
Abstract
Background
The present study focused to develop a simple and sensitive HPLC method for resolution and estimation process-related impurities of zuclopenthixol and further assessment of forced degradation behavior of zuclopenthixol.
Results
The chromatographic separation of drug substance, process-related impurities and its degradation products (DPs) was achieved on KNAUER C18 (250 mm × 4.6 mm, 5µ id) column at that was maintained at 35 °C temperature using 0.1 M sodium acetate buffer at pH 4.3 and methanol in 20:80 (v/v) as mobile phase A, 0.1% formic acid and acetonitrile in 75:25 (v/v) as mobile phase B. Equal volume of mobile phase A and B was pumped in isocratic elution at 0.8 mL/min. Detection wavelength was selected as 257 nm. In the proposed conditions, zuclopenthixol is identified at 6.91 and 1.91 min and 2.89 min, respectively, for impurity B and A min with acceptable system suitability and specificity. The method produces LOD at 0.009 for impurities with calibration range of 30–180 µg/mL for zuclopenthixol and 0.03–0.18 µg/mL for impurities. The other validation parameters were notices to be with in the acceptable levels for zuclopenthixol and its impurities. The drug was exposed to different stressed conditions (acid, base, peroxide, thermal and UV light) according to ICH Q1A (R2) guidelines. The DPs formed during the stress study were identified and characterized by LCMS/MS in ESI positive mode.
Conclusion
The analysis involved a comparison of collision-induced dissociation mass spectrometry data between the degradation products and zuclopenthixol. As a result, potential structures for six degradation compounds were suggested. The results from additional validation studies were similarly pleasing and demonstrated their suitability for the routine analysis of zuclopenthixol and its associated impurities in both bulk drug and pharmaceutical dosage forms. Additionally, these findings can be extended to assess the mechanism of stress degradation in zuclopenthixol.
Similar content being viewed by others
Background
The presence of pharmaceutical impurities in a pharmaceutical product can affect its quality and cause harmful effects. The pharmaceutical drug synthesis procedure, formulation selection as well as its storage conditions may be responsible for the formation of impurities [1]. In order to identify the impurities in a drug moiety, it was very essential to conduct impurity profiling. The organic impurities in a drug substance can be analyzed quantitatively using a stability-indicating HPLC (high-performance liquid chromatography) method [2]. It was very significant to evaluate the stability of drug molecule with the influence of various environment conditions with time. In order to evaluate the drug stability, pharmaceutical industries adopted International Conference on Harmonization (ICH) guidelines [3,4,5].
The dopamine 1 and 2 antagonist, 5-HT2 and α1-adrenergic drug zuclopenthixol was pharmacologically classified as antipsychotic neuroleptic drug and have thioxanthene group in its molecular structure. It was suggested for the treatment of mental illness such as schizophrenia and psychoses [6]. It exhibit high affinity toward 5-HT2 and alpha1-adrenergic receptors, weaker affinity to block histamine H1 receptor activity and very low affinity toward α2-adrenergic receptor and muscarinic cholinergic receptor [7]. The side effects of akathisia, somnolence, dry mouth, hyperkinesia and hypokinesia are most common, whereas vertigo, heart palpitations, tachycardia, accommodation disorder, salivary hypersecretion, abnormal vision, vomiting, constipation, dyspepsia and diarrhea are less common, and tinnitus, abdominal pain, hyperacusis, mydriasis, nausea, flatulence and thirst are uncommon during the usage of zuclopenthixol [8].
From review of literature, it come to know that one LCMS method reported for pharmacokinetic evaluation of zuclopenthixol along with fluphenazine [9]. One author [10] reported the involvement of zuclopenthixol on fatality using HPLC–DAD and HPLC–MS. One work reported [11] the fatal intoxication of zuclopenthixol during the process of psychiatric therapy. One author reported LCMS method [12] for quantification of zuclopenthixol along with its N-dealkylated metabolite in biological fluids.
