
Abstract
Background
Amyloidosis is the extracellular deposition of amyloid fibril protein in any tissue or organ. The clinical manifestations of pulmonary amyloidosis are variable and without specific symptoms. We report a rare case of diffuse alveolar septal amyloidosis which is an extremely rare pattern of involvement, with a very poor prognosis, to improve our understanding of the disease.
Case presentation
A 27-year-old man complained of shortness of breath and cyanosis. High-resolution computed tomography revealed diffuse ground-glass opacifications with interlobular septal thickening in both lungs. The immune-histochemistry showed monoclonal lambda light chains. This case also showed nephrotic syndrome and cardiac arrhythmia, suggesting an involvement of the kidney and the heart. Diagnosis: The diagnosis was finally established by tru-cut transthoracic sonar guided lung biopsy (TSLB), and histological examination revealed Congo red-positive amorphous eosinophilic deposits in the alveolar sept. Interventions: The patient was admitted to a respiratory intensive care unit and put on non-invasive ventilation, then discharged on domiciliary oxygen therapy, and started treatment with chemotherapy melphalan 2 mg daily plus prednisone 60 mg daily immediately after the result of histopathology. Outcomes: Three months after treatment, dyspnea and hypoxemia improved, and he continued treatment. The patient was in a good clinical condition after 10 months of follow-up, but he died suddenly.
Conclusion
As it is difficult to distinguish diffuse alveolar septal amyloidosis from other interstitial and granulomatous lung diseases because of their similar symptoms and imaging findings, thus, transthoracic sonar guided lung biopsy and histological examination is very important in the diagnosis of diffuse alveolar septal amyloidosis.
Similar content being viewed by others
Background
Amyloidosis is a heterogeneous group of diseases characterized by the deposition of insoluble misfolded proteins in the extracellular matrix of various tissues and organs (kidneys, nerves, vessels, liver, spleen, gastrointestinal tracts, heart, and respiratory tracts), eventually leading to organ failure and death, if left untreated. Amyloidosis can be acquired or hereditary, systemic or localized; systemic amyloidosis is listed among rare diseases. With an estimated incidence of ∼10 cases per million person-years [1]. All amyloid fibrils appear as faintly red on Congo red staining on microscopic examination and show a typical apple-green birefringence under polarized light. To date, 31 fibril proteins have been identified in humans, and it is now recommended that amyloidosis be named after these fibril proteins [2]. The most common forms of amyloidosis include systemic AL amyloidosis (formerly primary amyloidosis), systemic AA amyloidosis (formerly secondary amyloidosis), systemic wild-type ATTR amyloidosis (formerly age-related or senile systemic amyloidosis), systemic hereditary ATTR amyloidosis (formerly familial amyloid polyneuropathy), and localized AL amyloidosis [3]. Involvement of the lungs is relatively common but rarely symptomatic. Three different clinicopathologic forms of amyloidosis can appear in the lungs: diffuse alveolar septal amyloidosis, nodular pulmonary amyloidosis, and tracheobronchial amyloidosis. Among these, the diffuse parenchymal pulmonary amyloidosis, also known as diffuse alveolar septal amyloidosis, is an extremely rare case and the least common type of pulmonary amyloidosis, usually seen in only 3% cases of pulmonary amyloidosis [4], and its diagnosis ante-mortem is very unusual and the diagnosis of diffuse alveolar septal amyloidosis is usually by post-mortem examination or lung biopsy (either open, VATS, sonar guided, or TBB via FOB) [5]. The most common association is systemic AL amyloidosis, but cases of diffuse alveolar septal amyloidosis that are caused by systemic AA, systemic wild-type ATTR, and systemic hereditary ATTR amyloidosis have also been reported [6, 7]. Since the clinical characteristics of the different forms of amyloidosis are similar, but treatment options differ radically and the prognosis of various forms of amyloidosis can differ, precise identification of the disease process is vital to avoid therapeutic errors.
In Egyptian literature, there were no documented cases of pulmonary amyloidosis. In this article, we will describe the clinical characteristics of a patient with diffuse parenchymal pulmonary amyloidosis associated with respiratory failure diagnosed by tru-cut transthoracic sonar guided lung biopsy which also involved the kidneys and heart, with manifestations due to diffuse infiltration of the lung, nephrotic syndrome, and cardiac arrhythmia, to improve our understanding of this disease.
