
Abstract
Background
The aim of this article is to review the essential concepts, current terminologies and classification, management guidelines and the rationale of gender assignment in different types of differences/disorders of sexual development.
Main body
The basics of the present understanding of normal sexual differentiation and psychosexual development were reviewed. The current guidelines, consensus statements along with recommendations in management of DSD were critically analyzed to formulate the review. The classification of DSD that is presently in vogue is presented in detail, with reference to old nomenclature. The individual DSD has been tabulated based on various differential characteristics. Two schemes for analysis of DSD types, based on clinical presentation, karyotype and endocrine profile has been proposed here. The risk of gonadal malignancy in different types of DSD is analyzed. The rationale of gender assignment, therapeutic options, and ethical dimension of treatment in DSD is reviewed in detail.
Conclusion
The optimal management of different types of DSD in the present era requires the following considerations: (1) establishment of a precise diagnosis, employing the advances in genetic and endocrine evaluation. (2) A multidisciplinary team is required for the diagnosis, evaluation, gender assignment and follow-up of these children, and during their transition to adulthood. (3) Deeper understanding of the issues in psychosexual development in DSD is vital for therapy. (4) The patients and their families should be an integral part of the decision-making process. (5) Recommendations for gender assignment should be based upon the specific outcome data. (6) The relative rarity of DSD should prompt constitution of DSD registers, to record and share information, on national/international basis. (7) The formation of peer support groups is equally important. The recognition that each subject with DSD is unique and requires individualized therapy remains the most paramount.
Similar content being viewed by others

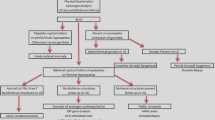

Background
The aim of this article is to review the essential concepts, current terminologies and classification, management guidelines, and the rationale of gender assignment in different types of differences/disorders of sexual development (DSD). The basics of the present understanding of normal sexual differentiation and psychosexual development were reviewed. The current guidelines, consensus statements along with recommendations in management of DSD were critically analyzed to formulate the review. The classification of DSD that is presently in vogue is presented in detail, with reference to old nomenclature. The individual DSD has been tabulated based on various differential characteristics. Two schemes for analysis of DSD types, based on clinical presentation, karyotype, and endocrine profile has been proposed here. The risk of gonadal malignancy in different types of DSD is analyzed. The rationale of gender assignment, therapeutic options, and ethical dimension of treatment in DSD is reviewed in detail.
Main text
The normal sexual differentiation
The normal pattern of human sexual development and differentiation that involves specific genetic activity and hormonal mediators [1, 2] is explained by the classical Jost’s paradigm; the essence of which is narrated below [3].
-
1.
The establishment of chromosomal sex (XX or XY) occurs at the time of fertilization. The variations in sex chromosome include XO, XXY or mosaicism as in XO/XY.
-
2.
Chromosomal sex influences the determination of the gonadal sex, thus differentiating the bipotential gonadal ridge into testis or ovary. (Variations in gonadal sex include ovotestis and streak gonad.) The SRY gene (referred to as the testis-determining gene) on the short arm of Y chromosome directs the differentiation into testes, with formation of Leydig and Sertoli cells [4, 5].
-
3.
The sex phenotype (internal and external genitalia) is determined by the specific hormones secreted by the testes, which translates the gonadal sex into phenotype. Testosterone secretion by Leydig cells promotes Wolffian duct differentiation into vas deferens, epididymis, and seminal vesicles. The Wolffian ducts regress in the absence of androgenic stimulation. Testosterone is converted to dihydrotestosterone (DHT), by 5-alpha reductase, which results in masculinization of external genitalia, closure of urethral folds, and development of the prostate and scrotum. In the absence of influence of SRY gene, the development of bipotential gonad will evolve along the female pathway. Thus, the Mullerian ducts develop (even without any obvious hormonal input) into the uterus, fallopian tubes, and the proximal 2/3 of vagina. DHT is also important for the suppression of development of the sinovaginal bulb, which gives rise to the distal 1/3 of vagina. The fact that internal duct development reflects the ipsilateral gonad (due to the paracrine effect of sex hormones) is an important consideration in the understanding of specific types of DSD. The anti-Mullerian hormone (AMH) from Sertoli cells of Testis is vital for the regression of Mullerian structures. Therefore, Wolffian structures will develop on one side, along with Mullerian duct regression, only in the presence of a fully functional testis. But, Mullerian duct structures develop on one side even in the presence of an ipsilateral streak gonad. The genital tubercle develops as a clitoris, the urethral folds form the labia minora, and the labioscrotal swellings form the labia majora [1, 2, 4,5,6].
