
Abstract
Background
Pulmonary cyst is a rounded lung parenchymal lucency, usually containing air or fluid. Approach to establishing the etiology of lung cyst includes determining their location, number, distribution, and presence or absence of associated computed tomography findings (associated with nodules or ground-glass attenuation). Multiple cysts may be seen in various diseases, and radiological imaging is the usual starting point in detecting lung cysts. Importance of adequate clinical examination accompanied with relevant investigations in securing the etiology of lung cysts cannot be overemphasized. We present a first case of a rare multiple cystic lung disease from the Kingdom of Bahrain that was successfully managed with oral corticosteroids.
Case presentation
A 42-year-old male, chronic smoker presented with progressive dyspnea and productive cough of 1 year duration. He was evaluated and found to have multiple variable sized thin and thick-walled cysts with bizarre shapes in both lungs. A diagnosis of pulmonary Langerhans’ cell histiocytosis was made, and the patient was treated with smoking cessation and oral corticosteroids for 9 months.
Conclusion
Patients with characteristic clinical and radiological features can be diagnosed as pulmonary Langerhans’ cell histiocytosis, without a tissue biopsy. A good response may be seen with smoking cessation and oral corticosteroids in selected group of patients.
Similar content being viewed by others


Background
Cyst is a round circumscribed space surrounded by epithelial or fibrous wall, varying in its thickness. Multiple cysts may be seen in lymphoid interstitial pneumonia (LIP), light chain deposition disease, Birt-Hogg-Dube syndrome, pulmonary Langerhans’ cell histiocytosis (PLCH), lymphangioleiomyomatosis (LAM), desquamative interstitial pneumonia, infections like pneumocystis jirovecii pneumonia, and others. Good clinical evaluation that includes relevant history and physical examination, radiological evaluation, and correlating them with laboratory findings including appropriate biopsies may be helpful in arriving at a proper diagnosis of lung cysts. One of the algorithmic approaches to evaluate a cystic lung disease is to first ascertain that true cysts are seen on imaging studies, and mimics like bronchiectasis, cavities, and emphysema have been excluded. The second step is to see the location of these lucencies on imaging. More often than not, sub-pleural location of these lucencies have bullae, paraseptal emphysema, and honeycombing as their differentials. The next step will be to determine whether these cysts have other radiographic abnormalities. If there are not any abnormalities, then it would be worthwhile to see if they are solitary or multiple. Determining the presence of associated radiological abnormalities like nodules or ground glass opacities help in further refining the diagnosis of cystic lung diseases. We present a case of multiple parenchymal lung cysts that was recently encountered in our center.
Case presentation
A 42-year-old man presented to our hospital with progressive dyspnea and cough with mucoid expectoration of 1-year duration. He denied other respiratory, systemic, and constitutional symptoms. The patient was a secondary school teacher and a smoker (40 pack years) with no history of alcohol consumption and drug abuse. There was no exposure to pets. On examination, vitals were normal except for respiratory rate of 25 breaths.min−1, and SpO2 was 93% on ambient air. He had grade 3 clubbing. Widespread crepitations were heard over both the infra-clavicular areas. Examination of the other systems was unremarkable.
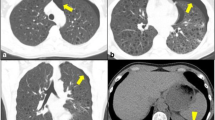
Investigations showed a normal hemogram and biochemical profile. Chest radiograph revealed reticular opacities and cysts in both the upper and mid zones and preservation of lung volumes (Fig. 1). Anti-nuclear antigen and rheumatoid factor were absent. Spirometry revealed forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) of 81%, FVC 3.05 L (74% predicted), and FEV1 2.86 L (71% predicted). There was no significant bronchodilator reversibility. Body plethysmography showed total lung capacity (TLC) of 5.04 L (98% predicted), residual volume (RV) 1.97 L (115% predicted), and RV/TLC 39.08%. Transfer factor of lung for carbon monoxide (TLCO) was 2.22 mmol/min/kPa (27% predicted), and transfer factor per unit lung volume (TL/VA) was 0.47 mmol/min/KPa/L (29% predicted). Arterial blood gas (ABG) showed PaO2 54 mm Hg, PaCO2 34 mm Hg, and 6-min walk test (6MWT) 89% (rest) and 77% (after exercise), and 6-min walk distance (6MWD) was 225 m. High-resolution computed tomography (HRCT) of the chest revealed variable sized cysts with thin and thick walls, with bizarre shapes (Fig. 2) predominantly in the upper lobes and sparing of the lung bases (Fig. 3 and Fig. 4). Fiberoptic bronchoscopy (FOB) was unremarkable. Broncho-alveolar lavage (BAL) for AFB and malignant cytology was negative. Anti-CD1a antibodies were negative. Echocardiography revealed normal left ventricular function (60%) with no ischemic changes, fair right ventricular function, and mild to moderate pulmonary hypertension (PH) with systolic pulmonary artery pressure (sPAP) of 55 mm Hg. Positron emission tomography-computed tomography (PET/CT) showed no signs of metabolic activity over the widespread lung cysts bilaterally. No other fluorodeoxyglucose (FDG)-avid lesions were detected in the rest of the scanned body. A diagnosis of PLCH was entertained based on the abovementioned classical clinical and radiological features. He was recommended to quit smoking and treated with oral corticosteroids at a dose of 40 mg/day with gradual tapering over 9 months.