Case presentation
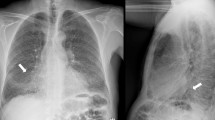
A 27-year-old male patient, electrical engineer, non-smoker, diabetic first discovered presented with dyspnea, which was progressive, without other symptoms such as cough, expectoration, fever, hemoptysis, or chest pain. On 20 June 2019, he sought medical advice by a pulmonologist and was evaluated to have bilateral almost symmetrical ground-glass opacity with inter/intra-lobar interstitial thickening of crazy paving appearance on high-resolution computed tomography (HRCT) of the chest (Fig. 1) and was treated as bronchopneumonia empirically with antibiotics for 3 weeks but without clinical improvement. On 10 July 2019, he was admitted to the Chest Department, Sohag University Hospital, with increasing dyspnea and respiratory failure. His physical examination was unremarkable apart from central cyanosis and received treatment for 2 days in the form of oxygen therapy and antibiotics with a slight improvement of dyspnea, then he was discharged on demand on domiciliary oxygen therapy. On 15 July 2019, the patient was admitted to Al-Houssine university Hospital in Cairo, where multiple investigations were performed to him including complete blood count (CBC) showing a normal count of white blood cell and platelet count and presence of polycythemia (WBC 10.2/mm3, RBC 8/mm3, PLT 293/mm3, HGB 17.7g/dl, and HCT 55), abdominal sonar, liver function tests, prothrombin time, concentration, and INR, which were all normal, and serology for hepatitis B and C and HIV was negative. Repeated sputum smear analysis for acid-fast bacilli (6 samples) was negative. Pulmonary function test (PFT) showed severely restrictive ventilation disorder and severely decreased diffusion capacity (DLCO). Arterial blood gases showed the presence of type I respiratory failure (PH 7.35, PCO2 40 mmHg, PO2 55 mmHg, O2 saturation 88%, and HCO 3:23 mEq/L), and the patient received oxygen by ordinary mask. Serology for immunological diseases was done including ANA IF, ADNA IF, ANCA C and ANCA P, anti-cardiolipin antibodies (IgM and IgG), anti-GBM, anti-Smith and RNP, anti-Smith antibodies, anti-SSA(RO) antibodies, anti-SS-B (LA), anti-JO.1, anti-SCL 70 antibodies, and anti-centromere antibodies, lupus anticoagulant (LA), all of them were negative. The patient received treatment in the form of oxygen therapy and antibiotics. Venesection was done for the patient with a slight improvement in the general condition. High-resolution chest CT (HRCT) repeated on 25 July 2019 showed the same findings as the previous one (Fig. 1) and suggested the possibility of alveolar proteinosis. On 27 July 2019, the patient was underwent fiberoptic bronchoscopy examination of the airways but no abnormality was detected either endobronchial or affection of the wall and bronchoalveolar lavage (BAL) was taken. TBLB was planned during performing FOB but it could not be performed due to the development of marked hypoxemia of the patient. Cytological examination of the BAL fluid showed the presence of inflammatory cells including neutrophils and lymphocytes; the background of the smears shows endobronchial columnar cells, epithelial cells, and homogenous pink proteinaceous material. Also, BAL fluid specimens were negative for routine bacterial, mycobacterial, and fungal examination. On 1 August 2019, a tru-cut transthoracic sonographic guided lung biopsy was performed, then the patient wanted to be discharged from the hospital on demand and waited for the result of the lung biopsy. On 8 August 2019, the histological examination of lung biopsy showed that the specimens contained amorphous acidophilic homogeneous hyaline material intersperse by small lymphoid cells and eosinophilic material stained positive with Congo red and displayed the apple-green birefringence typical of amyloid deposit under polarizing microscopy (Fig. 2). Two days after returning to Sohag governorate and on 14 August 2019, he returned back to Chest Department Sohag University Hospital with right-sided chest pain, fever, tachypnea, and palpitation. On physical examination, he had RR 35 cycle/min, HR 160 B/M, and on chest auscultation, bilateral inspiratory fine crepitations more basal were detected. He was admitted to CCU for 2 days due to supraventricular tachycardia and the heart rate was controlled on medical treatment but the patient was still tachypneic (RR 50 cycle/min) with the development of hemoptysis of a moderate amount of frank blood for 1 day. Arterial blood gas analysis (ABG) showed type I respiratory failure (PH 7.44, PCo2 35 mmHg, PO2 50 mmHg, O2 saturation 84%, and HCo3 28 mEq/L), and D dimer was 600 ng/ml, so to exclude the possibility of pulmonary embolism, CT angiography was done and there were no radiological signs of pulmonary embolism, it showed only the presence of the same findings in previous ones (Fig. 3). Then, the patient was admitted to respiratory ICU and put on NIVM (BiPAP) on 17 August 2019 with an improvement of oxygenation on FiO2 80%. After 5 days, he developed rapid atrial fibrillation (AF) without improvement on medical treatment (cordarone) due to the presence of intractable