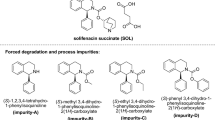
The comprehensive literature review suggests that there is no HPLC- or LCMS-based stability study method validated as per ICH Q2(R1) guideline [3,4,5] for investigating zuclopenthixol along with its impurities. Keep this in consideration, this study proposed to investigate degradation behavior of zuclopenthixol and identity its degradation products along with the stability of zuclopenthixol molecule in various stress studies. The EP (European Pharmacopoeia) impurity A and B of zuclopenthixol was used in the study. The decanoate form of zuclopenthixol was left over zuclopenthixol end product as impurity and was recognized as impurity A, whereas 2-chlorothioxanthone which was used during the process of synthesis of zuclopenthixol was left over zuclopenthixol final product which was recognized as impurity B by European Pharmacopoeia. The chemical structure of EP impurity A and B of zuclopenthixol is shown in Fig. 1.

Molecular structure of zuclopenthixol and impurities
Experimental procedure
Reagents and chemicals
The zuclopenthixol active pharmaceutical ingredient (API) with potency of 98.75% along with EP impurity 1 and 2 was brought from Lundbeck India Private Limited, Bengaluru, Karnataka, India. The Clopixol® brand injection formulation with 100 mg dosage was purchased in local pharmacy. The milli-Q® water, 0.2 µ nylon membrane filters and HPLC grade acetonitrile, methanol solvents were procured from Merck chemicals, Mumbai. The sodium acetate, formic acid, hydrogen peroxide, sodium hydroxide (NaOH) and hydrochloric acid (HCl) of AR (analytical reagent) grade reagents used in this study were procured from Fisher scientific, Mumbai.
Instrumentation
The method development and validation studied were performed on HPLC (1100 series) system with G1311 A model quaternary pump, G 1329A model temperature variable autosampler with 0.1–1500 μL injection capacity and G 1314 A model ultraviolet (UV) detector. Agilent ChemStation software was employed to perform the chromatographic integrations. The LCMS evaluation of stress degradation compounds was conducted on triple quadruple LCMS instrument (Waters, Japan) with MassLynx software.
Preparation of solutions
Diluent solution
The pure methanol solvent shows the maximum solubility of zuclopenthixol and its impurities in the study. Hence, HPLC grade methanol was selected as diluent in the study.
Buffer solution
In a 1-L volumetric flask containing 800 mL of milli-Q® water, 5.772 g of sodium acetate and 1.778 g of acetic acid was added. The flask was kept in an ultrasonic bath sonicator to dissolve the components completely in water. The pH of the solution was adjusted to 4.3 using 10 N hydrochloric acid. Then, it was filtered through 0.2 µ filter and the final volume made till mark using water.
Mobile phase
The mobile phase component A comprises 20:80 (v/v) portions of 0.1 M sodium acetate buffer at pH 4.3 and methanol which were measured accurately and mixed separately in a 100-mL volumetric flask, whereas the mobile phase component B comprises 75:25 (v/v) portions of 0.1% formic acid and acetonitrile which were measured accurately and mixed separately in a 100-mL volumetric flask. The equal volumes of mobile phase component A and B were mixed separately and were used as mobile phase in the study.
Zuclopenthixol and impurity solutions
The precisely weighed 25 mg of zuclopenthixol pure drug and its impurities were taken in a 25-mL flask which was already filled with 15 mL of diluent (pure methanol). Then, ultrasonic bath sonicator was used to dissolve weighed analytes in solvent and filter the solution through 0.2 µm filter. Then by utilizing the same solvent, the volume in the flask was made till mark to achieve 1000 µg/mL strength separate solutions of zuclopenthixol and its impurities. Necessary dilutions were made from this stock solution whenever required.
Formulation solution
The injection formulation containing 100 mg of zuclopenthixol with trade name Clopixol® was utilized for preparing formulation solution. One vial contains 1 mL of formulation solution and 100 mg of zuclopenthixol. One vial solution was precisely transfer to 100-mL volumetric flask which was half filled with methanol. The flask was sonicated to dissolve analytes formulation completely in solvent, and volume in the flask was brought till mark using same diluent and then filtered through 0.2 µ membrane filters to get 1000 µg/mL sample solution. Then, desired formulation concentration was prepared from this 1000 µg/mL solution.