Chest radiograph showing reticular opacities and cysts in both upper and mid zones and preservation of lung volumes

HRCT of the chest, axial image, revealing variable-sized cysts with thin and thick walls, bizarre shapes (white arrow) in upper lobes

HRCT of the chest, axial image, showing less numerous cysts in lower lobes

HRCT of the chest, coronal reconstruction image, demonstrating apicobasal gradient in distribution of cysts with predominant involvement of upper lobes and sparing of lung bases
The patient showed a good improvement in his general condition. Cough has completely subsided, and dyspnea has improved. Spirometry showed increase in FVC by 940 ml and 6MWD by 175 m, decrease in severity of PH (sPAP of 45 mm Hg), and stability of radiological changes over 1 year of follow-up. The patient has been kept under close observation and was strongly advised to abstain from smoking.
Discussion
PLCH belongs to a group of disorders associated with abnormal organ infiltration by Langerhans’ cells. It is an uncommon, smoking-related interstitial lung disease affecting young adults. More often than not, the lung is the only organ involved, although at times, the bone, skin, and pituitary gland may also be affected. It affects the Whites more than the Blacks or Asians. Its prevalence is 0.27 and 0.07 per 100,000 population, in men and women, respectively, as seen in a Japanese study [1]. There is no sufficient data on prevalence of PLCH in the Middle East. We believe this is the first published case of PLCH from Kingdom of Bahrain. Cigarette smoking is a near-universal association seen in adult PLCH. It induces recruitment and activation of Langerhans’ cells through the activation of cytokines like granulocyte-macrophage colony-stimulating factor, tumor necrosis factor-α, and osteopontin [2]. Langerhans’ cell is the pathologic cell type seen in PLCH which is characterized by the presence of intracytoplasmic inclusions like Birbeck granules (seen on electron microscopy) and CD1a antigens on cell surface. These cells are seen in clusters and along with eosinophils, lymphocytes, and neutrophils are distributed along the bronchovascular bundles in early disease. In advanced cases, interstitial fibrosis and cyst formation occurs with absence of Langerhans’ cells.
Presentation may be incidental detection on chest radiograph or with respiratory/constitutional symptoms or with pneumothorax [3]. Chest examination is usually unremarkable with digital clubbing and crepitations being uncommon. Imaging studies, especially HRCT, may reveal ill-defined or stellate nodules, cysts predominantly distributed in upper zones, with preservation of lung volumes and sparing costophrenic angles [4]. FDG-PET scans are positive in early nodular stage of disease, while later in the cystic stage of disease, they are usually negative, as was reflected in our patient. Predominantly nodular disease is associated with normal pulmonary function test (PFT), while cystic disease may show airflow limitation and hyperinflation. Typically, PFT findings include reduced FVC, normal or elevated RV, normal TLC, and increased or preserved RV/TLC. The TLCO maybe disproportionately reduced. In our patient, PFT revealed normal FEV1/FVC and TLC, reduced FVC with no hyperinflation. TLCO was disproportionately reduced. 6MWT showed a significant exercise related desaturation and reduced 6MWD. FOB may be utilized to exclude other causes of cystic lung diseases like hypersensitivity pneumonitis, LIP, LAM, and infections. BAL showing greater than 5% CD1a positive Langerhans’ cells strongly supports the diagnosis of PLCH [5] though it is poorly sensitive [6]. In advanced diseases, the number of CD1a positive cells in tissue specimens and BAL reduce significantly, bringing down the yield even further, as was observed in our case. Non-conclusive clinical and radiological findings need confirmation by surgical lung biopsy. Advanced cystic lung disease with altered pulmonary function may not be suitable candidates for surgical lung biopsy. Most of the authorities accept a diagnosis if the clinical and radiological features are sufficiently characteristic of PLCH, as was seen in our case.
