Sr, Armanmehr F, Azadi HG, Zahroodi HS, Ghalibaf AM, Bazzaz BSF et al (2023) A randomized, double‐blind placebo‐controlled add‐on trial to assess the efficacy, safety, and anti‐atherogenic effect of Spirulina platensis in patients with inadequately controlled type 2 diabetes mellitus. Phytother Res 37(4):1435–48
Hu B, Huang S, Yin L (2021) The cytokine storm and COVID-19. J Med Virol 93(1):250–6
Koupenova M, Kehrel BE, Corkrey HA, Freedman JE (2017) Thrombosis and platelets: an update. Eur Heart J 38(11):785–91
Johnson ED, Schell JC, Rodgers GM (2019) The D-dimer assay. Am J Hematol 94(7):833–9
Rostami M, Mansouritorghabeh H (2020) D-dimer level in COVID-19 infection: a systematic review. Exp Rev Hematol 13(11):1265–75
Li J, Liu Z, Wu G, Yi M, Chen Y, Li K et al (2020) D-Dimer as a prognostic indicator in critically ill patients hospitalized with COVID-19 in Leishenshan Hospital, Wuhan China. Front Pharmacol 11:600592
Bates SM, editor D-dimer assays in diagnosis and management of thrombotic and bleeding disorders. Seminars in thrombosis and hemostasis; 2012: Thieme Medical Publishers.
Palareti G, Legnani C, Cosmi B, Valdré L, Lunghi B, Bernardi F et al (2003) Predictive value of D-dimer test for recurrent venous thromboembolism after anticoagulation withdrawal in subjects with a previous idiopathic event and in carriers of congenital thrombophilia. Circulation 108(3):313–8
Samudrala PK, Kumar P, Choudhary K, Thakur N, Wadekar GS, Dayaramani R et al (2020) Virology, pathogenesis, diagnosis and in-line treatment of COVID-19. Eur J Pharmacol 883:173375
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MC et al (2020) Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemostasis 18(8):1995–2002
Paul E, Bunce P, Sasha M (2011) Pandemic H1N1 influenza infection and vascular thrombosis Clin. Infect Dis 52(2):e14
Asakura H, Takahashi H, Uchiyama T, Eguchi Y, Okamoto K, Kawasugi K et al (2014) DIC subcommittee of the Japanese Society on Thrombosis and Hemostasis. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care. 2:20
Asakura H (2014) Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care 2(1):1–7
Arachchillage DR, Laffan M (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemostasis 18(5):1233
Zhang T, Sun L, Feng R (2020) Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019. Zhonghua jie he he hu xi za zhi= Zhonghua Jiehe he Huxi Zazhi = Chines J Tuberculosis Respir Dis. 43(6):496–502
Levi M, Iba T (2021) COVID-19 coagulopathy: is it disseminated intravascular coagulation? Intern Emerg Med 16(2):309–12
Gomez-Mesa JE, Galindo-Coral S, Montes MC, Martin AJM (2021) Thrombosis and coagulopathy in COVID-19. Curr Problems Cardiol 46(3):100742
Mucha SR, Dugar S, McCrae K, Joseph D, Bartholomew J, Sacha GL, et al. Update to coagulopathy in COVID-19: manifestations and management. Cleve Clin J Med. 2020. https://www.ccjm.org/content/87/8/461.long
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395(10223):497–506
Levi M, Scully M (2018) How I treat disseminated intravascular coagulation. Blood 131(8):845–54
Hunt BJ, Levi M. Re The source of elevated plasma D‐dimer levels in COVID‐19 infection. Br J Haematol. 2020. https://onlinelibrary.wiley.com/doi/full/10.1111/bjh.16907?sid=nlm%3Apubmed
Sebuhyan M, Mirailles R, Crichi B, Frere C, Bonnin P, Bergeron-Lafaurie A et al (2020) How to screen and diagnose deep venous thrombosis (DVT) in patients hospitalized for or suspected of COVID-19 infection, outside the intensive care units. J Med Vasc 45(6):334–43
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–8
Liu H, Hu T, Zhang C, Chen X, Zhang S, Li M et al (2021) Mechanisms of COVID-19 thrombosis in an inflammatory environment and new anticoagulant targets. Am J Transl Res 13(5):3925–41
Zhu Y, Chen X, Liu X (2022) NETosis and neutrophil extracellular traps in COVID-19: immunothrombosis and beyond. Front Immunol 13:838011
Hani C, Trieu NH, Saab I, Dangeard S, Bennani S, Chassagnon G et al (2020) COVID-19 pneumonia: a review of typical CT findings and differential diagnosis. Diag Interv Imaging 101(5):263–8
