
Abstract
Background
Essential hypertension (EH) is an important risk factor for various cardiovascular, cerebral and renal disorders. It is a multi-factorial trait which occurs through complex interplay between genetic, epigenetic, and environmental factors. Even after advancement of technology and deciphering the involvement of multiple signalling pathways in blood pressure regulation, it still remains as a huge global concern.
Main body of the abstract
Genome-wide association studies (GWAS) have revealed EH-associated genetic variants but these solely cannot explain the variability in blood pressure indicating the involvement of additional factors. The etiopathogenesis of hypertension has now advanced to the level of epigenomics where aberrant DNA methylation is the most defined epigenetic mechanism to be involved in gene regulation. Though role of DNA methylation in cancer and other mechanisms is deeply studied but this mechanism is in infancy in relation to hypertension. Generally, 5-methylcytosine (5mC) levels are being targeted at both individual gene and global level to find association with the disease. But recently, with advanced sequencing techniques another methylation mark, N6-methyladenine (6mA) was found and studied in humans which was earlier considered to be absent in case of eukaryotes. Relation of aberrant 6mA levels with cancer and stem cell fate has drawn attention to target 6mA levels with hypertension too.
Conclusion
Recent studies targeting hypertension has suggested 6mA levels as novel marker and its demethylase, ALKBH1 as probable therapeutic target to prevent hypertension through epigenetic programming. This review compiles different methylation studies and suggests targeting of both 5mC and 6mA levels to cover role of methylation in hypertension in broader scenario.
Similar content being viewed by others

Background
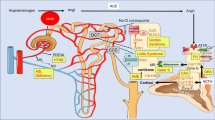
Hypertension, more commonly known as high blood pressure, occurs due to constantly high pressure on the walls of blood vessels. It is broadly classified into primary and secondary where primary or essential hypertension (EH) is more prevalent form affecting around 95% of adult patients having high blood pressure [1]. Primary hypertension is one which occurs without any known identifiable cause whereas secondary hypertension occurs secondarily to certain other primary ailment. Hypertension affects more than 1.5 billion people worldwide and is an important risk factor for various cardiovascular, cerebrovascular and other renal disorders causing considerable public health concern [2]. EH is a complex disorder [3] and occurs as a result of interplay between genetic and environmental factors (Fig. 1). Though association studies [4,5,6] targeting specific genes have found certain polymorphisms to be linked with the pathogenesis of hypertension but such association studies sometimes culminates into non-reproducible results in another population cohort. Additionally, genome-sequencing results have revealed presence of disease causing mutation without phenotypic presence [7]. Genome-wide association studies (GWAS) have identified different genetic loci to be associated with hypertension [8,9,10,11]. These genetic loci accounts for small fraction of heritability highlighting either the cumulative effect of such polymorphisms or role of some other non-genetic factor (epigenetic mechanisms) that can be modified through lifestyle or environmental factors [1] for disease outcome. This led to epigenome-wide association studies (EWAS) where DNA methylation at different CpG sites was found to be associated with blood pressure in individuals from varied ancestry [12, 13]. Epigenetics refer to the changes that occur in gene expression pattern without any change in the DNA sequence. Epigenetic modifications can be initiated by different factors that include environmental influence during foetal development, dietary habits, routine lifestyle, aging, and sometimes medical prescriptions [14]. Most commonly targeted epigenetic modifications (Fig. 2) include DNA methylation, histone modification and RNA-based mechanisms [15]. Out of these, DNA methylation is the vastly studied epigenetic mechanism affecting gene regulation in mammals [16]. It is best characterized epigenetic mark in relation to cancer but its role in hypertension is still in its infancy. Readers are suggested for detailed reviews [3, 17,18,19] on epigenetic mechanisms targeting blood pressure regulation as current article focuses specifically on the role of DNA methylation with emphasis on novel methylation mark in relation to hypertension.