hypoxemia so defibrillations were done by electrical cardioversion and the heart rate returned to sinus rhythm. During the patient stay in RICU, the following investigations were done: echocardiogram showed normal left ventricular dimensions with good global systolic function (EF of 69%) and there were no features of cardiac amyloidosis. Urine analysis showed the presence of proteinuria [a 24-h urine contained 320 mg of protein (normal 50–150 mg/24 h)], creatinine level was normal (0.9), albumin/creatinine ratio was 56.88, and urine protein-to-creatinine ratio was 1.17 (normal < 0.2). CBC showed leukocytosis with thrombocytopenia (WBCs 11.6/mm3, RBCs 6.5/mm3, and platelets 100/mm3), and thyroid function tests (free T3, freeT4, and TSH) were normal. Moreover, we performed immunohistochemistry (IHC) for subtyping amyloidosis, and immunohistochemical staining for amyloid protein revealed immunoglobulin light chains, lambda (λ) type (ALλ) light chain; therefore, he was diagnosed as systemic AL amyloidosis accompanied by diffuse parenchymal pulmonary, cardiac, and renal involvements. The patient was transferred again to Al Houssein Hospital where the patient started chemotherapy with melphalan 2 mg once daily and corticosteroid 60 mg/day along with symptomatic treatment (domiciliary oxygen). Then, his symptoms and radiology {(HRCT) of the lung} (Fig. 4) improved markedly and he returned to his work 3 months after the initiation of treatment. After 10 months of regular follow-up, the patient was in a good clinical condition and compliant with treatment. No adverse effects were observed. But on 15 July 2020, the patient deteriorated suddenly and arrested.

Chest high-resolution computed tomography (HRCT) showing bilateral almost symmetrical ground-glass opacity with inter/intra-lobar interstitial thickening of crazy paving appearance

Histopathology of lung biopsy. Congo red staining showed marked thickening of the alveolar wall and deposition of amorphous eosinophilic amyloid at the bronchial mucosa, pulmonary vessel wall, and interstitium

CT angiography showing normal findings

High-resolution chest CT after 3 months of treatment with melphalan and corticosteroids
Discussion
Amyloidosis is a disorder caused by misfolding of autologous protein and its extracellular deposition as fibrils, resulting in vital organ dysfunction and eventually death [8]. Based on immunohistochemistry, there are 5 major categories of amyloidosis: (1) primary or immunoglobulin light-chain (AL) amyloidosis due to immunoglobulin and light-chain deposition, (2) heritable amyloidoses due to mutant transthyretin (prealbumin) deposition, (3) AA amyloidosis/secondary amyloidosis due to serum AA protein deposition, (4) senile amyloidosis due to wild-type transthyretin deposition, and (5) dialysis-related amyloidosis due to beta 2-microglobulin deposition [9, 10]. In particular, AL fibril is a common type of amyloid deposit in the lung. AL/primary amyloidosis usually has male preponderance with the median age at diagnosis being 64 years and usual incidence of 6–10 cases/million/year. It usually occurs in plasma cell dyscrasias [11]. Pulmonary amyloidosis is a rare disease and may be localized or part of systemic amyloidosis [11]. Furthermore, pulmonary amyloidosis mainly comes in 3 forms according to the pathologist’s perspective; these include diffuse parenchymal/diffuse alveolar septal amyloidosis, nodular pulmonary amyloidosis, and tracheobronchial amyloidosis [12]. Tracheobronchial amyloidosis is an organ-limited type of amyloidosis that mainly represents multifocal submucosal plaques and usually presents localized AL amyloidosis without pulmonary parenchyma. Generally, it is symptomatic due to stenosis resulting from the amyloid deposits in the trachea and large bronchi [13]. Patients usually present typical symptoms, such as dyspnea, cough, chest tightness, and even hemoptysis. Nodular pulmonary amyloidosis is usually localized and usually presents with peripheral subpleural localizations of variable size under chest CT. It is defined as one or more nodular amyloid deposits involving the lung, which usually represents as localized AL or AL/AH (mixed immunoglobulin light/heavy chain) amyloidosis [8, 14]. Diffuse alveolar septal amyloidosis is usually a manifestation of systemic amyloidosis, is associated with systemic AL amyloidosis, and is characterized by the presence of amyloid deposits in the vessel walls and alveolar septa [15]. Pathologic examination of diffuse parenchymal pulmonary amyloidosis shows deposition of amorphous eosinophilic amyloid in the alveolar septa, especially around the capillary vessels [15]. Therefore, HRCT findings in such patients mainly comprise GGOs, interlobular septal thickening, intralobular reticular opacity, and nodules [9, 16]. Diffuse amyloidosis is sometimes accompanied by mediastinal lymphadenopathy. Pleural effusion may be present and occasionally dominate the clinical course. Multiple cysts and calcification probably resulting from fragile alveolar walls as a consequence of amyloid deposition both on alveolar walls and around capillaries have been described [4].
