
Abstract
Background
Diphallus (duplication of phallus) is rarely encountered in surgical practice with only 100 cases reported in literature. Some cases may be isolated but mostly associated with other anomalies, without clear data about its etiology.
Case presentation
We reported a 1-day-old newborn baby who was presented with complete duplication of the phallus, one of them being hypospadic associated with a high type imperforate anus, omphalocele, congenital pouch colon, sacral meningocele, and other congenital anomalies not reported before in such combinations.
Multiple stages surgical corrective procedures were performed over a period of 4 years with 4–6 months interval between each stage, starting with the management of omphalocele and colostomy, ended by excision of the abnormal phallus with abdominoplasty and closure of colostomy.
The outcome was evaluated, and literatures were reviewed in relation to types, presentations, and options for surgical correction with optimal outcomes.
Conclusion
A combination of diphallia and other abnormalities in our patient are not reported previously in such manner and were very difficult to be corrected.
Only expert pediatric surgeons should treat such conditions as every case is unique in nature. In all conditions, the abnormal phallus should be excised, and the final aim is to achieve a continent child with cosmetically acceptable genitalia.
Similar content being viewed by others

Background
Diphallia or penile duplication is an extremely rare abnormality with an incidence of 1 in 6,000,000 births [1,2,3], with approximately 100 cases reported since the first case reported by Weker in 1609 [4, 5].
It can be presented alone as isolated diphallia or in association with different anomalies [6, 7].
Schneider classified diphallia into 3 categories:
-
1.
Duplication of the glans
-
2.
Bifid diphallia
-
3.
Complete penis duplication with each penis having 2 corpora cavernosa and a corpus spongiosum [8]
Velanova et al. added a fourth category:
-
4.
Pseudodiphallia in which there is a rudimentary atrophic penis existing independently from the normal penis [9]
Hallowel and Wilson suggested that complete diphallia could result from longitudinal duplication of the infraumbilical cloacal membrane before the fourth week of gestation, and this can explain the association of genitourinary and intestinal anomalies [10,11,12].
It is believed that diphallia in fetuses occurs when injury, chemical agents, or malfunctioned genome which deeply disturbs the caudal cell mass of fetal mesoderm [7].
A recent report by Karna and Kapur suggested that chromosomal abnormalities may be the etiological factors responsible for this malformation and chromosomal analysis of their own patients shows balanced reciprocal chromosomal translocation 46,XY, t(1:14) (p 36,3: q 24,3) [13].
The surgical corrective management plan for penile duplication should be specific for each patient according to the associated anomalies, degree of penile duplication, functional quality of organs, and cosmetic appearance so as to preserve continence and erectile function [4,5,6].
Case presentation
A male newborn who was delivered with a cesarean section was transferred to our pediatric surgical center with an abnormal lower abdominal swelling in addition to abnormal genitalia. The patient’s parents are phenotypically normal with no family history of anomalies.
The mother was exposed to some diagnostic radiation in the first trimester. The weight of the baby was 2.7 kg with a normal APGAR score and passed clear urine in the first 24 h.
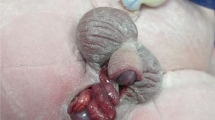
Physical examination demonstrated normal cardiac and respiratory systems, but there were multiple anomalies involving the lower abdomen, pelvis, and lower limbs (Figs. 1 and 2):
-
1.
A big infraumbilical omphalocele 10–12 cm in diameter
-
2.
Completely well formed, widely separated phallus on each side, one of them with hypospadias and dribbling of urine from both phallus on crying
-
3.
Separated hemiscrotum with each phallus with bilateral undescended testis and hernia
-
4.
High type imperforate anus
-
5.
Two separated anal dimple and cleft
-
6.
Abnormal bony pelvis with widely separated pubic symphysis.
-
7.
Sacrococcygeal swelling proved to be a meningocele later on
-
8.
Right-sided club foot

Hypogastric omphalocele, club foot, widely separated pubic symphysis, bilateral inguinal testis, and hernia