Gene-environment interaction for phenotypic manifestation of hypertension

Major epigenetic regulators
Main text
DNA methylation
DNA methylation plays a crucial role in regulating many cellular processes like embryonic development, X-chromosome inactivation (inactivation of one of the female X-chromosome for dosage compensation), and genomic imprinting (epigenetic marks control gene expression in parent-specific manner where either of paternally or maternally inherited allele is active rather than activation of both alleles) [20]. DNA methylation in mammals mainly occurs at cytosine in cytosine-guanine dinucleotides (CpG) which involves the transfer of methyl group (CH3) from S-adenosyl-methionine (SAM) to the 5 carbon position of cytosine resulting in the formation of 5-methylcytosine (5mC) [15]. The “P” identifies the presence of phosphodiester bond between cytosine and guanine nucleotides [21]. CpG islands are mostly located within the promoter region or in the end 5′ region and are generally unmethylated [22]. CpG dinucleotide sites other than the promoter region have different methylation status and are methylated [23]. Methylation profile of cytosine in promoter CpG islands affects expression pattern of the gene. Promoters can be differentiated as high, intermediate, and low CpG promoters and anti-correlation has been found between CpG density and DNA methylation in genome wide DNA methylation analysis [24]. DNA methylation is functionally linked to suppression of gene transcription [14] (Fig. 3). This is achieved either directly by preventing the binding of transcriptional activators to promoter region or indirectly by attachment of methyl-CpG binding proteins (MBP) [25]. Mammalian DNA methylation is carried by DNA methyltransferases (DNMTs) including DNMT1, DNMT3a, and DNMT3b (Fig. 3). DNMT1 is mainly responsible for maintaining DNA methylation pattern in the newly synthesized DNA strand whereas DNMT3a and DNMT3b are responsible for de novo DNA methylation [26]. Pathway for active DNA demethylation involves ten-eleven-translocases (TET) family (Fig. 3) of proteins [27] that converts 5mC to 5-hydroxymethlycytosine (5hmC) and further to 5-formylcytosine (5fC) which subsequently is oxidised to form 5-carboxylcytosine (5caC) [1, 28]. These TET dependant oxidation products are considered as demethylation intermediates of 5mC playing crucial role in transcription, chromatin remodelling and DNA repair [29]. Both 5fC and 5caC are recognized and excised by thymine-DNA glycosylase (TDG) resulting in replacement by unmodified cytosine [1]. Effect of DNA methylation on gene expression is vastly studied in relation to cancer but its role with context to hypertension has recently gained interest. Pioneer work on rats highlighted the role of DNA methylation on blood pressure regulating genes.

Role of DNA methylation on gene expression
DNA methylation and hypertension
One of the pioneer studies targeting the effect of diet on blood pressure was done by Bogdarina et al. [30]. Low protein diet in pregnant rats yielded hypertensive offsprings by upregulation of AT1b gene through promoter hypomethylation. Similarly, maternal low protein diet (MLPD) altered expression of RAS genes in fetal brains where promoter hypomethylation increased expression of ACE gene [31]. In similar manner, MLPD during gestation led to hypertension in sexually dimorphic pattern in the offsprings where females were severely affected and males were moderately hypertensive [32]. Further studies on rats showed enhanced expression of AT1a in offsprings of nicotine administered pregnant rats as a result of promoter hypomethylation resulting in hypertension [33]. Promoter hypomethylation heightened AT1a expression causing increased blood pressure in spontaneously hypertensive rats (SHR) compared to Wistar-Kyoto (WKY) rats [34]. Similarly, hypomethylation-related upregulation of Na-K-Cl cotransporter I gene (NKCC1) caused hypertension in SHR [35]. Riviere et al. [36] performed in vitro and in vivo studies targeting the role of DNA methylation on promoter CpG islands of ACE gene. Recently epigenetic studies on human genes regulating blood pressure have also come up where methylation status at global and individual gene level is being targeted. Lower 5mC levels in DNA of hypertensive individuals was found by Smolarek et al. [37] whereas increased global DNA methylation levels were found in pre-eclampsia group by Kulkarni et al. [38] in comparison to controls. Genome-wide methylation analysis validated significant CpG sites in sulfatase gene (SULF1) and prolylcarboxypeptidase gene (PRCP) [39]. Methylation analysis between cases and controls showed significant methylation difference in the promoter CpG site of SULF1 gene where hypermethylation was observed in the hypertensive cases. Similarly, trans-ancestry genome wide association on East Asian, European, and South Asian individuals found genetic variants at 12 new loci in the genes involved in vascular smooth muscle and renal function to be associated with blood pressure [40]. Sentinel blood pressure SNPs from this study were found to be affected by methylation status of nearby CpG sites highlighting the role