The concept of psychosexual development was added to the above sequence by Money et al. [7]. The brain undergoes sexual differentiation consistent with the other characteristics of sex. It is proposed that androgens organize the brain in early development and pubertal steroids activate the same, leading to masculine behavior. The sexual differentiation of genitalia occur in first 2 months of pregnancy, while sexual differentiation of brain occurs in the second half of pregnancy, and hence these processes can be influenced independently. Therefore, the extent of virilization of genitalia may not reflect the extent of masculinization of brain [8, 9].
Psychosexual development is a complex and multifactorial process influenced by brain structure, genetics, prenatal and postnatal hormonal factors, environmental, familial, and psychosocial exposure [10,11,12]. Psychosexual development is conceptualized as three components: (1) gender identity is defined as the self-representation of a person as male, female or even, neither. (2) Gender role (sex-typical behavior) describes behavior, attitudes and traits that a society identifies as masculine or feminine. (3) Sexual orientation denotes the individual responsiveness to sexual stimuli, which includes behavior, fantasies, and attractions (hetero/bi/homo-sexual).
Psychosexual development is influenced by various factors such as Androgen exposure, sex chromosome genes, brain structure, family dynamics and social structure. With reference to altered psychosexual development, two conditions are important to be recognized and differentiated. (1) Gender dissatisfaction denotes unhappiness with the assigned sex, the etiology of which is poorly understood. (With respect to subjects with DSD, it has to be remembered that homo-sexual orientation or cross-sex interest is not considered an indication of incorrect gender assignment.) (2) Gender dysphoria (GD) is characterized by marked incongruence between the assigned gender and experienced/expressed gender, which is associated with clinically significant functional impairment. (It can occur in the presence or absence of DSD) [12,13,14].
The term “disorders/differences of sex development” (DSD) is defined as congenital anomalies in which development of chromosomal, gonadal, or phenotypic sex (including external genitalia/internal ductal structures) is atypical. In a wider perspective, DSD includes all conditions where chromosomal, gonadal, phenotypical, or psychological sex are incongruent. The three components of psychosexual development also may not always be concordant in DSD [15, 16].
A greater understanding of underlying genetic and endocrine abnormalities has necessitated refinement in terminologies and classification of DSD. The newer classification of DSD aims to be more precise, specific, flexible, and inclusive of advances in genetic diagnosis, while being sensitive to patient concerns (Table 1). Terms such as intersex, hermaphrodite, pseudohermaphrodite, and sex reversal are avoided, to this end, in diagnostic terminologies. Presently, a specific molecular diagnosis is identified only in about 20% of all DSD. The majority of virilized 46 XX infants will have CAH, but only 50% of 46 XY DSD will have a definitive diagnosis [16, 17].
For the purpose of understanding of the basic pathology and ease of comprehension, DSD can be classified as follows:
-
1.
Sex chromosomal DSD: here, the sex chromosome itself is abnormal. This includes XO (Turner syndrome), XXY (Klinefelter’s syndrome), mosaic patterns of XO/XY (Mixed Gonadal Dysgenesis and Partial Gonadal Dysgenesis), XX/XY (Ovotesticular DSD), and even SRY-positive XX in 46 XX testicular DSD (de la Chapelle syndrome). These are essentially genetic anomalies characterized by a varying degrees of gonadal dysgenesis/abnormal gonadal differentiation secondary to the sex chromosome defect and in certain situations, associated systemic abnormalities and increased risk of malignancies. The phenotypic sex (internal ductal structures and external genitalia) reflects the gonadal sex.
-
2.