Smoking cessation is the most important intervention in resolution or stabilization of disease, effective as a sole therapy in up to 60% cases [7]. Oral steroids are recommended in treatment of symptomatic PLCH and in those who progress despite smoking cessation [8]. Doses of 0.5–1 mg/kg/day of prednisone tapered over 6–12 months results in clinical and radiological improvement with benefits observed in patients with nodular lesions [9]. Almost a third of patients are managed with oral corticosteroids [10]. These may be ineffective in advanced diseases, and its use may be associated with diminished survival, although, in this study, the observation was attributed to selection bias [11]. Our patient was managed with smoking cessation and steroid therapy, to which, the patient demonstrated a good clinical response as evidenced by symptomatic improvement, increase in FVC and 6MWD, decrease in severity of PH, and stability of radiological changes over 1 year of follow-up. Cladribine use has been reported to achieve a good clinic-radiological improvement in various case series. There is no well-established role of treatment of PH with vasodilators and is therefore managed symptomatically. Supplemental oxygen is used to correct hypoxemia. Lung transplantation is a final option for advanced cases of PLCH.
Conclusion
PLCH is a rare smoking-related ILD associated with multiple variable cysts and nodules preferentially distributed in upper lung lobes sparing the costophrenic angles. Patients with characteristic clinical and radiological features can be diagnosed as PLCH, without a tissue biopsy, and the diagnosis can be cemented once the patient shows a good response to smoking cessation. It needs to be managed with appropriate therapeutic measures based on the clinical condition, and further studies are required to optimize management decisions.
Availability of data and materials
Not applicable
References
Watanabe R, Tatsumi K, Hashimoto S, Tamakoshi A, Kuriyama T (2001) Respiratory failure research group of Japan. Clinico-epidemiological features of pulmonary histiocytosis X. Intern Med 40:998–1003
Suri H, Yi E, Nowakowski G, Vassallo R (2012) Pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis 7:16
Radzikowska E, Blansinska- Przerwa K, Wiatr E, Bestry I, Langfort R, Roszkowski-Sliz K (2018) Pneumothoax in patients with pulmonary Langerhans cell histiocytosis. Lung 196:715–720
Kulwiec E, Lynch D, Aguayo S, Schwarz M, King T (1992) Imaging of pulmonary histiocytosis X. Radiographics 12:515–526
Harari S, Comel A (2001) Pulmonary Langerhan cell histiocytosis. Sarcoidosis Vasc Diffuse Lung Dis 18:253–262
Harari S, Torre O, Cassandro R, Taveira-DaSilva A, Moss J (2012) Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med 106:1286–1292
Elia D, Torre O, Cassandro R, Caminati A, Harari S (2015) Pulmonary Langerhan cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med 26:351–356
Lorillon G, Tazi A (2017) How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev 26:170070
Tazi A, Soler P, Hance A (2000) Adult pulmonary Langerhans’ cell histiocytosis. Thorax 55:405–416
Mason R, Foley N, Branley H, Adamali H, Hetzel M, Maher T et al (2014) Pulmonary Langerhans cell histiocytosis (PLCH): a new UK register. Thorax 69:766–767
Delobbe A, Durieu J, Duhamel A (1996) Wallaert B and the Groupe d’Etude en Pathologie Interstitielle de la Société de Pathologie Thoracique du Nord. Eur Respir J 9:2002–2006
Acknowledgements
None
Funding
There were no funds received from any source.
Author information
Authors and Affiliations
Contributions
(1) Substantial contributions to the conception and design, acquisition, analysis, and interpretation of the data: all authors (AP and MR); (2) drafting the article and revising the article critically for important intellectual content: corresponding author (AP); (3) final approval of the manuscript version to be published: all authors (AP and MR)
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Yes
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Panjwani, A., Salman, M.R. Multiple cystic lung disease in a smoker. Egypt J Intern Med 34, 93 (2022). https://doi.org/10.1186/s43162-022-00184-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43162-022-00184-y