In our case, a high-resolution CT of the lung showed bilateral ground-glass opacities (GGOs) with interlobular septal thickening in both lungs. A pulmonary function test showed a severe restrictive pattern and severe reduction of diffusion capacity (DLCO). Bronchoscopic examination showed no abnormal findings. Our case showed cardiac arrhythmia and normal left ventricular EF on echocardiography and also showed nephrotic syndrome suggesting an involvement of the kidneys. Transthoracic sonar guided biopsy revealed diffuse hyaline thickening of the alveolar septum and blood vessel wall on a light microscope, along with apple-green birefringence on a polarizing microscope following Congo red staining. Immunohistochemistry showed positive lambda light chain. Finally, pulmonary amyloidosis was diagnosed based on histological examination, and the characteristics of this patient were completely consistent with diffuse alveolar septal amyloidosis. Fortunately, we could confirm the diagnosis of diffuse alveolar septal amyloidosis without serious complications.
Diffuse parenchymal pulmonary amyloidosis has a remarkably different, more clinical presentation. Such patients may develop symptoms of coughing and shortness of breath secondary to the amyloid deposits. It is characterized by widespread amyloid deposition involving small vessels and the interstitium [17]. This is reflected by lung function tests showing a restrictive pattern with reduced diffusion capacity of carbon monoxide and hypoxemia upon exertion [18]. Affected individuals are more likely to progress to pulmonary hypertension and respiratory failure [16]. Tissue biopsy is the gold standard for the diagnosis and typing of pulmonary amyloidosis. Diagnosis of amyloidosis is confirmed by the presence of apple-green birefringence under polarized light of a tissue biopsy stained with Congo red [12, 19], which is very important and helpful for differentiating it from other diseases. Therefore, the differential diagnosis of pulmonary amyloidosis is quite broad and mainly includes infectious, neoplasms (i.e., lymphangitic carcinomatosis), pneumoconiosis, pulmonary sarcoidosis, interstitial, and granulomatous lung diseases [19, 20]. The diagnosis of pulmonary amyloid can be made by fiberoptic bronchoscopy, VATLB, and open thoracotomy. Although transbronchial lung biopsy (TBLB) is useful in some amyloidosis cases, it may be of less efficacy in establishing diagnosis due to the limit of the amount of biopsy sampling. The lung VATLB is highly specific and sensitive. The biopsy of a clinically suspected organ is an invasive procedure and may be associated with complications including hemorrhage. Because amyloidosis is a systemic disease, routine biopsies from non-symptomatic sites including the rectal mucosa, abdominal fat pad, and labial salivary glands are more commonly used [21].
Scala et al. [22] confirm the rarity of intra-thoracic amyloidosis having a frequency of about 1.0% among cases of diffuse interstitial lung diseases that referred to center during the study time (from 2007 to 2012); only three cases of amyloidosis with involvement of the respiratory system were identified among the 298 patients: the first case was tracheobronchial amyloidosis, the second case was primary systemic AL amyloidosis with cardiac and pleural involvement (as bilateral pleural effusion) confirmed by the mean of tissue specimen examination obtained with the Cope needle pleural biopsy, and the third case was a 53-year-old male patient who presented with severe dyspnea associated with severe non-hypercapnic hypoxemia (PaO2/FiO2 ratio 120; PaCO2 28.5 mmHg, pH 7.485) to ambulatory and subsequently was admitted in the respiratory intensive care unit, and as the hypoxemia was refractory to high-flow oxygen therapy, a trial of non-invasive ventilation (NIV) delivered via a full-face mask was attempted (mode pressure-support, PS = 20 cm H2O; PEEP 8 cm H2O; FiO2 0.80); then, after 20 h, endotracheal intubation and invasive mechanical ventilation resulted mandatory due to NIV failure. Fiberoptic bronchoscopy with BAL and TBLB was performed during NIV; the histological exam showed a pathologic pattern of unspecific neutrophilic capillaritis. As the patient refused to undergo surgical biopsy, empiric therapy with an intravenous pulsed bolus of cyclophosphamide and steroids was given. But he died after a worsening of his clinical conditions, radiological findings, and lung gas exchange, and the autopsy reported systemic AL amyloidosis with prevalent lung and renal involvement.