Disorders of gonadal development: these are characterized by abnormal gonadal development, in the absence of any obvious sex chromosomal abnormality, i.e., Karyotype is either 46 XX or 46 XY. It includes 46 XY complete gonadal dysgenesis (Swyer syndrome), 46 XY partial gonadal dysgenesis, 46 XY ovotesticular DSD, 46 XX pure gonadal dysgenesis (Finnish syndrome) and 46 XX ovotesticular DSD. Here also, the phenotypic sex reflects the gonadal sex (streak or dysgenetic gonads/ovotestis).
-
3.
Abnormalities in phenotypic sex secondary to hormonal defects: these are characterized by normal chromosomal sex (46 XX or 46 XY) and gonadal sex (testes/ovaries), but abnormal phenotype (internal ductal and/or external genital) due to defects in hormonal function. In 46 XY DSD, this can be due to defects in synthesis or action of androgens or less commonly, AMH. In 46 XX DSD, this is due to androgen excess, as in Congenital Adrenal Hyperplasia, or less commonly, gestational hyperandrogenism.
-
4.
Primary endocrine abnormalities: These are characterized by a severe underlying endocrine abnormality, as in congenital hypogonadotropic hypogonadism or pan-hypopitutarism.
-
5.
Malformation syndromes: these are characterized by the presence of genital abnormalities due to severe congenital anomalies including persistent cloaca, cloacal exstrophy, Mullerian agenesis/MRKH syndrome, or vaginal atresia.
The common pattern of correlation of gonadal sex with internal duct structure development is summarized in Table 2. The cardinal characteristics of chromosomal, gonadal, and phenotypic sex in the individual types of DSD is summarized in Table 3.
The genetic testing in DSD
For a sex chromosome DSD, no further genetic analysis is required. However, a DSD with 46 XX or 46 XY karyotype, the underlying etiology may be a monogenic disorder where the candidate gene has to be analyzed. The algorithm of genetic analysis of DSD is defined according to the results of sex chromosome complement (karyotyping/array CGH or SNP array) and presence of regions of Y chromosome (FISH/QFPCR). The next step is to study specific genes involved in gonadal development by techniques including Sanger sequencing combined with MLPA to assess specific genetic defects. Further analysis includes evaluation for causes of monogenic DSD or analysis of copy number variations (CNV) or both. Panels for candidate genes (CYP21A2 in CAH, AR in androgen insensitivity syndrome) provide rapid and reliable results. The evolving use of whole exome sequencing (WES) and whole genome sequencing (WGS) aim to identify previously unrecognized genetic etiology of DSD.
The further characterization of 46 XY DSD
The further characterization of individual types of 46 XY DSD based on endocrine and genetic evaluation is summarized in Table 4. The selective use of the following investigations is required in 46 XY DSD to arrive at a specific diagnosis of the subtype:
-
1.
Assay of serum testosterone, LH and FSH.
-
2.
hCG stimulation test, to assess response in testosterone levels.
-
3.
Assay of AMH, to detect the presence of functioning testicular tissue.
-
4.
Testosterone: dihydrotestosterone (DHT) ratio.
-
5.
Testosterone: androstenedione ratio.
-
6.
ACTH test, for the diagnosis of testosterone biosynthesis defects.
-
7.
Specific substrates like progesterone, 17-OHP, and 1-OH pregnenelone, for typing of Androgen biosynthesis defects.
-
8.
Ultrasound scan/MRI and laparoscopy for the detection of Mullerian structures.
-
9.
Gonadal biopsy for the diagnosis of ovotesticular DSD and gonadal dysgenesis.
-
10.
Genetic testing including screening of androgen receptor gene for mutations, Molecular testing for 5-alpha reductase-2 gene mutations, androgen receptor expression, and androgen binding study in genital skin fibroblasts.
The further characterization of 46 XX DSD is summarized in Table 5. The classification of the major types of DSD based on the different clinical manifestations is summarized in Table 6.
Gonadal dysgenesis syndromes
There are five common patterns of gonadal dysgenesis syndromes, in addition to the dysgenetic ovotestis which is found in 46 XX or 46 XY ovotesticular DSD.
-
a.
46 XY complete gonadal dysgenesis (Swyer syndrome)
-
b.
46 XY partial gonadal dysgenesis (Noonan syndrome)
-
c.
45 XO/46 XY mixed gonadal dysgenesis
-
d.
46 XX pure gonadal dysgenesis (Finnish syndrome)
-
e.