Sacral meningocele, left hypospadic phallus, double anal cleft, and raphae
On admission, the baby was stable; plain x-ray in AP view and lateral invertogram suggest extra pelvic bone inside with high type imperforate anus.
On exploration, a transverse colostomy was performed with excision of the omphalocele sac, and congenital cystic transverse colon was found. The baby recovered well postoperatively and was discharged home 7 days later with functioning colostomy to be lost for follow-up.
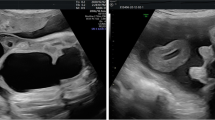
Two years later, the parents returned for advice to reconstruct the remaining anomalies. Evaluation of the anatomy was done by ultrasound, MRI, IVP, and MCU, which showed an abnormally shaped urinary bladder and left-sided grade 1 vesicoureteric reflux.
Multiple stages of reconstructive surgeries were performed within 4 months interval:
-
1.
Excision of the prolapsed cystic colon and refashioning of colostomy
-
2.
Excision of the sacral meningocele
-
3.
Correction of the lower limb deformity
-
4.
Posterior sagittal anorectoplasty and closure of the rectourethral fistula to the hypospadic phallus
-
5.
Excision of the hypospadic phallus with lower abdominoplasty and approximation of hemiscrotum with orchidopexy (Figs. 3 and 4).
-
6.
Closure of colostomy