Chu et al. [23] reported 13 cases of isolated pulmonary amyloidosis between 1990 and 2011: 7 (7/13) patients with tracheobronchial amyloidosis; 3 (3/13) patients two with nodular amyloidosis, one with diffuse shadows and nodules; one (1/13) lung consolidation; and two (2/13) with mediastinal lymph node amyloidosis and bilateral pleural effusion. Three patients died of pneumonia and respiratory failure, one patient with endotracheal amyloidosis, after 4 years (age 48 years), and two patients with mediastinal lymph node amyloidosis and bilateral pleural effusion, 5 and 9 months, after original treatment (ages 72 years and 49 years), and one patient with diffuse and multiple nodular pulmonary amyloidosis died of respiratory failure 1 year later (age 55 years).
Utz et al. [6] retrospectively analyzed the clinical record of patients seen at the Mayo Clinic over a 13-year period (1980–1993), and of 55 patients with pathologically proven amyloidosis, only 11 had the disease confined to the respiratory system.
Kim et al. [24] reported a case of pulmonary diffuse alveolar septal amyloidosis confirmed by transbronchial lung biopsy, which also involved the kidneys, heart, peripheral nerves, and gastrointestinal tracts, manifested by diffuse infiltration of the lungs, nephrotic syndrome, cardiac arrhythmia, congestive heart failure, a tingling sensation in both hands and multiple nodules in the gastrointestinal tracts, and sudden death occurred and was related to cardiac amyloidosis, especially with conduction system involvement that was evident by ECG and Holter monitoring also a granular sparkling appearance of the myocardium on echocardiography.
Kline et al. [25] described three cases of pulmonary parenchymal amyloidosis diagnosed by transbronchial lung biopsy via the flexible fiberoptic bronchoscope. Although there was a report that serious complications appeared during bronchoscopy and transbronchial lung biopsy in one patient with amyloidosis [26], on the whole, bronchoscopy and transbronchial lung biopsy are considered relatively safe methods when they are carried out with caution. We recommend that bronchoscopy should be carried out under the monitoring of ECG and oxygen saturation and that an emergency operation team for open thoracotomy stand by during the procedure to control any massive bleeding.
In English literature, there are six cases that reported of diffuse alveolar septal amyloidosis for which ante-mortem diagnosis was made by transbronchial lung biopsy up until 1987 [25, 26].
When the diagnosis of AL amyloidosis in lungs and bronchi is made, the classification and differentiation between localized and systemic AL amyloidosis is important as therapy and prognosis differ [1]. The principles in the treatment of amyloidosis are to inhibit the synthesis of amyloid and its extracellular deposition, reduce the production of amyloid precursors, and promote the degradation of amyloid [27]. Chemotherapy is the treatment of choice for AL amyloidosis, and regression has been demonstrated earlier systematically. O’Regan et al. [13] reported that systemic chemotherapy consisting of 0.15 mg/kg/day melphalan plus 20 mg/day prednisone for 4 weeks (MP regime) is one of the most commonly used treatments and is believed to inhibit the production and deposition of amyloid. Kyle et al. [28] treated patients with primary systemic amyloidosis with colchicine (n = 72), MP regimen (n = 77), or MP regimen in combination with colchicine (n = 71) and reported median survival times of 8.5 months, 18 months, and 17 months, respectively. Also, Chu et al. [23] treated one patient with massive pulmonary consolidation with the MP regimen with good results. Lung transplantation for isolated pulmonary amyloidosis has been reported [29]. The AL amyloid then contributed to pulmonary hypertension (PH) with severe symptoms necessitating lung transplantation. Ellender et al. [30] described a case with PH from amyloidosis secondary to systemic lupus erythematosus and Sjögren’s syndrome, the patient received bilateral lung transplantation and remained stable after 7 years post lung transplantation. Lung transplantation for isolated pulmonary amyloidosis or combined with PH may be performed in highly selected patients with good long-term outcome.
Our patient was treated with melphalan 2 mg daily plus prednisone 60 mg daily under complete aseptic conditions and close observation of the patient for detection of any side effect of the medication for 10 months. Fortunately, the patient was in good clinical condition during the follow-up period. On 16 July 2020, the patient died suddenly, most probably due to cardiac cause. Although there was no pathologic confirmation, it is evident that this case involved the heart, and sudden death was related to cardiac amyloidosis, especially with conduction system involvement that was evident by events of supraventricular tachycardia and rapid atrial fibrillation. Cardiovascular events account for two-thirds of fatal causalities in AL and TTRwt cardiac amyloidosis [31]. Arrhythmias are common in cardiac AL and may lead to acute hemodynamic compromise. Sudden cardiac death, often due to pulseless electrical activity, is an important cause of early mortality [32].