45 XO Turner’s syndrome.
Gender assignment in DSD
The classical “optimal gender policy” involved early sex assignment and surgical correction of genitalia and hormonal therapy, with the objective of an unambiguous gender of rearing, that will influence the future gender identity and gender role [7, 11]. The genital phenotype (characteristics of genitalia) has historically been the guide for gender assignment, considering esthetic, sexual, and fertility considerations. This perspective, which assumes psychosexual neutrality at birth, has been challenged now, with the present focus shifting to the importance of prenatal and genetic influences on psychosexual development. In addition to the progress in the diagnostic techniques and therapeutic modalities, there has been greater understanding of the associated psychosocial issues and acceptance of patient advocacy [19,20,21].
Factors to be considered for gender assignment in DSD
-
1.
The most common gender identity outcome, observed incidence of GD, and requirement of gender reassignment in the specific type of DSD from available data.
-
2.
The most common pattern of psychosexual development in the particular DSD, consistent with established neurological characteristics.
-
3.
The requirement of genital reconstructive surgery to conform to the assigned sex.
-
4.
The estimated risk of gonadal malignancy and need for gonadectomy (Table 7).
-
5.
The requirement, possible response, and timing of HRT.
-
6.
The expected post-pubertal cosmetic and functional outcome of genitalia, after reconstruction where required.
-
7.
The potential for fertility, even with the presumed aid of assisted reproduction techniques.
Though GD in patients with DSD influences, the choice of gender assignment (and reassignment), sexual orientation, and gender-atypical behavior do not affect the decision-making process in gender assignment of DSD [22].
Gender assignment in neonates should be done only after expert evaluation. The evaluation, therapy, and long-term follow-up should only be done at a centre with an experienced multidisciplinary team. The multidisciplinary team for management of DSD should include pediatric subspecialists in endocrinology, surgery/urology, genetics, gynecology, and psychiatry along with pediatrician/neonatologist, psychologist, specialist nurse, social worker, and medical ethicist. The core group will vary according to the type of DSD. All individuals with DSD should receive the appropriate gender assignment [22,23,24,25]. The patient and family should be able to have an open communication and participation in the decision-making process. The concerns of patients and their families should be respected and addressed in strict confidence.
The rationale of gender assignment in different clinical conditions of DSD
The usually recommended gender assignment guidelines in different clinical types of DSD is summarized in Table 8.
46 XX DSD—congenital adrenal hyperplasia (CAH)
In CAH, female gender identity is the most common outcome despite markedly masculinized gender-related behavior. Patients diagnosed in the neonatal period, particularly with lower degrees of virilization, should be assigned and reared as female gender, with early feminizing surgery. GD is rare when female gender is assigned. Those with delayed diagnosis and severely masculinized genitalia need evaluation by a multidisciplinary team. Evidence supports the current recommendation to rear such infants, even with marked virilization, as females [18, 19, 22, 23, 26]. A psychological counseling for children with CAH and their families, focused on gender identity and GD, is recommended.
46 XY complete gonadal dysgenesis
It is recommended to rear these children as female, due to following considerations: (a) these patients have typical female psychosexual development. (b) Reconstructive surgery is not required for the external genitalia to be consistent with female gender. (c) Hormonal replacement therapy (HRT) is required at puberty as streak gonads should be removed in view of high risk of gonadal malignancy. (d) Pregnancy is feasible with implantation of fertilized donor eggs and hormonal therapy [19, 22, 23].
Complete androgen insensitivity syndrome (CAIS)
It is recommended that subjects with CAIS should be reared as female, due to the following considerations: (a) they have well documented female-typical core psychosexual characteristics, with no significant GD, in accordance with the proposed absence of androgenization of the brain. (b) Surgical reconstruction of the genitalia is not required for consistency with female gender, though vaginoplasty may be necessary. (c) HRT is required with estrogens after gonadectomy, but testosterone replacement is untenable due to androgen resistance [18, 19, 22, 23, 26].