Grillet F, Behr J, Calame P, Aubry S, Delabrousse E (2020) Acute pulmonary embolism associated with COVID-19 pneumonia detected with pulmonary CT angiography. Radiology 296(3):E186–E8
Cohen AT, Dobromirski M, Gurwith MM (2014) Managing pulmonary embolism from presentation to extended treatment. Thromb Res 133(2):139–48
Moore AJE, Wachsmann J, Chamarthy MR, Panjikaran L, Tanabe Y, Rajiah P (2018) Imaging of acute pulmonary embolism: an update. Cardiovasc Diagn Ther 8(3):225–43
Abbasi B, Kahani N, Ghalibaf AM, Layegh P, Niroumand S, Akhavan R et al (2022) Evaluating the diagnostic value of multi-detector brain CT angiography in diagnosing acute cerebral venous thrombosis. Sci Rep 12(1):18685
Abbasi B, Seyed Hosseini M, Moodi Ghalibaf A, Akhavan R, Emadzadeh M, Bolvardi E (2022) Evaluating anemia on non-contrast thoracic computed tomography. Sci Rep 12(1):21380
Rotzinger D, Beigelman-Aubry C, Von Garnier C, Qanadli S (2020) Pulmonary embolism in patients with COVID-19: time to change the paradigm of computed tomography. Thromb Res 190:58–9
Revel M-P, Parkar AP, Prosch H, Silva M, Sverzellati N, Gleeson F et al (2020) COVID-19 patients and the radiology department–advice from the European Society of Radiology (ESR) and the European Society of Thoracic Imaging (ESTI). Eur Radiol 30(9):4903–9
Subramaniam R, Blair D, Gilbert K, Sleigh J, Karalus N (2006) Computed tomography pulmonary angiogram diagnosis of pulmonary embolism. Australas Radiol 50(3):193–200
Silva BV, Jorge C, Plácido R, Mendonça C, Urbano ML, Rodrigues T et al (2021) Pulmonary embolism and COVID-19: a comparative analysis of different diagnostic models performance. Am J Emerg Med 50:526–31
Patel P, Patel P, Bhatt M, Braun C, Begum H, Wiercioch W et al (2020) Systematic review and meta-analysis of test accuracy for the diagnosis of suspected pulmonary embolism. Blood Adv 4(18):4296–311
Ramadan L, Koziatek CA, Caldwell JR, Pecoriello J, Kuhner C, Subaiya S et al (2021) Pulmonary thromboembolism in COVID-19: evaluating the role of D-dimer and computed tomography pulmonary angiography results. Am J Emerg Med 46:786–7
Pontone G, Scafuri S, Mancini ME, Agalbato C, Guglielmo M, Baggiano A et al (2021) Role of computed tomography in COVID-19. J Cardiovasc Comput Tomogr 15(1):27–36
Gervaise A, Bouzad C, Peroux E, Helissey C (2020) Acute pulmonary embolism in non-hospitalized COVID-19 patients referred to CTPA by emergency department. Eur Radiol 30(11):6170–7
Paliogiannis P, Mangoni AA, Dettori P, Nasrallah GK, Pintus G, Zinellu A (2020) D-dimer concentrations and COVID-19 severity: a systematic review and meta-analysis. Front Public Health 8:432
Ventura-Díaz S, Quintana-Pérez JV, Gil-Boronat A, Herrero-Huertas M, Gorospe-Sarasúa L, Montilla J et al (2020) A higher D-dimer threshold for predicting pulmonary embolism in patients with COVID-19: a retrospective study. Emerg Radiol 27(6):679–89
Rodriguez-Sevilla JJ, Rodó-Pin A, Espallargas I, Villar-García J, Molina L, Terán PP et al (2020) Pulmonary embolism in patients with COVID-19 pneumonia: the utility of D-dimer. Archivos de bronconeumologia 56(11):758
Zhan H, Chen H, Liu C, Cheng L, Yan S, Li H et al (2021) Diagnostic value of D-dimer in COVID-19: a meta-analysis and meta-regression. Clin Appl Thromb Hemost 27:10760296211010976
Demelo-Rodríguez P, Cervilla-Muñoz E, Ordieres-Ortega L, Parra-Virto A, Toledano-Macías M, Toledo-Samaniego N et al (2020) Incidence of asymptomatic deep vein thrombosis in patients with COVID-19 pneumonia and elevated D-dimer levels. Thromb Res 192:23–6
Cui S, Chen S, Li X, Liu S, Wang F (2020) Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost 18(6):1421–4
Vivan MA, Rigatti B, da Cunha SV, Frison GC, Antoniazzi LQ, de Oliveira PHK et al (2022) Pulmonary embolism in patients with COVID-19 and D-dimer diagnostic value: a retrospective study. Braz J Infect Dis 26(6):102702
Artifoni M, Danic G, Gautier G, Gicquel P, Boutoille D, Raffi F et al (2020) Systematic assessment of venous thromboembolism in COVID-19 patients receiving thromboprophylaxis: incidence and role of D-dimer as predictive factors. J Thromb Thrombolysis 50(1):211–6