The prognosis of pulmonary amyloidosis varies according to its forms and individuals. Since primary diffuse alveolar septal amyloidosis is extremely rare, it is very difficult to be confirmed in living patients and very few case series were published earlier [33, 34]. Thompson et al. reported four cases and Hui et al. reported six cases. The average survival period in patients with systemic amyloidosis with lung involvement is 16 months only, and with the development of heart failure, the survival duration decrease to less than 4 months [5, 18]. Recently, Gandham et al. [35] reported a patient with diffuse alveolar septal amyloidosis, who was treated with chemotherapy (cyclophosphamide), dexamethasone, and bortezomib and was well with symptomatic improvement in the 6-month follow-up. Moreover, O’Regan et al. [14] reported ten patients referred to the Amyloid Program at Boston University over the past 15 years and found that 30% of tracheobronchial amyloidosis patients died within 7 to 12 years after diagnosis and all having proximal or severe mid-airway disease. In general, the long-term prognosis of nodular pulmonary amyloidosis is the best, but diffuse alveolar septal amyloidosis is the worst. Gradual worsening of pulmonary function and symptoms is typical. Death usually occurs due to refractory hypoxemia in acute respiratory distress syndrome (ARDS), massive hemoptysis due to the arterial rupture caused by amyloid deposition in the vessels’ wall, massive lung embolism due to the thrombosis of the inferior cava vein, and cardiac involvement. Less frequently, death occurs for chylothorax due to the thoracic duct involvement or for gas embolism, as a complication of the biopsy [26, 36]. Mortality is higher in case of misdiagnosis and consequent delayed treatment.
Overall, amyloidosis of the lower respiratory tract is very rare but may reflect a significant clinical problem in either organ-limited or systemic amyloidosis. The most fundamental treatment options for amyloidosis are based on the fibril protein type. Targeting different pathogenic mechanisms, such as the unequivocal identification of the amyloid type, is vital to avoid therapeutic errors [15, 20]. Therefore, to achieve precise treatment, all new patients require unequivocal amyloid typing and complete assessment to determine their optimal treatment. Although there is still no proven drug therapy for pulmonary amyloidosis, luckily, several new drugs that can accelerate the clearance of tissue amyloid deposits, interfere with amyloid fibrillogenesis, and stabilize the amyloid precursor proteins in the pipeline could potentially benefit patients with pulmonary amyloidosis.
There are 2 main characteristics of this case. First, our patient is the youngest patient (27 years old) who presented with pulmonary amyloidosis and complaining of dyspnea, tachypnea, tachycardia, and central cyanosis, which may, for a long time, be falsely diagnosed as interstitial or granulomatous lung diseases [20, 21], and second, there is no specific treatment for diffuse alveolar septal amyloidosis but it is usually treated by chemotherapy and corticosteroids; moreover, our case needs treatment in RICU with NIMV due to respiratory failure and CCU due to supraventricular tachycardia. Therefore, as in the literature, there were limited studies on the diffuse alveolar septal amyloidosis and ages of all reported cases were above 45 years, but as regards diffuse alveolar septal amyloidosis presented with respiratory failure and treated in RICU, after a search, we found only a case of systemic AL amyloidosis presenting with diffuse pulmonary involvement and respiratory failure reported by Scala et al. [23] but patient age was 54 years old and diagnosed post-mortem as mentioned above; therefore, we want to consider this case as the only reported case of systemic AL amyloidosis with diffuse alveolar septal amyloidosis and RF in a young aged patient.
Conclusion
Knowledge about pulmonary amyloidosis is important due to nonspecific findings in CT chest, and poor prognosis and treatment depends on the pattern of involvement; therefore, amyloidosis should be considered among the complex differential diagnostic pathways of diffuse interstitial lung diseases. Diffuse parenchymal pulmonary amyloidosis is a fatal disorder that is rare and often undiagnosed. Tissue biopsy is the gold standard to achieve the correct diagnosis as soon as possible with the use of subcutaneous fat pad and lung biopsy.
Availability of data and materials
All data and material of the case are available.
