
Abstract
Background
Susac syndrome is a rare disease with multisystem manifestations. While the exact pathogenesis is not known, it has been proposed to be an autoimmune endotheliopathy affecting the microvasculature of the brain, retina, and inner ear. The disease is characterized by a triad of encephalopathy, vision loss, and hearing loss. However, patients may not have the triad at initial presentation and present with only a single finding.
Case presentation
A 25-year-old male resident of Panu Akil presented to Combined Military Hospital Lahore with complaints of dizziness and vertigo associated with severe migraine like headaches. He experienced sudden painless loss of vision in his right eye and was admitted to the medical intensive care unit. On examination, he had right-beating nystagmus and diplopia along with scintillating scotoma. After his admission, contrast-enhanced magnetic resonance imaging (MRI) was done which showed numerous enhancing bilateral white matter internal capsule micro-infarcts indicating typical “string of pearls” sign and a snowball lesion on the corpus callosum. His fundus fluorescein angiography (FFA) did not exhibit any branched retinal artery occlusions (BRAO). Fundoscopy showed the presence of drusen spots. His pure tone audiometry was unremarkable. Based on the highly characteristic findings present on the MRI, a diagnosis of Susac syndrome was made. He was started on injection methylprednisolone 1 gm IV in 500 ml normal saline over 1 h once a day for 5 days and then once a week for 8 weeks. He was also started on tablet mycophenolate mofetil 500 mg once daily for 7 days. Patient showed marked clinical improvement afterwards.
Conclusions
Susac syndrome is a rare multisystem illness with an often insidious presentation. Patients can be misdiagnosed due to the nonspecific nature of the early complaints present in the disease. High index of suspicion is required for timely diagnosis and adequate management. Although no specific guidelines exist, management consists mainly of immunosuppressants.
Similar content being viewed by others


Background
Susac syndrome was first described in 1979 by J. O Susac after whom it is named. He reported the condition in two young women [1]. It is characterized by a clinical triad consisting of encephalopathy, branched retinal artery occlusion, and sensorineural hearing loss [2]. It is an autoimmune encephalopathy primarily affecting women from the ages of 20–40 years. However, male patients have been reported with the age range extending from 7 to 72 years [2]. From the first reported cases in 1979, the number of reported cases increased to 200 worldwide by 2012 [3] and to 500 by 2020 [4]. The disease is extremely rare, and attempts have been made to calculate its incidence in different populations. Annual incidence was calculated to be 0.024/100,000 (95% CI: 0.010–0.047) in a Central European population [5].
Susac syndrome has a wide array of clinical manifestations. However, the triad of encephalopathy, branched retinal artery occlusion (BRAO), and sensorineural hearing loss is characteristic. Early diagnosis is often difficult due to the rarity of the illness and the absence of all three classic symptoms at onset [6]. Attempts have been made to establish a diagnostic criterion for Susac syndrome, and although certain differences are found in each, the involvement of the brain, retinal artery, and ear is present in all established diagnostic criteria.
The exact pathogenesis is still not known, but the involvement of anti-endothelial cell antibodies (AECA) in the development of Susac syndrome has been proposed in 2007 [6], and since then, other studies have been done which suggest a similar involvement of AECA in its pathogenesis [7]. However, further studies are still needed to verify definitive involvement of these antibodies.
The classical triad is diagnostic of the condition; however, very few numbers of patients present with all 3 symptoms initially with only 13% of patients presenting with the classical triad at onset [8]. Initially, central nervous system (CNS) involvement is most common, followed by ocular and then ear involvement [6]. The earliest sign of encephalopathy is headache, and it may precede the development of the other symptoms by approximately 6 months. This headache has been proposed to occur due to involvement of the leptomeningeal vessels [9]. Patients may also present with memory loss, seizures, and dementia. The initial presenting headache, although a very nonspecific symptom is the most common early presenting complaint, present in up to 80% patients and can vary in severity and nature [6]. Due to the nonspecific early symptoms of the disease, patients are frequently misdiagnosed as multiple sclerosis, acute disseminated encephalomyelitis, and Behcet’s disease [10].
The classic ocular finding is branched retinal artery occlusion which can present as vision loss with central or paracentral scotomas. However, if the peripheral retina is involved, patients may be asymptomatic. BRAO involvement can often be bilateral. In addition to BRAO, Gass plaques may also be present which appear as yellow refractile lesions resembling emboli occurring due to lipid extravasation from retinal vessels [6].
Ear involvement presents as hearing loss which is often sudden and causes severe debilitation. It may occur overnight and has been referred to as “bang-bang hearing loss.” Hearing loss may also be accompanied by tinnitus and vertigo [9].
SS has been classified depending on its clinical course as monocyclic, polycyclic and chronic continuous. Monocyclic is self-limiting and resolves by itself in approximately 2 years, polycyclic extends beyond 2 years and chronic continuous has a variable clinical picture with flares occurring at unpredictable times [9].
Diagnosis is based on involvement of all three systems (CNS, eye, ear). MRI of the brain in SS patients showed corpus callosum involvement (snowball lesions considered pathognomonic) with peri- and supraventricular hyperintensities of variable sizes. A more recent finding involving the internal capsule has been reported showing multiple infarcts in a “string of pearls” configuration [3]. Fundus fluorescein angiography revealed BRAO in a majority of patients which was found to be both unilateral or bilateral. Audiometric evaluation revealed sensorineural hearing loss, which was also found to be both unilateral and bilateral [11].
Since the condition is proposed to occur due to autoimmune pathology, the mainstay of treatment has been immunosuppression. Treatment depends on the severity of the presenting illness. Although the dosage may vary, most commonly administered medications are corticosteroids and immunoglobulins. In extreme cases and those refractory to corticosteroids, cyclophosphamide and mycophenolate mofetil have been used [10, 12].
However, no randomized clinical trials have been done which can provide definitive evidence as to the effectiveness of current treatment modalities.
Case presentation
A 25-year-old male resident of Panu Akil presented to Combined Military Hospital Lahore on 19 May 2022 with complaints of dizziness and vertigo associated with severe migraine like headache. He had multiple episodes of vomiting and gait ataxia. He experienced sudden painless loss of vision in his right eye 2 days ago with the vision starting to recover after 2 h of the initial attack. The patient was admitted to the medical intensive care unit with the diagnosis of acute disseminated encephalomyelitis (ADEM) based on the clinical picture.
On examination, he had right-beating nystagmus and diplopia along with scintillating scotoma. His best-corrected visual acuity on presentation at our setup was 6/9 (right eye) and 6/9 (left eye). After his admission, contrast-enhanced MRI was done which showed numerous enhancing bilateral white matter internal capsule micro-infarcts indicating typical “string of pearls” sign and a snowball lesion on the corpus callosum which can be seen in Figs. 1 and 2, respectively.