Method development
The process of developing the method commenced with the identification of an appropriate wavelength for detecting zuclopenthixol and its impurities, utilizing a UV detector. UV–visible spectrophotometer employed to pinpoint wavelength for both zuclopenthixol and its impurities. A standard solution containing zuclopenthixol and its impurities at a concentration of 10 µg/mL was individually scanned over 400 to 200 nm range. The superimposed UV absorption spectra of zuclopenthixol and its impurities confirmed iso-absorption wavelength that was found suitable for the accurate detection of both zuclopenthixol along with impurities. We assessed various stationary phase configurations from different manufacturers to attain optimal resolution for zuclopenthixol and its impurities. Through systematic experimentation involving diverse solvent combinations, pH ranges, and flow rates, we fine-tuned the mobile phase composition. The conditions that successfully resolve zuclopenthixol and its impurities was selected and carried forward for subsequent validation procedures.
Method validation
The applicability of method proposed for its intended purpose was determined by performing method validation studies and was conducted by utilizing ICH guidelines [3,4,5] as well as method reported in the literature [13,14,15,16,17,18,19,20,21].
The signal (s) and noise (n) ratio approach with specification of 3:1 and 10:1 (s:n) was utilized for evaluating detection limit (LOD) and quantification limit (LOQ), respectively. LOD and LOQ give the information related to the method sensitivity and lower the results higher the method sensitivity. The sequential analysis of the lowest concentration of impurities confirms the method sensitivity.
LOQ of impurities was considered while preparing calibration curve standard dilutions of zuclopenthixol along with impurities. The LOQ concentration of impurities was treated as minimum calibration curve concentration, and zuclopenthixol concentration was considered as it contains 0.1% of impurity. The arranged solutions were evaluated in the proposed method and calibration curve constructed by considering area (y-axis) against its strength (x-axis). Then, the linear equation was derived by adopting least squares regression procedure.
The degree of repetition as well as reproducibility of test findings in the proposed method was evaluated in precision. The zuclopenthixol solution contains 0.1% impurities was prepared and analyzed in same day (n = 6), two alternate days (n = 3) and two different analysts (n = 3) to evaluate intraday, interday precision and ruggedness, respectively. Robustness was tested by intentionally introducing slight deviations in the proposed method conditions. Precision level solution was evaluated in all varied conditions, and % change in response was evaluated and % change of < 2 was treated as permissible. Method accuracy was determined by adding pre-analyzed samples at four levels in linearity range, i.e., LOQ, 50%, 100% and 150%. Recovery percentage in every analysis and % RSD of every studied level was tabulated, and results were observed carefully to evaluate method accuracy.
The stress degradation studies were performed to evaluate specificity and stability-indicating power method optimized in this study. The placebo and zuclopenthixol injection solution was mixed to 5 mL of HCl (1 N), 5 mL NaOH (1 N) and 5 mL hydrogen peroxide (3%), respectively, in acid, base and peroxide degradation separately. The stress exposed samples were neutralized and bring up to precision level prior to analysis. The placebo and zuclopenthixol injection solution was exposed to UV light at 103,959 Wh/m2 energy and 80 °C for 24 h in UV and thermal degradation, respectively. These samples were bringing up to precision level and was analyzed in the proposed method. The stress study chromatograms was observed carefully for evaluating the stress degradation products and chromatographic response were utilized for % stability evaluation by comparing stability chromatographic response with calibration curve.
The degradation compounds (DPs) generated during stress expose and identified in the stress degradation chromatogram were characterization using LCMS/MS analysis. The eluents detected in UV detector were directed to mass detector for mass spectral characterization of DPs. In this, care should be taken such that 40% of eluents were entered in the mass detected with help of a splitter.
The analytical HPLC method proposed in this study was studied to identify and quantify impurities of zuclopenthixol in injection formulations. Sample solution derived from Clopixol® injection solution directly and spiked with known concentration of studied impurities was evaluated this method. The noticed chromatograms along with its response were utilized for evaluating method applicability.