Abbreviations
- HRCT:
-
High-resolution computed tomography
- TSLB:
-
Transthoracic sonar guided lung biopsy
- RICU:
-
Respiratory intensive care unit
- NIV:
-
Non-invasive ventilation
- AL:
-
Amyloid light chain
- AA:
-
Amyloid A protein
- ATTR:
-
Amyloid transthyretin
- VATS:
-
Video-assisted thoracoscopic surgery
- TBB:
-
Transbronchial biopsy
- FOB:
-
Fiberoptic bronchoscopy
References
Baumgart JV, Stuhlmann-Laeisz C, Hegenbart U, Nattenmüller J, Schönland S, Krüger S, Behrens HM, Röcken C (2018) Local vs. systemic pulmonary amyloidosis-impact on diagnostics and clinical management. Virchows Arch 473(5):627–637. https://doi.org/10.1007/s00428-018-2442-x
Andras K, Thomas V (2017) Colby: Amyloidosis of the lung. Arch Pathol Lab Med. 141:247–254. https://doi.org/10.5858/arpa.2016-0102-RA
Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJM, Westermark P (2014) Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 21(4):221–224. https://doi.org/10.3109/13506129.2014.964858
Berk JL, O'Regan A, Skinner M (2002) Pulmonary and tracheobronchial amyloidosis. Semin Respir Crit Care Med. 23(2):155–165. https://doi.org/10.1055/s-2002-25304
Poh SC, Tjia TS, Seah HC (1975) Primary diffuse alveolar septal amyloidosis. Thorax. 30(2):186–191. https://doi.org/10.1136/thx.30.2.186
Utz JP, Swensen SJ, Gertz MA (1996) Pulmonary amyloidosis: the Mayo Clinic experience from 1980 to 1993. Ann Intern Med. 124(4):407–413. https://doi.org/10.7326/0003-4819-124-4-199602150-00004
Ueda M, Ando Y, Haraoka K, Katsuragi S, Terasaki Y, Sugimoto M, Sun X, Uchino M (2006) Aging and transthyretin-related amyloidosis: pathologic examinations in pulmonary amyloidosis. Amyloid. 13(1):24–30. https://doi.org/10.1080/13506120500537194
Milani P, Basset M, Russo F et al (2017) The lung in amyloidosis. Eur Respir Rev. 26(145):1–8
Khoor A, Colby TV (2017) Amyloidosis of the lung. Arch Pathol Lab Med. 141(2):247–254. https://doi.org/10.5858/arpa.2016-0102-RA
Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med. 349(6):583–596. https://doi.org/10.1056/NEJMra023144
Kyle RA, Gertz MA (1995) Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 32:45–59
de Almeida RR, Zanetti G, Pereira E et al (2015) Respiratory tract amyloidosis. State-of-the-art review with a focus on pulmonary involvement. Lung. 193(6):875–883. https://doi.org/10.1007/s00408-015-9791-x
O’Regan A, Fenlon HM, Beamis JFJr, et al. (2000) Tracheobronchial amyloidosis. The Boston University experience from 1984 to 1999. Medicine. 79(2):69–79. https://doi.org/10.1097/00005792-200003000-00001
Grogg KL, Aubry MC, Vrana JA, Theis JD, Dogan A (2013) Nodular pulmonary amyloidosis is characterized by localized immunoglobulin deposition and is frequently associated with an indolent B-cell lymphoproliferative disorder. Am J Surg Pathol. 37(3):406–412. https://doi.org/10.1097/PAS.0b013e318272fe19
Yamada M, Takayanagi N, Yamakawa H, Ishiguro T, Baba T, Shimizu Y, Okudela K, Takemura T, Ogura T (2020) Amyloidosis of the respiratory system: 16 patients with amyloidosis initially diagnosed ante mortem by pulmonologists. ERJ Open Res 6(3):00313–02019
Czeyda-Pommersheim F, Hwang M, Chen SS, Strollo D, Fuhrman C, Bhalla S (2015) Amyloidosis: modern cross-sectional imaging. Radiographics. 35(5):1381–1392. https://doi.org/10.1148/rg.2015140179
Poletti V, Costabel U, Casoni GL, Bigliazzi C, Drent M, Olivieri D (2004) Rare infiltrative lung diseases: a challenge for clinicians. Respiration. 71(5):431–443. https://doi.org/10.1159/000080625
Sato H, Ono A, Okada F, Maeda T, Saburi Y, Urabe S, Mori H (2015) A case of diffuse alveolar septal amyloidosis associated with multiple myeloma. J Thorac Imaging. 30(6):W73–W75. https://doi.org/10.1097/RTI.0000000000000183
Guasch M, Ojanguren A, Gomez JR (2020) Pulmonary amyloidosis: a diagnostic challenge. Cir Esp 98(1):50–52. https://doi.org/10.1016/j.ciresp.2019.03.008
Shenin M, Xiong W, Naik M, Sandorfi N (2010) Primary amyloidosis causing diffuse alveolar hemorrhage. J Clin Rheumatol. 16(4):175–177. https://doi.org/10.1097/RHU.0b013e3181df8345
Hachulla E, Grateau G (2002) Diagnostic tools for amyloidosis. Joint Bone Spine. 69(6):538–545. https://doi.org/10.1016/S1297-319X(02)00449-9