of DNA methylation on blood pressure regulation. Along with such studies on global methylation difference, role of DNA methylation pattern in altering gene expression is also studied at individual gene level. Enzyme 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2) is involved in the conversion of cortisol to cortisone and inactivity of this enzyme results in cortisol-based activation of mineralocorticoid receptors resulting in sodium retention and elevated blood pressure. Study on epigenetic control of HSD11B2 gene coding for this enzyme showed that promoter methylation resulted in hypertension by gene downregulation [41]. Promoter hypomethylation at 5 CpG sites in the promoter of alpha-adducin gene (ADD1) coding for α-adducin was found to be associated with hypertension. Gender-based segregation related hypomethylation at CpG1 with increased risk of EH in females whereas hypomethylation at CpG2-5 sites increased the risk in males [42]. Similarly, methylation pattern in CpG island of glucokinase (GCK) gene showed association with EH. Patients with EH showed hypomethylation at CpG1-3 sites and hypermethylation at CpG4 site of the studied gene [43]. Additionally, genes in which role of promoter methylation in EH is targeted includes angiotensin II type 1 receptor gene, AGTR1 [44, 45], aldosterone synthase gene-Cytochrome P450 family 11 subfamily B member 2, CYP11B2 [46], sodium channel epithelial 1 beta subunit gene, SCNN1B [47], interleukin-6 gene, IL-6 [48], toll-like receptor 2 gene, TLR2 [49], and mitofusin 2 gene, Mfn2 [50]. Association between promoter hypermethylation of beta-3-adrenoreceptor gene (ADRB3) with dyslipidemia and blood pressure was targeted by Guay et al. [51].
N6-methyladenine as new epigenetic mark
Three type of methylated adenine bases are considered biologically relevant where 1-methyladenine (1mA) and 3-methyladenine (3mA) occurs due to alkylation damage in DNA and 6-methyladenine (6mA) is mainly attributed by enzymatic activity [52]. 6mA and m6A refers to methylated adenine in DNA and RNA respectively. In contrast to 5-methylcytosine (5mC), N6-methyladenine (6mA) modification of DNA is more prevalent in case of prokaryotes where it plays crucial role from DNA replication and repair to viability of certain bacterial strains [53,54,55]. Earlier studies failed to detect DNA 6mA modification in eukaryotes which led to consideration of absence of such modification in the eukaryotic system including humans [56]. But with the technological breakthroughs and development of deep sequencing, eukaryotic species were also reported to possess 6mA DNA modification including Chlamydomonas reinhardti, Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana, fungi, Mus musculus, Oryza sativa, zebra fish, and pig [53, 57, 58]. In addition to other chemical modifications of RNA, m6A RNA modification of human mRNA plays pivotal role in RNA splicing, mRNA stability and gene expression [53, 57]. Presence of 6mA DNA modification was considered to be absent in humans in previous studies [56] but advanced sequencing techniques found presence of 6mA even in human genomic DNA [59]. 6mA DNA modification sites were identified in the human genome which accounted for approximately 0.051% of the total adenines in the human genome and results indicated [G/C]AGG[C/T] as most significant motif associated with 6mA modification [53]. This group further presented data on density distribution of 6mA across all human chromosomes and found presence of 6mA modification sites mostly in the intronic and intergenic regions with no significant enrichment of 6mA around transcription start sites (TSSs) and transcription terminal sites (TTSs) in the human genome [53]. The functional effects of N6-methyladenine modification is regulated by methyltransferases, demethylases and binding proteins [55]. Methyltransferase responsible for human 6mA DNA modification is N-6 adenine-specific DNA methyltransferase 1 (N6AMT1) as observed through silencing and overexpression experiments [53]. Similarly, methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14) methyltransferase complex in association with adaptor proteins like Wilms’ tumor 1 associating-protein (WTAP), RNA binding protein motif 15 (RBM15) and KIAA1429 is responsible for m6A modification where adaptor proteins contribute to functional activity of methyltransferase complex [55, 60, 61]. m6A RNA readers recognize and bind to m6A sites in RNA regulating downstream functions by affecting splicing, export, stability, and translation of mRNA [55, 62, 63]. These m6A RNA readers include YTH family of proteins comprising of YTHDC1, YTHDC2 and YTHDF (YTHDF1, YTHDF2, and YTHDF3) family and IGF2BP family proteins (IGF2BP1, IGF2BP2, and IGF2BP3) [55, 64]. YTH family of proteins recognize through YTH RNA-binding domain [55, 65] and IGF2BP family proteins recognize and bind GG (m6A)C sequence through K homology domains [55]. Demethylation of DNA 6mA is performed by ALKBH1 enzyme [53,54,55] whereas demethylases involved in removal of m6A methylation include fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) [55, 57, 61] (Fig. 4). Involvement of m6A modification is found in circadian