5-alpha reductase deficiency
Male gender assignment is usually recommended due to the following considerations: (a) the genital tissue is responsive to androgens. (b) The potential for fertility. (c) The reported high incidence of subjects requesting female-to-male gender reassignment after puberty*. (d) HRT is not required at puberty for patients reared as male, if testes are not removed. (e) As the risk of gonadal malignancy is low, testes can potentially be retained. (f) They are very likely to have a male gender identity.*(As most neonates with this disorder have female external genitalia at birth, they are reared as females. Profound virilization occurs at puberty, with a gender role change from female to male during adolescence in up to 63% cases.) About 60% of these patients, assigned female in infancy and virilizing at puberty, and all who are assigned male, live as males. When the diagnosis is made in infancy, the combination of male gender identity in the majority and the potential for fertility, should be considered for gender assignment [19, 22, 23].
17-beta-HSD-3 deficiency
Classical features are that of an undervirilized male. Some of the affected patients with feminine genitalia at birth are reared as females. Virilization occurs at puberty, with gender role change from female to male in up to 64% cases. They are highly likely to identify as males. Male gender assignment is recommended in partial defects. But there is no strong data to support male gender assignment, as in 5-alpha reductase deficiency. The other considerations against male gender assignment are the lack of reported cases of fertility and the intermediate risk of germ cell tumors. Hence, regular testicular surveillance is required for those reared as male, with retained testes. Therefore, gender assignment should be made considering all the above factors [18, 19, 22, 23, 26].
Partial androgen insensitivity syndrome (PAIS)
Infants with PAIS are assigned to male/female gender, depending partially on the degree of undervirilization. The virilization at puberty is also variable and incomplete. The response to hCG stimulation test/testosterone therapy can serve as a guide to the possible sex of rearing. The phenotype is highly variable in PAIS, which is correspondingly reflected in the sex of rearing. The gender identity has considerable fluidity in PAIS, though gender identity is usually in line with the gender of rearing. Though fertility is possible if the testes are retained, it should be remembered that there is an intermediate risk of gonadal germ cell tumors. Hence, gender assignment in these patients is a complex, multifactorial process [18, 19, 22, 23, 26].
47 XXY Klinefelter’s syndrome and variants
They usually report a male gender identity, but with a putative high incidence of GD, which needs to be elaborated in larger series.
Mixed gonadal dysgenesis
The genital phenotype is highly variable. The prenatal androgen exposure, internal ductal anatomy, testicular function at and after puberty, post-puberty phallic development, and gonadal location have to be considered to decide the sex of rearing.
Ovotesticular DSD
These entities were previously referred to as “true hermaphroditism”, signifying the presence of both testicular and ovarian tissue, though dysgenetic, in the same subject. The three patterns seen are as follows:
-
a.
46 XX/XY–33% of ovotesticular DSD, with testis and ovary/ovotestis.
-
b.
46 XX–33% of ovotesticular DSD, with dysgenetic ovotestis.
-
c.
46 XY–7% of ovotesticular DSD, with dysgenetic ovotestis.
This is characterized by ambiguity of genitalia or severe hypospadias at birth, with secondary sexual changes at puberty, corresponding to the relative predominance of ovarian/testicular tissue. The management depends on the age at diagnosis and anatomical differentiation. Either sex assignment is appropriate when the diagnosis is made early, prior to definition of gender identity. The sex of rearing should be decided considering the potential for fertility, based on gonadal differentiation and genital development. It should be ensured that the genitalia are, or can be made, consistent with the chosen sex [19, 22,23,24,25].
General guidelines for surgery and HRT in DSD
Feminizing genitoplasty
Surgery for correction of virilization (clitoral recession, with conservation of neurovascular and erectile structures, and labioplasty) should be carried out in conjunction with the repair of the common urogenital sinus (vaginoplasty). The current recommendation is to perform early, single-stage feminizing surgery for female infants with CAH. It is opined that correction in first year of life relieves parental distress related to anatomic concerns, mitigates the risks of stigmatization and gender identity confusion, and improves attachment between the child and parents. The current recommendation is the early separation of vagina and urethra, the rationale of which includes the beneficial effects of estrogen for wound healing in early infancy, limiting the postoperative stricture formation and avoidance of possible complications from the abnormal connection between the urinary tract and peritoneum through the Fallopian tubes. Surgical reconstruction in infancy may require refinement at puberty. Vaginal dilatation should not be undertaken before puberty. An absent or inadequate vagina, requiring a complex reconstruction of at high risk of stricture formation, may be appropriately delayed. But, the need for complete correction of urogenital sinus, prior to the onset of menstruation, is an important consideration [19, 22,23,24,25,26].