Results
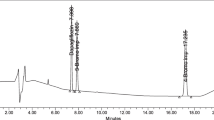
The effective separation of zuclopenthixol and its impurities was attained utilizing a KNAUER C18 column (250 mm × 4.6 mm, 5µ id), which was maintained at a temperature of 35 °C. The chromatographic conditions included the use of a mobile phase consisting of 0.1 M sodium acetate buffer at pH 4.3 and methanol in 20:80 (v/v) as mobile phase A, while mobile phase B comprised 0.1% formic acid and acetonitrile in proportion of 75:25 (v/v). An isocratic elution mode was employed, with both mobile phase A and B being pumped in equal volumes at 0.8 mL/min flow rate. For detection purposes, a wavelength of 257 nm was chosen based on observations indicating optimal detector response in comparison with other wavelengths for all analytes. Figure 2 presents the chromatograms obtained for placebo, the standard zuclopenthixol solution spiked with impurities, and the pattern for impurity separation and detection. Based on these chromatograms, the specificity of the method for effectively isolating and identifying process-related impurities of zuclopenthixol was confirmed.

Specificity chromatograms in the optimized method. Chromatogram observed while analyzing placebo solution (A) and precision level solution of zuclopenthixol spiked with impurities (0.1%)
The system suitability data demonstrates suitability of system, with the tailing factor measuring below 1.5, resolution between any adjacent eluting analyte peaks exceeding 2.5 and theoretical plates of all analyte peaks exceeding 2500. This outcome underscores the method’s strong selectivity. Sensitivity evaluation was performed by signal-to-noise (s/n) approach, and the outcomes were expressed as LOD and LOQ. The calculated detection limit was 0.009 µg/mL, while the LOQ was determined to be 0.03 µg/mL for both impurity A and impurity B. These results underscore the method’s elevated sensitivity, particularly suitable for quantification of impurities.
The LOQ concentration of impurities, i.e., 0.03 µg/mL was taken as initial concentration for constructing the calibration curve for impurities. The zuclopenthixol standard solution was prepared so the solution have 0.1% of each impurity, and a precise fitting calibration curve was established within the concentration range of 30–180 µg/mL for zuclopenthixol and 0.03–0.18 µg/mL for the impurities investigated in the study. The calibration curve exhibited a strong linear relationship, with notably high correlation coefficients observed for both impurities and standard zuclopenthixol.
The obtained peak area values exhibited %RSD (relative standard deviation) below the threshold for both zuclopenthixol and the two impurities in various precision studies including intraday, interday precision, precision at the LOQ and ruggedness assessments. These findings underscore the method’s commendable precision. To assess accuracy, recovery studies were conducted by spiking concentrations of 50%, 100%, and 150% of the target, equating to 60 µg/mL for zuclopenthixol and 0.09 µg/mL for impurities A and B. % Recovery was calculated for standard and all impurities in each analysis, with %RSD values determined for each spiked level. Incorporating zuclopenthixol and the studied impurities, the achieved % recovery fell within the acceptable range of 98–102%, affirming the accuracy of the method. Additionally, %RSD values at each spiked level remained below 2%, aligning with the acceptable limit and further confirming the method’s accuracy. The summarized outcomes encompassing in validation of proposed method are presented in Table 1.
No notable alteration in the chromatographic response or system suitability was observed when the experiment was conducted with slight deviations in the proposed method conditions. Resolutions between consecutive analytes consistently exceeded 2.0, and tailing factors for all analytes remained within acceptable limits. The variability in zuclopenthixol and impurity estimation stayed below the acceptable threshold of 2, confirming the method’s robustness. The results of the robustness study conducted in the developed method are detailed in Table 2.