Scala R, Maccari U, Madioni C et al (2015) Amyloidosis involving the respiratory system: 5-years’ experience of a multi-disciplinary group’s activity. Ann Thorac Med 10(3):212–216
Chu H, Zhao L, Zhang Z, Gui T, Yi X, Sun X (2012) Clinical characteristics of amyloidosis with isolated respiratory system involvement: a review of 13 cases. Ann Thorac Med. 7(4):243–249. https://doi.org/10.4103/1817-1737.102186
Kim CH, Kim S, Kwon OJ et al (1990) Pulmonary diffuse alveolar septal amyloidosis: diagnosed by transbronchial lung biopsy. Korean J Intern Med. 5(1):63–68. https://doi.org/10.3904/kjim.1990.5.1.63
Kline LR, Dise CA, Ferro TJ, Hansen-Flaschen JH (1985) Diagnosis of pulmonary amyloidosis by transbronchial biopsy. Am Rev Respir Dis. 132(1):191–194. https://doi.org/10.1164/arrd.1985.132.1.191
Strange C, Heffner JE, Collins BS, Brown FM, Sahn SA (1987) Pulmonary hemorrhage and air embolism complicating transbronchial biopsy in pulmonary amyloidosis. Chest. 92(2):367–369. https://doi.org/10.1378/chest.92.2.367
Falk RH, Comenzo RL, Skinner M (1997) The systemic amyloidoses. N Engl J Med. 337(13):898–909. https://doi.org/10.1056/NEJM199709253371306
Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM (1997) A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 336(17):1202–1207. https://doi.org/10.1056/NEJM199704243361702
Ware LB, Keith FM, Gordon RL, Ries CA, Seitz RF, Gold WM, Golden JA (1998) Lung transplantation for pulmonary amyloidosis: a case report. J Heart Lung Transplant. 17(11):1129–1132
Ellender CM, McLean C, Williams TJ, Snell GI, Whitford HM (2015) Autoimmune disease leading to pulmonary AL amyloidosis and pulmonary hypertension. Respirol Case Rep. 3(2):78–81. https://doi.org/10.1002/rcr2.104
Escher F, Senoner M, Doerler J, Zaruba MM, Messner M, Mussner-Seeber C, Ebert M, Ensinger C, Mair A, Kroiss A, Ulmer H (2020) When and how do patients with cardiac amyloidosis die? Clinical Research in Cardiology. 109(1):78–88. https://doi.org/10.1007/s00392-019-01490-2
Grogan M, Dispenzieri A (2015) Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 20(2):155–162. https://doi.org/10.1007/s10741-014-9464-5
Gómez AT, Alonso RM, García MM et al (2008) Thoracic amyloidosis: high-resolution computed tomographic findings in 3 cases. J Comput Assist Tomogr. 32(6):926–928. https://doi.org/10.1097/RCT.0b013e31815a3420
Wang Q, Chen H, Wang S (2014) Laryngo-tracheobronchial amyloidosis: a case report and review of literature. Int J Clin Exp Pathol. 7(10):7088–7093
Gandham AK, Gayathri AR, Sundararajan L (2019) Pulmonary amyloidosis: a case series. Lung India. 36:229–232
Road JD, Jacques J, Sparling JR (1985) Diffuse alveolar septal amyloidosis presenting with recurrent hemoptysis and medial dissection of pulmonary arteries. Am Rev Resp Dis. 132(6):1368–1370. https://doi.org/10.1164/arrd.1985.132.6.1368
Acknowledgements
The authors would like to present their appreciation to the residents who helped us in data collection.
Funding
Nil.
Author information
Authors and Affiliations
Contributions
There is contribution of all authors. SN was responsible for the collection of the data about the patient, his follow-up during his stay in Sohag University Hospital, and the writing of the manuscript. MA was responsible for taking the lung biopsy from the patient and his follow-up during his stay in Al Houssein Hospital. HM was responsible for the revision of the manuscript and preparation for publication. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was taken from the patient who participated in the case study.
Consent for publication
Written informed consent was taken from the patient who participated in the case study for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohammadien, H.A., Morsi, S.N. & Al Shahat, M.A. Systemic AL amyloidosis presenting with diffuse alveolar septal involvement and respiratory failure: a case report and review of the literature. Egypt J Bronchol 15, 23 (2021). https://doi.org/10.1186/s43168-021-00070-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43168-021-00070-1