MRI of the brain showing white matter internal capsule micro-infarcts as “string of pearls” appearance (white arrows)

“Snowball lesion” on the corpus callosum (white arrow)
These infarcts were also noted on cerebellar regions as well. These findings favored the diagnosis of Susac syndrome (SS).
His visual symptoms also raised the suspicion of BRAO (branch retinal artery occlusion) sequelae which is one of the hallmarks of SS. His ocular coherence tomography (OCT) macula was ordered which showed detached retinal pigmentary epithelium in the right eye (Fig. 3). The left eye did not show any detachment (Fig. 4).

Ocular coherence tomography of the right eye showing detached retinal pigmentary epithelium (white arrow)

Ocular coherence tomography of the right eye showing intact retinal pigmentary epithelium
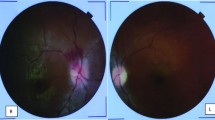
His fundus fluorescein angiography (FFA) however did not exhibit any BRAO. Fundoscopy showed the presence of only drusen spots. This can be attributed to early recognition of the condition before further complications could develop. While the triad of encephalopathy, BRAO, and hearing loss are characteristic, it is not often present early on, and thus, we were able to diagnose the patient before further progression of the illness. FFAs of right and left eyes are shown in Figs. 5 and 6, respectively.

Fundus fluorescein angiography of the right eye

Fundus fluorescein angiography of the left eye
His pure tone audiometry was unremarkable. He had a run of other investigations done in which his cerebrospinal fluid routine exam was normal; the autoimmune encephalitis profile including anti-myelin oligodendrocyte glycoprotein (MOG) antibodies and anti-aquaporin-4 antibodies was unremarkable. Anti-endothelial cell IgG antibodies (AECA) were not available at our hospital, and so the test could not be performed. Plasma exchange was started, of which he had 5 sessions, and his condition started to improve after the third session. He was started on injection methylprednisolone 1 gm IV in 500 ml normal saline over 1 h once a day for 5 days and then once a week for 8 weeks. He was also started on tablet mycophenolate mofetil 500 mg once daily for 7 days. After that, the dose was increased to twice a day for 7 days and then to two tablets twice a day for 7 days. The patient showed marked improvement and has been advised monthly follow-up at our neurology clinic.
Conclusions
Susac syndrome is a rare multisystem illness with an often-insidious presentation. Classical triad of encephalopathy, vision and hearing loss, is diagnostic; however, patients do not present with the triad early in the disease so they may not often be diagnosed. Due to the nonspecific nature of the early complaints, adequate management can be delayed. High index of suspicion is required for timely diagnosis and adequate management. Although no large-scale studies have been done owing to the extreme rarity of the illness, treatment primarily consists of immunosuppressants and has shown clinical improvement in patients.
Availability of data and materials
Available
Abbreviations
- SS:
-
Susac syndrome
- MRI:
-
Magnetic resonance imaging
- FFA:
-
Fundus fluorescein angiography
- BRAO:
-
Branched retinal artery occlusion
- IV:
-
Intravenous
- AECA:
-
Anti-endothelial cell antibodies
- CNS:
-
Central nervous system
- OCT:
-