Forced degradation studies performed to evaluate the effectiveness of the method for resolution of degradation compounds, and the study was conducted in acid, base, peroxide, thermal and UV light degradation conditions. There is no considerable degradation was noticed in UV light and thermal degradation conditions with % assay of 97.49% and 97.76%, respectively. Among the degradation conditions, high % degradation was noticed in acid degradation study with a % degradation of 8.96%. The chromatogram observed in this study (Fig. 4A) show well-resolved DPs at tR of 2.67 min, 5.20 min and 6.01 min and were named as DP 1, DP 4 and DP 5, respectively. The chromatogram identified in peroxide degradation study (Fig. 4B) clearly resolve two degradation products at tR of 3.98 min and 8.66 min and were designated as DP 3 and DP 6, respectively, with a % degradation of 7.95%. The peak corresponds to impurity B at tR of 1.98 min was also noticed in peroxide degradation chromatogram. The % assay of zuclopenthixol in base degradation was calculated to be 92.37% with mass balance of 99.13%. The chromatogram clearly resolves two DPs at tR of 3.43 min and 8.68 min and these impurities were marked as DP 2 and DP 6, respectively. The results of the peak purity test, as determined by the PDA detector, validated the purity and homogeneity of the zuclopenthixol peak across all examined stress samples. The mass balance for stressed samples fell within the range of 98.93% to 99.71%. These peak purity test outcomes consistently confirmed the homogeneity and purity of the zuclopenthixol peak within the analyzed stress samples. The assay of zuclopenthixol exhibited negligible variation in the presence of impurities, and the peak purity results of the stress samples further substantiate the specificity and capability of the developed method to indicate stability. Comprehensive details are provided in Table 3, while Fig. 3 illustrates representative chromatograms observed during the forced degradation study.

Forced degradation chromatograms of zuclopenthixol. A Acidic stress study chromatogram of zuclopenthixol visualizing DP 1, 4 and 5; B Basic stress study chromatogram of zuclopenthixol visualizing DP 2 and 6; C Peroxide degradation chromatogram of zuclopenthixol visualizing DP 3 and 6
Characterization of DPs by LCMS/MS
The DPs generated due to stress effect on zuclopenthixol pure drug were characterized through LCMS/MS analysis. The LC conditions optimized in the study were utilized without any change and the mass operating conditions optimized such that the condition produce maximum detection of each mass fragment with very less or no noise. The mass detector operated 3600 V of capillary voltage, 60 V of fragmentor voltage and 65 V of skimmer voltage, 6 L/H flow of drying (nitrogen) gas at 350 °C and 40 Psi of nebulizer gas. The same experiment condition was monitored throughout the analysis and an average of 20–30 scans were conducted. The preliminary test confirms that the positive ion mode was suitable for optimum and maximum detection of all DPs.
The ESI MS spectrum of DP 1 depicted in Fig. 10A, identified at tR of 2.67 min, exhibits a prominent parent ion at m/z 211 (m + 1), suggesting a plausible molecular formula of C14H10S. Additionally, the spectrum displays less abundant product ions at m/z 161 (m + 1) with a molecular formula of C10H8S. Based on the observed fragmentation pattern, the compound is recognized as 9-methylidene-9H-thioxanthene, characterized by a molecular formula of C14H10S and a molecular mass of 210 g/mol. The proposed mass fragmentation pattern of DP 1 is illustrated in Fig. 4.

Mass fragmentation pattern of DP 1
The mass fragmentation spectra of DP 2 (Fig. 10B) reveal a dominant parent ion at m/z 357 (m + 1) when observed under positive ionization mode. Additionally, the spectrum displays fragment ions at m/z 137 (m + 1) resulting from the loss of C12H13ClN. Through accurate mass measurements, the elemental compositions of the molecular ion of DP 2 and all its fragmented ions have been verified. Based on the data obtained, DP 2 has been definitively identified as 1-[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]piperazine, possessing a molecular formula of C20H21ClN2S. The proposed mass fragmentation pattern of DP 2 is depicted in Fig. 5.