Léonard-Lorant I, Delabranche X, Séverac F, Helms J, Pauzet C, Collange O et al (2020) Acute pulmonary embolism in patients with COVID-19 at CT angiography and relationship to d-dimer levels. Radiology 296(3):E189-e91
Maatman TK, Jalali F, Feizpour C, Douglas A 2nd, McGuire SP, Kinnaman G et al (2020) Routine venous thromboembolism prophylaxis may be inadequate in the hypercoagulable state of severe coronavirus disease 2019. Crit Care Med 48(9):e783–e90
Tuck AA, White HL, Abdalla BA, Cartwright GJ, Figg KR, Murphy EN et al (2021) To scan or not to scan - D-dimers and computed tomography pulmonary angiography in the era of COVID-19. Clin Med (Lond) 21(2):e155–e60
Bledsoe JR, Knox D, Peltan ID, Woller SC, Lloyd JF, Snow GL et al (2022) D-dimer thresholds to exclude pulmonary embolism among COVID-19 patients in the emergency department: derivation with independent validation. Clin Appl Thromb/Hemostasis 28:10760296221117996
Revel M-P, Beeker N, Porcher R, Jilet L, Fournier L, Rance B et al (2022) What level of D-dimers can safely exclude pulmonary embolism in COVID-19 patients presenting to the emergency department? Eur Radiol 32(4):2704–12
Nadeem I, Anwar A, Jordon L, Mahdi N, Rasool MU, Dakin J et al (2021) Relationship of D-dimer and prediction of pulmonary embolism in hospitalized COVID-19 patients: a multicenter study. Future Microbiol 16(12):863–70
Alonso-Fernández A, Toledo-Pons N, Cosío BG, Millán A, Calvo N, Ramón L et al (2020) Prevalence of pulmonary embolism in patients with COVID-19 pneumonia and high D-dimer values: a prospective study. PLoS One 15(8):e0238216
Planquette B, Le Berre A, Khider L, Yannoutsos A, Gendron N, de Torcy M et al (2021) Prevalence and characteristics of pulmonary embolism in 1042 COVID-19 patients with respiratory symptoms: a nested case-control study. Thromb Res 197:94–9
Loffi M, Regazzoni V, Toselli M, Cereda A, Palmisano A, Vignale D et al (2021) Incidence and characterization of acute pulmonary embolism in patients with SARS-CoV-2 pneumonia: a multicenter Italian experience. PLoS One 16(1):e0245565
Mouhat B, Besutti M, Bouiller K, Grillet F, Monnin C, Ecarnot F, et al. Elevated D-dimers and lack of anticoagulation predict PE in severe COVID-19 patients. Eur Respir J. 2020;56(4). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7487272/
Taccone FS, Gevenois PA, Peluso L, Pletchette Z, Lheureux O, Brasseur A et al (2020) Higher intensity thromboprophylaxis regimens and pulmonary embolism in critically ill coronavirus disease 2019 patients. Crit Care Med 48(11):e1087–e90
Whyte MB, Kelly PA, Gonzalez E, Arya R, Roberts LN (2020) Pulmonary embolism in hospitalised patients with COVID-19. Thromb Res 195:95–9
Hadid T, Kafri Z, Al-Katib A (2021) Coagulation and anticoagulation in COVID-19. Blood Rev 47:100761
Goudot G, Chocron R, Augy J-L, Gendron N, Khider L, Debuc B, et al. Predictive factor for COVID-19 worsening: insights for high-sensitivity troponin and D-dimer and correlation with right ventricular afterload. Front Med. 2020;7. https://www.frontiersin.org/articles/10.3389/fmed.2020.586307/full
Naymagon L, Zubizarreta N, Feld J, van Gerwen M, Alsen M, Thibaud S et al (2020) Admission D-dimer levels, D-dimer trends, and outcomes in COVID-19. Thromb Res 196:99–105
Acknowledgements
“Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).”
Funding
There was no funding support for this study.
Author information
Authors and Affiliations
Contributions
M.P and A.M conducted the main idea of the study and supervision. S.B, F.F, A.S, E.H.N, M.H, F.F, A.S, and A.M drafted the manuscript. All authors reviewed and accepted the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beidollahkhani, S., Fayedeh, F., Shoja, A. et al. d-dimer as a biomarker for COVID-19-associated pulmonary thromboembolism: a narrative review from molecular pathways to the imaging findings. Egypt J Bronchol 17, 44 (2023). https://doi.org/10.1186/s43168-023-00221-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43168-023-00221-6