Ocular coherence tomography
- MOG:
-
Anti-myelin oligodendrocyte glycoprotein
References
Susac JO, Hardman JM, Selhorst JB (1979) Microangiopathy of the brain and retina. Neurology 29(3):313–316. https://doi.org/10.1212/wnl.29.3.313
Kleffner I, Duning T, Lohmann H, Deppe M, Basel T, Promesberger J et al (2012) A brief review of Susac syndrome. J Neurol Sci 322(1-2):35–40. https://doi.org/10.1016/j.jns.2012.05.021
Rennebohm R, Susac JO, Egan RA, Daroff RB (2010) Susac’s syndrome--update. J Neurol Sci 299(1-2):86–91. https://doi.org/10.1016/j.jns.2010.08.032
Susac Syndrome (2020) The portal for rare diseases and orphan drugs. https://www.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=EN&Expert=838. Accessed 9 Aug 2020
Seifert-Held T, Langner-Wegscheider BJ, Komposch M, Simschitz P, Franta C, Teuchner B et al (2017) Susac’s syndrome: clinical course and epidemiology in a Central European population. Int J Neurosci 127(9):776–780. https://doi.org/10.1080/00207454.2016.1254631
Pereira S, Vieira B, Maio T, Moreira J, Sampaio F (2020) Susac’s syndrome: an updated review. Neuro-ophthalmology (Aeolus Press) 44(6):355–360. https://doi.org/10.1080/01658107.2020.1748062
Jarius S, Neumayer B, Wandinger KP, Hartmann M, Wildemann B (2009) Anti-endothelial serum antibodies in a patient with Susac’s syndrome. J Neurol Sci 285(1-2):259–261. https://doi.org/10.1016/j.jns.2009.07.002
Dörr J, Krautwald S, Wildemann B, Jarius S, Ringelstein M, Duning T et al (2013) Characteristics of Susac syndrome: a review of all reported cases. Nat Rev Neurol 9(6):307–316. https://doi.org/10.1038/nrneurol.2013.82
Greco A, De Virgilio A, Gallo A, Fusconi M, Turchetta R, Tombolini M et al (2014) Susac’s syndrome—pathogenesis, clinical variants and treatment approaches. Autoimmunity Rev 13(8):814–821. https://doi.org/10.1016/j.autrev.2014.04.004
Heng LZ, Bailey C, Lee R, Dick A, Ross A (2019) A review and update on the ophthalmic implications of Susac syndrome. Survey Ophthalmol 64(4):477–485. https://doi.org/10.1016/j.survophthal.2019.01.007
Karahan SZ, Boz C, Saip S, Kale N, Demirkaya S, Celik Y et al (2019) Susac syndrome: clinical characteristics, diagnostic findings and treatment in 19 cases. Multiple Sclerosis Related Disord 33:94–99. https://doi.org/10.1016/j.msard.2019.05.018
Egan RA (2019) Diagnostic criteria and treatment algorithm for Susac syndrome. J Neuro Ophthalmol 39(1):60–67. https://doi.org/10.1097/WNO.0000000000000677
Author information
Authors and Affiliations
Contributions
SA and TK made the diagnosis and followed the patient. SA, TK and TL wrote the paper. TL, UI and HS revised the manuscript, UI and HS were the referral opthalmologists. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the patient for publication of this case report and accompanying data. It was approved by the Institutional Review Board for Clinical Research Committee of Pak Emirates Military Hospital. All procedures were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent for publication
Written consent to publish was obtained from the patient prior to submission and is available.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmed, S., Khan, T.A., Liaqat, T. et al. A young soldier with Susac syndrome: a case report. Egypt J Intern Med 34, 82 (2022). https://doi.org/10.1186/s43162-022-00170-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43162-022-00170-4