rhythm, viral replication, embryogenesis and development of human diseases (obesity, cancer and neuronal disorders) [58, 61, 62]. Recent data have shown involvement of DNA 6mA modification in the development and progression of cancer. Increased activity of demethylase AKLBH1 and decreased activity of N6AMT1 methyltransferase was reported in cancer cells that resulted in decreased abundance of 6mA in targeted cancer tissues [53]. Contrary to this, higher levels of DNA 6mA were observed in glioblastoma stem cell (GSC) and primary glioblastoma specimens compared to normal human astrocytes relating higher 6mA levels with disease progression [66]. Another major role of DNA 6mA modification was related with DNA damage repair [67] where N6-methyladenine minimizes incorporation of 8-oxo-2’-deoxyguanosine (8-oxoG) against adenine by DNA polymerase. Thereby, preventing the chance of mutation through oxidative damage by avoiding mispairing of 8-oxoG with adenine. Additionally, the role of m6A modification in regulating fate of stem cell was also reported [68]. Depletion of ALKBH1 demethylase in mice resulted in deformities in eyes, skeletal and neural system and even early embryonic lethality [69,70,71]. Knockout of ALKBH1 gene resulted in undifferentiated state of embryonic stem cells (ESCs) suggesting possible role of m6A in transcriptional silencing [72]. Contradictory results of m6A level on stem cell fate also exists where inhibition of METTL3 methyltransferase complex in embryos of zebrafish prevented progression of cell cycle and resulted in developmental defects [73].

Regulation of m6A modification and role in RNA processing
N6-methyladenine modification and hypertension
Role of novel DNA methylation modification, N6-methyladenine (6mA) in the prognosis of hypertension is largely unclear. Recently, handful of papers has found relation between 6mA DNA level and hypertension. Reduced 6mA methylation level was found in the leukocytes of hypertensive patients which returned back to normal after successful treatment of hypertension [74]. Results similar to clinical investigation were observed by them in both mouse and rat hypertension models which showed reduced 6mA DNA methylation level in the leukocytes. Increased ALKBH1 level reduced global 6mA DNA level in human aortic smooth muscle cells (HASMCs) treated with different concentrations of Ang II and endothelin 1 (ET-1). Results were confirmed through siRNA transfection where silencing of ALKBH1 reduced the reduction in 6mA DNA level in the treated HASMCs [74]. Similarly, elevated AKLBH1 level was found to be associated with reduced 6mA DNA level in the vascular smooth muscle cells (VSMCs) of hypertension models through in vitro and in vivo studies. Further experiments identified hypoxia inducible factor 1 α (H1F1α) that is required for Ang II-induced vascular remodelling as the target of ALKBH1. Relation between them was found as elevation in H1F1α level was inhibited by silencing of ALKBH1. Their findings suggested 6mA as sensitive marker for diagnosis of hypertension and ALKBH1 as novel therapeutic target to prevent hypertension through epigenetic reprogramming because ALKBH1 mediates cross talk between 6mA DNA level and H1F1α activity. In contrary to this, earlier reports found increased m6A levels due to decreased expressed or mutations of another demethylase gene, FTO to be associated with hypertension and cardiovascular diseases [75, 76]. On the basis of these results, Paramasivan et al. [77] suggested targeting of m6A levels through methyltransferase (METTL3) and demethylase (FTO) enzyme as potential therapeutic strategy for hypertension. Pulmonary artery hypertension (PAH) disease is characterized by pulmonary vascular remodelling culminating in right heart failure [78]. Role of methylation in disease intervention of PAH has been observed [79] but the role of internal modification of mRNA, N6-methyladenine (m6A) in PAH was recently studied [64]. Experiments were performed in mouse model with SET domain containing 2 histone lysine methyltransferase (SETD2)-specific knockout of smooth muscle cells (SMCs). SETD2 enzyme catalyzes trimethylation of lysine 36 on histone 3 (H3K36me3) which is a histone marker for active transcription region [80]. Lack of SETD2 in SMCs protected mice from hypoxia-induced PAH and further reduced level of methyltransferase like 14 (METTL14) and methylation of m6A RNA [64]. Involvement of SETD2-mediated H3K36me3 in PAH was observed where clear differences were found in protein levels of SETD2 and H3K36me3 in SMCs and SETD2-deficient SMCs. SETD2-deficient SMCs further showed impaired protein level of METTL14 and diminished m6A RNA levels [64]. Their results demonstrated that SETD2 mediated m6A RNA modification through H3K36me3 and METTL14 to result into hypoxia-induced PAH in mice. They further suggested inhibition of SETD2/METTL14 activity as a possible strategy for treatment of pulmonary artery hypertension in clinical settings. These few ongoing studies provides a hint towards potential role of 6mA level in the progression of hypertension which can be targeted in future studies deciphering the role of methylation in disease manifestation.