Male genital reconstruction
The standard timing and techniques of operative procedures for correction of ventral curvature and urethral reconstruction, along with selective use of pre-operative testosterone supplementation is advised when male sex of rearing is adopted. The complexity of phallic reconstruction later in life, compared to infancy, is an important consideration in this regard. There is no evidence that prophylactic removal of discordant structures (utriculus/pseudovagina, Mullerian remnants) that are asymptomatic, is required. But symptoms in the future may mandate surgical removal. In patients with symptomatic utriculus, removal can be attempted laparoscopically, though it may not be practically feasible to preserve the continuity of vas deferens [19, 22,23,24,25].
Gonadectomy
The gonads at the greatest risk of malignancy are both dysgenetic and intra-abdominal. The streak gonad in a patient with MGD, raised male should be removed by laparoscopy in early childhood. Bilateral gonadectomy (for bilateral streak gonads) is done in early childhood for females with gonadal dysgenesis and Y chromosome material, which should be detected by techniques like FISH and QFPCR. In patients with defects of Androgen biosynthesis raised female, gonadectomy is done before puberty. The testes in patients with CAIS and those with PAIS, raised as females, should be removed to prevent malignancy in adulthood. Immunohistochemical markers (IHM) that can serve to identify gonads at risk of developing malignancy include OCT 3/ 4, PLAP, AFP, beta-Catenin and CD 117. Early removal at the time of diagnosis (along with estrogen replacement therapy) also takes care of the associated hernia, psychological problems associated with the retained testes and risk of malignancy. Parental choice allows deferment until adolescence, in view of the fact that earliest reported malignancy in CAIS is at 14 years of age. A scrotal testis in gonadal dysgenesis is at risk of malignancy. Current recommendations are surveillance with testicular biopsy at puberty to detect premalignant lesions, which if detected, is treated with local low-dose radiotherapy (with preliminary sperm banking). Also, patients with bilateral ovotestes are potentially fertile from the functioning ovarian tissue. Separation of ovarian and testicular tissue, though challenging, is preferably done early in life [19, 22,23,24,25,26].
Hormonal therapy/sex steroid replacement
Hormonal induction at puberty in hypogonadism should attempt to replicate normal pubertal maturation to induce secondary sexual characteristics, pubertal growth spurt, optimal bone mineral accumulation together with psychosocial support for psychosexual maturation. Treatment is initiated at low doses and progressively increased. Testosterone supplementation in males (initiated at bone age of 12 years) and estrogen supplementation in females (initiated at bone age of 11 years) is given accordingly for established hypogonadism. In males, exogenous testosterone is generally given till about 21 years, while the same in females is variable. Also, in females a progestin is added after breakthrough bleeding occurs, or within 1–2 years of continuous estrogen. No evidence of benefit exists for addition of cyclical progesterone in females without uterus [22,23,24,25].
The advances in molecular diagnosis of DSD
The advent of advanced tools for genetic diagnosis has enabled specific diagnosis to be made by molecular studies. WES and WGS represent evolving translational research that help to identify novel genetic causes of DSD. The techniques for identification of novel genetic factors in DSD have evolved from the use of CGH and custom array sequencing to the use of next generation sequencing (NGS) which mainly includes polymerase-based and ligase-based techniques. The importance of molecular diagnosis in DSD lies in the guidance of management in relation to possible gender development, assessment of adrenal and gonadal function, evaluation of the risk of gonadal malignancy, assessment of the risk of familial recurrence, and prediction of possible morbidities and long-term outcome. Hence, the advances in molecular diagnosis of DSD constitute a rapidly evolving frontier in the understanding and therapy of DSD.
The ethical dimension in DSD
The predominant ethical considerations in management of DSD are twofold. Firstly, when the components of biological sex (the sexual profile of genome, gonads, phenotype, endocrine and neurological status) align strongly, prediction of gender identity and recommendations for sex assignment can be made accordingly. The more discordant the determinants of biological sex, more variation in subsequent components of psychosexual development. Secondly, irreversible anatomic and physiologic effects of surgical assignment of sex have to be avoided, especially when the components of biological sex do not strongly align. The objective in such situations should be to delay such treatment till the appropriate age [24,25,26].