Mass fragmentation pattern of DP 2
The ESI–MS spectrum of DP 3 (presented in Fig. 10C), observed at a retention time of 3.98 min, displays a parent ion at m/z 329 (m + 1) alongside a prominent fragment ion at m/z 244 (m + 1). The parent ion’s molecular formula is identified as C18H17ClN2S, and this corresponds to the fragment ion with a molecular formula of 243 (resulting from the loss of C4H9N2). Further analysis reveals that the compound is characterized as (2E)-2-{[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]imino}ethanamine. The structural details of this compound, as well as its associated fragmentation mechanism, are illustrated in Fig. 6.

Mass fragmentation pattern of DP 3
The ESI–MS spectrum, observed at a retention time of 5.20 min (Fig. 10D), exhibits a parent ion at m/z 311, corresponding to the [M + H]+ of DP 4, which forms under acidic stress conditions. Within the spectrum, there are abundant product ions at m/z 160 (m + 1). The integrity of DP 4’s molecular structure is validated through both peak purity testing and CID studies. The collection of these product ions, in conjunction with the parent ion, serves to affirm that DP 4 is indeed N-methyl-N-[3-(9H-thioxanthen-9-ylidene)propyl]ethane-1,2-diamine, having a molecular formula of C19H22N2S. A representation of its structure, alongside the fragmentation mechanism is presented in Fig. 7.

Mass fragmentation pattern of DP 4
The ESI MS spectrum of DP 5 (Fig. 10E) displayed notable product ions at m/z 288 [M + H]+. A significant product ion at m/z 161 possibly resulted from the loss of C6H8S from m/z 284. The elemental compositions of the molecular ion of DP 5 and all its fragmented ions were confirmed through precise mass measurements. Based on these analyses, DP 5 was identified as (3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propan-1-amine, with a molecular mass of 287 g/mol and a chemical formula of C16H14ClNS. Figure 8 present its molecular structure and fragmentation mechanism. The characterization of DP 5 in this study aligned with the oxidative degradation product reported by Thummar et al., 2014 [16].

Mass fragmentation pattern of DP 5
Based on the ESI MS spectrum of DP 6 as presented in Fig. 10F, the fragmentation pattern was proposed as presented in Fig. 9, and the compound was finalized as N,N-dimethyl-3-(9H-thioxanthen-9-ylidene)propan-1-amine with molecular formula of C18H19NS and molecular mass of 280 g/mol. This DP 6 was identified in the chromatogram of both base and peroxide stress studies (Fig. 10).

Mass fragmentation pattern of DP 6

Mass spectra of DPs observed in forced degradation study. Mass spectra identified at tR of 2.67 min for DP 1 (A), 3.43 min for DP 2 (B), 3.98 min for DP 3 (C), 5.20 min for DP 4 (D), 6.01 min for DP 5 (E) and 8.68 min for DP 6 (F)
The developed HPLC technique was put into practice to quantify pharmacopeia-defined impurities of zuclopenthixol within a pharmaceutical formulation. The formulation sample underwent direct analysis to assess the impurities present within it. Additionally, a formulation sample spiked with impurities was analyzed to gauge the method’s efficacy in separating and quantifying impurities within the formulation. The chromatogram acquired from the formulation solution spiked with impurities (depicted in Fig. 11) distinctly exhibited peaks corresponding to the impurities under investigation. In contrast, the chromatogram from the un-spiked formulation solution displayed no peaks associated with the studied impurities. This observation implies that the amount of impurity in the sample fell below the detection limit. Consequently, it confirms that the impurity quantity in the sample remained below permissible levels. This substantiates the successful applicability of the proposed method for the accurate quantification of process-related impurities in zuclopenthixol.

Formulation analysis chromatogram of zuclopenthixol. Chromatogram noticed for formulation solution spiked with impurities (A) and with no impurities spiked (B)
Discussion
In the absence of any documented analytical approach in the literature for quantifying pharmacopeial impurities associated with zuclopenthixol, the primary objective of this investigation was to devise a straightforward HPLC technique. This method’s purpose was to enable the evaluation of EP impurity A and B present in zuclopenthixol samples. The optimization of method was performed on different configurations of columns including octadecylsilane (ODS), phenyl–hexyl, cyano and amino columns for best resolution of analytes. The mobile phase composition with suitable pH range was optimized by varying different solvent compositions including various strengths of buffers. The hydrophilic ionizable functional groups present in zuclopenthixol and its impurities were effectively resolved with mobile phase containing pH buffers and hence various buffers with different pH range were studied for effective resolution.