Conclusion
N6-methyladenine modification of DNA is prominently found in prokaryotes but considered to be absent in case of eukaryotes. With the development of deep sequencing it was found to be present in limited number of eukaryotes too. Though m6A modification of mRNA plays crucial role in RNA splicing, export, stability, decay, and gene expression in humans but presence of 6mA in human genomic DNA was recently identified through single-molecule real-time (SMRT) sequencing technology. Role of methyltransferase and demethylase responsible for affecting 6mA and m6A levels was recently found in different diseases and mechanisms involving cancer and stem cell fate. As a result, research targeting role of these enzymes in hypertension found relation of 6mA level with hypertension progression. Current research suggests targeting of 6mA level as novel diagnostic marker and demethylases, AKLBH1, and FTO as potential therapeutic strategy against hypertension.
Availability of data and materials
There is no availability of data and materials.
Abbreviations
- ADD1 :
-
Alpha-adducin gene
- AGTR1 :
-
Angiotensin II type 1 receptor gene
- ALKBH5:
-
AlkB homolog 5
- ADRB3 :
-
Beta-3-adrenoreceptor gene
- 11 beta-HSD2:
-
11 beta-hydroxysteroid dehydrogenase type 2
- 5caC:
-
5-carboxylcytosine
- CYP11B2 :
-
Cytochrome P450 family 11 subfamily B member 2
- DNMTs:
-
DNA methyltransferases
- EH:
-
Essential hypertension
- ET1:
-
Endothelin 1
- EWAS:
-
Epigenome-wide association studies
- 5fC:
-
5-formylcytosine
- FTO:
-
Fat mass and obesity associated protein
- GWAS:
-
Genome-wide association studies
- GSC:
-
Glioblastoma stem cell
- GCK :
-
Glucokinase gene
- HASMCs:
-
Human aortic smooth muscle cells
- 5hmC:
-
5-hydroxymethlycytosine
- H1F1α:
-
Hypoxia inducible factor 1 α
- IL-6 :
-
Interleukin-6
- MLPD:
-
Maternal low protein diet
- 1mA:
-
1-methyladenine
- 3mA:
-
3-methyladenine
- 6mA:
-
6-methyladenine
- 5mC:
-
5-methylcytosine
- METTL3:
-
Methyltransferase-like 3
- METTL14:
-
Methyltransferase-like 14
- Mfn2 :
-
Mitofusin 2 gene
- N6AMT1:
-
N-6 adenine-specific DNA methyltransferase 1
- NKCC1 :
-
Na-K-Cl cotransporter I gene
- 8-oxoG:
-
8-oxo-2′-deoxyguanosine
- PRCP :
-
Prolylcarboxypeptidase gene
- PAH:
-
Pulmonary artery hypertension
- RBM15:
-
RNA binding protein motif 15
- SAM:
-
S-adenosyl-methionine
- SCNN1B :
-
Sodium channel epithelial 1 beta subunit gene
- SETD2 :
-
SET domain containing 2 histone lysine methyltransferase
- SMRT:
-
Single-molecule real-time
- SMCs:
-
Smooth muscle cells
- SHR:
-
Spontaneously hypertensive rats
- SULF1 :
-
Sulfatase gene
- TET:
-
Ten-eleven-translocases
- TDG:
-
Thymine-DNA glycosylase
- TLR2 :
-
Toll-like receptor 2 gene
- TSSs:
-
Transcription start sites
- TTSs:
-
Transcription terminal sites
- VSMCs:
-
Vascular smooth muscle cells
- WTAP:
-
Wilms’ tumor 1 associating-protein
- WKY:
-
Wistar-Kyoto rats
References
Demura M, Saijoh K (2017) The role of DNA methylation in hypertension. Adv Exp Med Biol 956:583–598
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J (2005) Global burden of hypertension: analysis of worldwide data. Lancet 365:217–223
Wise IA, Charchar FJ (2016) Epigenetic modifications in essential hypertension. Int J Mol Sci 17:451
Wu CK, Tsai CT, Chang YC, Luo JL, Wang YC, Hwan JJ et al (2009) Genetic polymorphisms of the angiotensin II type 1 receptor gene and diastolic heart failure. J Hypertens 27:502–507
Niu S, Zhang B, Zhang K, Zhu P, Li J, Sun Y et al (2015) Synergistic effects of gene polymorphisms of the renin-angiotensin-aldosterone system on essential hypertension in Kazakhs in Xinjiang. Clin Exp Hypertens 13:1–8
Singh M, Singh AK, Singh S, Pandey P, Chandra S, Gambhir IS (2016) Angiotensin-converting enzyme gene I/D polymorphism increases the susceptibility to hypertension and additive diseases: A study on North Indian patients. Clin Exp Hypertens 38:305–311
Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H et al (2014) Clinical interpretation and implications of whole-genome sequencing. JAMA 311:1035–1045
Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI et al (2011) Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478:103–109
Franceschini N, Fox E, Zhang Z, Edwards TL, Nalls MA, Sung YJ et al (2013) Genome-wide association analysis of blood pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am J Hum Genet 93:545–554
Ehret GB, Caulfield MJ (2013) Genes for blood pressure: an opportunity to understand hypertension. Eur Heart J 34:951–961
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H et al (2018) Genetic analysis of over one million people identifies 535 new loci associated with blood pressure traits. Nat Genet 50:1412–1425
Richard MA, Huan T, Ligthart S, Gondalia R, Jhun MA, Brody JA et al (2017) DNA methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet 101:888–902
Kazmi N, Elliott HR, Burrows K, Tillin T, Hughes AD, Chaturvedi N et al (2020) Associations between high blood pressure and DNA methylation. PLoS One 15:e0227728
Raftopoulos L, Katsi V, Makris T, Tousoulis D, Stefanadis C, Kallikazaros I (2015) Epigenetics, the missing link in hypertension. Life Sci 129:22–26
Han L, Liu Y, Duan S, Perry B, Li W, He Y (2016) DNA methylation and hypertension: emerging evidences and challenges. Brief Funct Genomics 15:460–469
Feinberg AP (2008) Epigenetics at the epicenter of modern medicine. JAMA 299:1345–1350
Wang J, Gong L, Tan Y, Hui R, Wang Y (2015) Hypertensive epigenetics: from DNA methylation to microRNAs. J Hum Hypertens 29:575–582
Stoll S, Wang C, Qiu H (2018) DNA methylation and histone modification in hypertension. Int J Mol Sci 19:1174
Arif A, Sadayappan S, Becker RC, Martin LJ, Urbina EM (2019) Epigenetic modification: a regulatory mechanism in essential hypertension. Hypertens Res 42:1099–1113
Huang JB, Liang J, Zhao XF, Wu WS, Zhang F (2013) Epigenetics: novel mechanism of pulmonary hypertension. Lung 191:601–610
Millis RM (2011) Epigenetics and hypertension. Curr Hypertens Rep 13:21–28
Miranda TB, Jones PA (2007) DNA methylation: the nuts and bolts of repression. J Cell Physiol 213:384–390
Zhao M, Wang Z, Yung S, Lu Q (2015) Epigenetic dynamics in immunity and autoimmunity. Int J Biochem Cell Biol 67:65–74
Jeltsch A, Broche J, Bashtrykov P (2018) Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes 9:566
Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070
Denis H, Ndlovu MN, Fuks F (2011) Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep 12:647–656
Rasmussen KD, Helin K (2016) Role of TET enzymes in DNA methylation, development and cancer. Genes Dev 30:733–750
Liang M (2018) Epigenetic mechanisms and hypertension. Hypertension 72:1244–1254
Kumar S, Chinnusamy V, Mohapatra T (2018) Epigenetics of modified DNA bases: %-Methylcytosine and Beyond. Front Genet 9:640
Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ (2007) Epigenetic modification of the renin angiotensin system in the fetal programming of hypertension. Circ Res 100:520–526
Goyal R, Goyal D, Lietzke A, Gheorghe C, Longo LD (2010) Brain Renin-Angiotensin System: Fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci 17:227–238
Goyal R, Longo LD (2013) Maternal protein deprivation: sexually dimorphic programming of hypertension in the mouse. Hypertens Res 36:29–35
Xiao D, Dasgupta C, Li Y, Huang X, Zhang L (2014) Perinatal nicotine exposure increases angiotensin II receptor mediated vascular contractility in adult offspring. PLoS One 9:e108161
Pei F, Wang X, Yue R, Chen C, Huang J, Li X et al (2015) Differential expression and DNA methylation of angiotensin type 1A receptors in vascular tissues during genetic hypertension development. Mol Cell Biochem 402:1–8
Lee HA, Baek I, Seok YM, Yang E, Cho HM, Lee DY et al (2010) Promoter hypomethylation upregulates Na+-K+-2Cl- cotransporter 1 in spontaneously hypertensive rats. Biochem Biophys Res Commun 396:252–257