The arguments favoring recognition of DSD as an alternate gender, with delayed sex assignment and deferred surgical therapy has gained ground over the past decades, highlighted by certain judicial interventions across the globe. In this regard, it has to be emphasized that a transgender state, without incongruity of biological sex, has to be clearly distinguished from a DSD. Though differences in psychosexual development can occur in DSD, the vast majority of clinically diagnosed DSD (CAH, MGD, 46 XY DSD) have the anatomic and physiological consequences of altered components of biological sex. The issues in these subjects are not only confined to the genitalia, but also include problems that can include life-threatening cortisol deficiency, features of hypogonadism and urogenital sinus, and even the risk of gonadal malignancy. The early identification and correction of each issue is vital, and the best available window for the same is limited and usually, early in life. It is some of the less frequently encountered types of DSD (ovotesticular DSD, 17-BHSD deficiency, PAIS) that invariably require a more complex decision-making process. The diagnostic and therapeutic approach in the majority of clinically encountered DSD requires a structured scientific approach, with due consideration of the intricacies of psychosexual development.
Conclusion
The optimal management of different types of DSD in the present era requires the following considerations: (1) establishment of a precise diagnosis, employing the advances in genetic testing and endocrine evaluation. (2) A multidisciplinary team is required for the diagnosis, evaluation, gender assignment and follow-up of these children, and during their transition to adulthood. (3) Deeper understanding of the issues in psychosexual development in DSD is vital for therapy. (4) The patients and their families should be an integral part of the decision-making process. (5) Recommendations for gender assignment should be based upon the specific outcome data. (6) The relative rarity of DSD should prompt constitution of DSD registers, to record and share information, on national/international basis. (7) The formation of peer support groups is equally important. The recognition that each subject with DSD is unique and requires individualized therapy remains the most paramount.
Availability of data and materials
Available on request.
Abbreviations
- DSD:
-
Disorders of sexual differentiation
- CAH:
-
Congenital adrenal hyperplasia
- MGD:
-
Mixed gonadal dysgenesis
- CAIS:
-
Complete androgen insensitivity syndrome
- PAIS:
-
Partial androgen insensitivity syndrome
- FSH:
-
Follicular stimulating hormone
- LH:
-
Leutinizing hormone
- hCG:
-
Human chorionic gonadotropin
- FISH:
-
Fluorescence in situ hybridization
- QFPCR:
-
Quantitative fluorescence polymerase chain reaction
- CGH:
-
Comparative genomic hybridization
- MLPA:
-
Multiplex ligand-dependent probe amplification
References
Kim SS, Kolon TF. Hormonal abnormalities leading to disorders of sexual development. Expert Rev Endocrinol Metab. 2009;4:161–72.
Hiort O, Birinbaun W, Marshall L, Wunsch L, Werner R, Schroder T, et al. Management of disorders of sex development. Nat Rev Endocrinol. 2014;10:520–9.
Jost A. A new look at the mechanism controlling sex differentiation in mammals. Johns Hopkins Med J. 1972;130:38–53.
Achermann JC, Hughes IA. Disorders of sex development. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams textbook of endocrinology. 11th ed. Philadelphia: Elsevier Saunders; 2008.
Migeon CJ, Wisniewski AB. Human sex differentiation and its abnormalities. Best Pract Res Clin Obstet Gynaecol. 2003;17:1–18.
Pasterski V. Disorders of sex development and atypical sex differentiation. In: Rowland DL, Incrocci L, editors. Handbook of sexual and gender identity disorders. Hoboken: Wiley; 2008. p. 354–74.
Money J, Hampson JG, Hampson JL. An examination of some basic sexual concepts: the evidence of human hermaphroditism. Bull Johns Hopkins Hosp. 1955;97:301–19.
Gooren L. The endocrinology of sexual behavior and gender identity. In: Jameson L, de Groot LJ, editors. Endocrinology—adult and pediatric, 6th edn. Philadelphia: Elsevier Saunders; 2010.
Swaab DF. Sexual differentiation of the brain and behavior. Best Pract Res Clin Endocrinol Metab. 2007;21:431–44.