Excision of the abnormal phallus

Scrotal approximation and abdominoplasty
The child developed a small urinary fistula at the site of the excised phallus which was closed later.
The child is 6 years old now, still using diapers but partially continent for stool and urine.
Regular follow-up and assessment demonstrates improvement in his vesicoureteric reflux, but further evaluation is needed for his covered exstrophy, cosmetic appearance, and continence.
Discussion
Neugebauer in 1898 and Nesbit and Bromme in 1933 were reviewed cases of diphallus in literature [14], after the first case reported by Weker in 1609 [4].
Although only 100 cases were reported in literatures, we believe that this is merely an underestimation as many of these are unrecorded or not reported to the surgical practice.
Ravi et al. and Wilson et al. classified diphallus as glandular, bifid, concealed, complete, hemidiphallus, and triple penises [11, 15]. We found that the Schnieder and Velanova classification is more appropriate and applicable to our case [8, 9].
In complete diphallia, the most common additional anomalies are intestinal and genitourinary, with the most common intestinal anomaly being an imperforate anus and the least common being rectal duplication [16, 17].
Karka et al. reported a case of complete diphallia with hypogastric omphalocele that had not been reported previously [5]. In our case, a big hypogastric (infraumbilical) omphalocele was the presenting anomaly and it is the second case reported to date, and it was successfully primarily repaired.
In addition to that, there was a feature of covered or pseudo-exstrophy in the form of wide pubic diastasis and low set umbilicus in our case similar to a case reported by Chadha et al. but without omphalocele [18]. This covered exstrophy is believed to result from persistence without rupture of an overdeveloped infraumbilical cloacal membrane [19, 20].
In addition to omphalocele and covered exstrophy, all other associated anomalies in our case were not reported in such association in previous literatures, particularly the presence of congenital cystic colon, sacral meningocele, and the extra pelvic bone inside.
Chadha et al. suggested that it is difficult to explain the completely well-developed duplicated penis in his case and other reported cases [18]. It may be due to an alteration of embryonic development, but it is not completely understood [2]. In our case, such a severe type of diphallus and associated anomalies may be contributed to an injury or malfunctioning gene [7], which may be secondary to radiation exposure to the mother in the first trimester.
Life-threatening anomalies should be treated first [4]; therefore, our management was concentrated primarily to the associated omphalocele and imperforate anus as priority, leaving diphallus to be treated electively.
All literatures recommended the removal of one of the duplicated phallus with its urethra and selecting the abnormal phallus [2, 4,5,6]; we excised the hypospadic phallus, but we faced difficulties in approximating the widely separated hemiscrotum and testes in the midline. This will require future refashioning to be cosmetically acceptable.
Although all surgical procedures were done successfully in our case, the patient still calls for good, regular follow-up, and plans for further intervention is needed for more acceptable functional and cosmetic results.
Conclusion
Complete diphallia is a rare anomaly of unclear causes, demanding careful assessment because of a high incidence of multiple life-threatening anomalies association that should be treated first.
Combination of diphallia and other abnormalities in our case is not reported previously in such manner and was very difficult to be corrected.
Only expert pediatric surgeons should treat such conditions in stages as every case is unique in nature.
In all conditions, the abnormal phallus should be excised, and the final aim is to achieve a continent child with cosmetically acceptable genitalia.
Availability of data and materials
All data and materials are available.
References
Viswanatha RT, Chandrasekharam V. Diphallus with duplications of cloacal derivatives, report of a rare case. J Urol. 1980;124:555–8.
Alireza M, Fatollah R, Shahnaz S, Leily M, Shaghyegh H. Diphallus with imperforate anus and complete duplication of rectosigmoid colon and lower urinary tract. Iran J Pediatr. 2010;2(2):229–32.
Adair EL, Lewis EL. Ectopic scrotum and diphallia: Report of a case. J Urol. 1960;84:115–7.
Tarik IA. Case report: supernumery penis (Diphellia terrata). Int J Collab Res Intern Med Public Health. 2012;4(5):502–6.
Karak I, Erdal T, Basak AU, Derya Y, Gulcin I, Derya E. Surgical management of complete penile duplication accompanied by multiple anomalies. Canadian Assoc Urol J. 2014;8(9-10):741–3.
Kekler S, Stephany HA, Splide TL, Snyder CL. Isolated diphallia: case report and literature review. Eur J Pediatr Surg. 2009;19:254–5.
Kardasevic M, Begic F, Sivic M. Penile duplication in newborn with multiple anomalies. Med Arh. 2012;66:206–8.
Schneider P. Cited by Lamitter JK, Uson AC, Melicow AC: The male genital tract. In: Mustard WT, Ravitch MM, Snyder WH, et al., editors. Pediatric Surgery vol 2. 2nd ed. Chicago: Year Book Medical Publishers; 1969. p. 1263. (chap 77).
Vilanova X, Raventos A. Pseudodiphallia - a rare anomaly. J Urol. 1954;71:338–46.
Hallowell JG, Witherington R, Ballagas AJ. Embryologic considerations of diphallus and associated anomalies. J Urol. 1977;117:728–32.
Wilson JSP, Horton C, editors. Diphallus plastic and reconstructive surgery of genital area; 1973. p. 1888–91.
Djordjevic ML, Perovic SV. Complex penile joining in a case of wide penile duplication. J Urol. 2005;173:587–8.
Karna P, Kapur S. Diphallus and associated anomalies with balanced autosomal chromosomal translocation. Clin Genet. 1994;46:209–11.
Abdulkadir T, Mert A, Unsal O, et al. Complete diphallus in a 14 year old body. Marmara Med J. 2007;20(3):190–2.
Ravi K, Madan MS. Complete duplication of bladder, urethra and external genitalia. J Urol. 1987;137:1243–5.
Dewan PA, Lawrence MJ, Pip A, et al. Diphallus associated with partial caudal duplication. Pediatr Surg Int. 1998;14:131–3.
Bakheet MA, Refaei M. Penile duplication and two anal openings: report of a very rare case. Iran J Pediatr. 2012;22:133–6.
Chadha R, Bagga D, Gupta S, Mahajan JK. Complete diphallia associated with features of covered exstrophy. J Pediatr Surg. 2001;36(7):1–3.
Marshall VF, Muecke EC. Variations in exstrophy of the bladder. J Urol. 1962;88:766–96.
Boemers TML, de Jong TPVM, Rovekamp MH, et al. Covered exstrophy associated with an anorectal malformation: a rare variant of classical bladder exstrophy. Pediatr Surg Int. 1994;9:438–40.
Acknowledgements
I would like to express my sincere thanks and gratitude to all medical and paramedical staff at Al khansaa Teaching Hospital for assisting and helping me during the long period of management of this difficult case and also to thank the parents who were so patient and cooperative with me to do all the requirements for managing the difficult anomalies of their child.
Funding
No funding (not applicable)
Author information
Authors and Affiliations
Contributions
The sole author wrote the manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. The institute only gives it to articles but not case reports.
Consent for publication
Both parents of my patient agreed for publication verbally, and recently, the parents sent me an agreement for publication electronically; nothing in the manuscript refers to the identity of the child.
Competing interests
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Abbasi, B.K. Complete diphallia associated with unusual multiple congenital anomalies: case report and review of literatures. Ann Pediatr Surg 18, 7 (2022). https://doi.org/10.1186/s43159-021-00141-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43159-021-00141-4