Riviere G, Lienhard G, Andrieu T, Vieau D, Frey BM, Frey FJ (2011) Epigenetic regulation of somatic angiotensin converting enzyme by DNA methylation and histone acetylation. Epigenetics 6:478–489
Smolarek I, Wyszko E, Barciszewska AM, Nowak S, Gawronska I, Jablecka A et al (2010) Global DNA methylation changes in blood of patients with essential hypertension. Med Sci Monit 16:149–155
Kulkarni A, Chavan GP, Mehendale S, Yadav H, Joshi S (2011) Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol 30:79–84
Wang X, Falkner B, Zhu H, Shi H, Su S, Xu X et al (2013) A genome wide methylation study on essential hypertension in young African American males. PLoS One 8:e53938
Kato N, Loh M, Takeuchi F, Verweji N, Wang X, Zhang W et al (2015) Trans-ancestry genome- wide association study identifies 12 genetic loci influencing blood pressure and implicated a role for DNA methylation. Nat Genet 47:1282–1293
Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V et al (2008) Epigenetic control of 11 β-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 199:323–327
Zhang LN, Liu PP, Wang L, Yuan F, Xu L, Xin Y et al (2013) Lower ADD1gene promoter DNA methylation increases the risk of essential hypertension. PLoS One 8:e63455
Fan R, Wang WJ, Zhong QL, Duan SW, Xu XT, Hao LM et al (2015) Aberrant methylation of GCK gene body is associated with the risk of essential hypertension. Mol Med Rep 12:2390–2394
Fan R, Mao S, Zhong F, Gong M, Yin F, Hao L et al (2015) Association of AGTR1 promoter methylation levels with essential hypertension risk: A matched case-control study. Cytogenet Genome Res 147:95–102
Chaudhary M, Chaudhary S (2019) Functional relevance of promoter CpG island of human angiotensin II type 1 receptor (AT1R) gene. Mol Cell Biochem 457:31–40
Gu T, Mao S, Fan R, Zhong F, Zhu F, Hao L et al (2016) Interactions between CYP11B2 promoter methylation and smoking increase risk of essential hypertension. Biomed Res Int 2016:1454186
Zhong Q, Liu C, Fan R, Duan S, Xu X, Zhao J et al (2016) Association of SCNN1B promoter methylation with essential hypertension. Mol Med Rep 14:5422–5428
Mao SQ, Sun JH, Gu TL, Zhu FB, Yin FY, Zhang LN (2017) Hypomethylation of Interleukin-6 (IL-6) gene increases the risk of essential hypertension: a matched case-control study. J Hum Hypertens 31:530–536
Mao S, Gu T, Zhong F, Fan R, Zhu F, Ren P et al (2017) Hypomethylation of the Toll-like receptor-2 gene increases the risk of essential hypertension. Mol Med Rep 16:964–970
Jin F, Li X, Wang Z, Liu Y, Liu J, Sun D, Jin Y et al (2018) Association of mitofusin 2 methylation and essential hypertension: a case-control study in a Chinese population. Hypertens Res 41:605–613
Guay SP, Brisson D, Lamarche B, Biron S, Lesscelleur O, Biertho L et al (2014) ADRB3 gene promoter DNA methylation in blood and visceral adipose tissue is associated with metabolic disturbances in men. Epigenomics 6:33–43
Bochtler M, Fernandes H (2021) DNA adenine methylation in eukaryotes: Enzymatic mark or a form of DNA damage? Bioessays 43:e2000243
Xiao CL, Zhu S, He M, Chen D, Zhang Q, Chen Y et al (2018) N6-methyladenine DNA modification in the human genome. Mol Cell 71:306–318
Alderman MH, Xiao AZ (2019) N(6)-methyladenine in eukaryotes. Cell Mol Life Sci 76:2957–2966
Liang Z, Kidwell RL, Deng H, Xie Q (2020) Epigenetic N6-methyladenosine modification of DNA and RNA regulates cancer. Cancer Biol Med 17:9–19
Ratel D, Ravanat JL, Berger F, Wion D (2006) N6-methyladenine: the other methylated base of DNA. Bioessays 28:309–315
Li Z, Zhao P, Xia Q (2019) Epigenetic methylations on N6-adenine and N6-adenosine with the same input but different output. Int J Mol Sci 20:2931
Wu KJ (2020) The epigenetic roles of DNA N6-methyladenine (6mA) modification in eukaryotes. Cancer Lett 494:40–46