Meyer-Bahlburg HF. Sex steroids and variants of gender identity. Endocrinol Metab Clin N Am. 2013;42:435–52.
Money J. The concept of gender identity disorder in childhood and adolescence after 39 years. J Sex Marital Ther. 1994;20:163–77.
Zucker KJ. Intersexuality and gender identity differentiation. J Pediatr Adolesc Gynecol. 2002;15:3–13.
Hughes IA. Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metabol. 2008;22:119–34.
Houk CP, Lee PA. Approach to assigning gender in 46, xx congenital adrenal hyperplasia with male external genitalia: replacing dogmatism with pragmatism. J Clin Endocrinol Metab. 2010;95:4501–8.
Bao AM, Swaab DF. Sexual differentiation of the human brain: relation to gender identity, sexual orientation and neuropsychiatric disorders. Front Neuroendocrinol. 2011;32:214–26.
Fisher AD. Disorders of sex development. In: Kirana PS, Tripodi F, Reisman Y, Porst H, editors. The EFS and ESSM syllabus of clinical sexology. Amsterdam: Medix publishers; 2013.
Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554–63.
Guerrero-Fernández J, Azcona San Julián C, Barreiro Conde J, de la Vega JA B, Carcavilla Urquí A, Castaño González LA, et al. Guía de actuación en las anomalías de la diferenciación sexual (ADS)/desarrollo sexual diferente (DSD) [Management guidelines for disorders/different sex development (DSD)]. An Pediatr (Barc). 2018;89(5):315 e1-315.e19.
Houk CP, Hughes IA, Ahmed SF, Lee PA. Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference. Pediatrics. 2006;118:753–7.
van der Zwan YG, Callens N, van Kuppenveld J, Kwak K, Drop SL, Kortmann B, et al. Long-term outcomes in males with disorders of sex development. J Urol. 2013;190:1038–42.
Warne G, Grover S, Hutson J, Sinclair A, Metcalfe S, Northam F, et al. A long-term outcome study of intersex conditions. J Pediatr Endocrinol Metab. 2005;18:555–67.
Fisher AD, Ristori J, Fanni E, Castellini G, Forti G, Maggi M. Gender identity, gender assignment and reassignment in individuals with disorders of sex development: a major of dilemma. J Endocrinol Investig. 2016;39(11):1207–24.
Hughes IA, Houk C, Ahmed SF, Lee PA. Lawson Wilkins PediatricEndocrine society/European society for Paediatric Endocrinol-ogy consensus group. Consensus statement on management ofintersex disorders. J Pediatr Urol. 2006;2:148–62.
Lee PA, Houk CP, Ahmed SF, HugheRs IA. International ConsensusConference on intersex organized by the Lawson Wilkins pediatric Endocrine Society & the European Society for Paediatric Endocrinology. Consensus statement on management of inter-sex disorders International Consensus Conference on Intersex. Pediatrics. 2006;118:e488–500.
Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. Global DSD Update Consortium. Global disorders of sex development update since 2006: perceptions approach andcare. Horm Res Paediatr. 2016;85:158–80.
Douglas G, Axelrad ME, Brandt ML, Crabtree E, Dietrich JE, French S, et al. Consensus in guidelines for evaluation of DSD by the Texas Children's Hospital multidisciplinary gender medicine team. Int J Pediatr Endocrinol. 2010;2010:919707.
Acknowledgements
The sincere guidance and help provided by Dr. K Sivakumar, Professor and Head, Department of Pediatric Surgery, Government Medical College, Thiruvananthapuram, Kerala, is gratefully acknowledged.
Funding
No funding has been procured for the study.
Author information
Authors and Affiliations
Contributions
The conceptualization, preparation, and revision of the manuscript have been done by the author, Dr. Vivek Parameswara Sarma. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable, as this is a Review article.
Consent for publication
Not applicable. The manuscript is an academic review article without any study of human subjects or any patient data.
Competing interests
The author declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sarma, V.P. A review of the essential concepts in diagnosis, therapy, and gender assignment in disorders of sexual development. Ann Pediatr Surg 18, 13 (2022). https://doi.org/10.1186/s43159-021-00149-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43159-021-00149-w