Ye P, Luan Y, Chen K, Liu Y, Xiao C, Xie Z (2017) MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res 45:D85–D89
Wang X, Huang J, Zou T, Yin P (2017) Human m6A writers: two subunits, two roles. RNA Biol 14:300–304
Visvanathan A, Somasundaram K (2018) mRNA traffic control reviewed: N6-methyladenosine (m6A) takes the driver’s seat. Bioessays 40. https://doi.org/10.1002/bies.201700093
Maity A, Das B (2016) N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J 283:1607–1630
Kim GW, Siddiqui A (2021) The role of N6-methyladenosine modification in the life cycle and disease pathogenesis of hepatitis B and C viruses. Exp Mol Med 53:339–345
Zhou XL, Huang FJ, Li Y, Huang H, Wu QC (2021) SEDT2/METTL14-mediated m6A methylation awakening contributes to hypoxia-induced pulmonary arterial hypertension in mice. Aging 13:7538–7548
Berlivet S, Scutenaire J, Deragon JM, Bousquet-Antonelli C (2019) Readers of the m6A epitranscriptomic code. Biochim Biophys Acta Gene Regul Mech 1862:329–342
Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q et al (2018) N6-methyladenine DNA modification in glioblastoma. Cell 175:1228–1243
Zhang X, Blumenthal RM, Cheng X (2021) A role for N6-methyladenine in DNA damage repair. Trends Biochem Sci 46:175–183
Wu Y, Zhou C, Yuan Q (2018) Role of DNA and RNA N6-adenine methylation in regulating stem cell fate. Curr Stem Cell Res Ther 13:31–38
Pan Z, Sikandar S, Witherspoon M, Dizon D, Nguyen T, Benirschke K et al (2008) Impaired placental trophoblast lineage differentiation in Alkbh1(-/-) mice. Dev Dyn 237:316–327
Nordstrand LM, Svard J, Larsen E, Nilsen A, Ougland R, Furu K et al (2010) Mice lacking Alkbh1 display sex-ratio distortion and unilateral eye defects. PLoS One 5:e13827
Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM et al (2012) ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells 30:2672–2682
Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K et al (2016) DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 532:329–333
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A et al (2017) m6A-dependant maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Guo Y, Pei Y, Li K, Cui W, Zhang D (2020) DNA N6-methyladenine modification in hypertension. Aging 12:6276–6291
Marcadenti A, Fuchs FD, Matte U, Sperb F, Moreira LB, Fuchs SC (2013) Effects of FTO rs9939906 and MC4R rs17782313 on obesity, type 2 diabetes mellitus and blood pressure in patients with hypertension. Cardiovasc Diabetol 12:103
Olza J, Ruperez AI, Gil-Campos M, Leis R, Fernandez-Orth D, Tojo R et al (2013) Influence of FTO variants on obesity, inflammation and cardiovascular disease risk biomarkers in Spanish children: a case-control multicentre study. BMC Med Genet 14:123
Paramasivam A, Priyadharsini JV, Raghunandhakumar S (2020) N6-adenosine methylation (m6A): a promising new molecular target in hypertension and cardiovascular diseases. Hypertens Res 43:153–154
Gerges C, Gerges M, Friewald R, Fesler P, Dorfmuller P, Sharma S et al (2020) Microvascular disease in chronic thromboembolic pulmonary hypertension: Hemodynamic phenotyping and histomorphometric assessment. Circulation 141:376–386
Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN et al (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573:281–286
Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB et al (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51:844–856
Acknowledgements
The author is thankful to the institute.
Funding
No funding was obtained to draft this review article.
Author information
Authors and Affiliations
Contributions
MC gathered information, compiled, and drafted the manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The author declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaudhary, M. Novel methylation mark and essential hypertension. J Genet Eng Biotechnol 20, 11 (2022). https://doi.org/10.1186/s43141-022-00301-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00301-y