The method utilizes a KNAUER C18 column (250 mm × 4.6 mm, 5µ id), which was maintained at a temperature of 35 °C. The chromatographic conditions included the use of a mobile phase consisting of 0.1 M sodium acetate buffer at pH 4.3 and methanol in 20:80 (v/v) as mobile phase A, while mobile phase B comprised 0.1% formic acid and acetonitrile in proportion of 75:25 (v/v). An isocratic elution mode was employed, with both mobile phase A and B being pumped in equal volumes at 0.8 mL/min flow rate. For detection purposes, a wavelength of 257 nm was chosen based on observations indicating optimal detector response in comparison with other wavelengths for all analytes.
Under proposed condition, the peak shapes observed for zuclopenthixol and its impurities exhibited symmetry in the resulting chromatogram. The differentiation between adjacent peaks was found to exceed a value of 2. Identification of constituents within the reference solution was accomplished by analyzing standard solutions individually and then correlating their retention times with established standard. Specifically, the retention times noted were 6.91 min for zuclopenthixol, 2.89 min for impurity A, and 1.91 min for impurity B. Regarding column efficiency, the evaluation indicated that for both zuclopenthixol and its associated impurities, the tailing factor fell below the acceptable threshold. Conversely, the theoretical plates and resolution values surpassed the acceptable limit.
The method was noticed to be very sensitive that can detect up to a very low concentration of 0.009 µg/mL that facilitates a very sensitive calibration curve range of 0.03–0.18 µg/mL for impurities. The effectiveness of the method for resolution and identification of stress degradation compounds were evaluated. The fragmentation pattern was analyzed systematically for the evaluation of stress degradation compounds. The stress degradation compounds were identified as 9-methylidene-9H-thioxanthene (DP 1), 1-[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]piperazine (DP 2), (2E)-2-{[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]imino}ethanamine (DP 3), N-methyl-N-[3-(9H-thioxanthen-9-ylidene)propyl]ethane-1,2-diamine (DP 4), (3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propan-1-amine (DP 5) and N,N-dimethyl-3-(9H-thioxanthen-9-ylidene)propan-1-amine (DP 6). The method can successfully prove for its applicability for quantification of pharmacopeia impurities of zuclopenthixol in formulations.
Conclusion
Forced degradation studies, following the stipulated conditions outlined by the ICH, were conducted on zuclopenthixol. The drug displayed remarkable stability under thermal and UV light degradation conditions, while revealing susceptibility to degradation in acidic, basic, and peroxide environments. The thermal and UV light degradation conditions did not lead to significant degradation. Throughout various stress conditions, a total of six degradation products (DPs) emerged. Among these, DP 6 was consistently identified in both basic and peroxide conditions. Utilizing LCMS/MS analysis, the characterization of these six distinct DPs, formed during the forced degradation, was effectively achieved, specifically in the ESI positive mode. The mechanisms and pathways underlying the formation of each DP of zuclopenthixol were outlined and discussed. The outcomes of this study hold significant potential in the identification of process-related impurities and potential degradation products, even at trace levels, in bulk drugs. This analytical method stands as a valuable tool for quality control sample assessment, serving regular and stability studies of zuclopenthixol and its associated process-related impurities.
Availability of data and materials
The research work has been carried out by us, and we assure you that it can be provided to you whenever required.
Abbreviations
- HPLC:
-
High-performance liquid chromatography
- LCMS/MS:
-
Liquid chromatography with tandem mass spectrometry
- ESI:
-
Electrospray ionization
- DPs:
-
Degradation products
- UV:
-
Ultra-violet
- DAD:
-
Diode array detector
- EP:
-
European Pharmacopoeia
References
Weaver RJ, Valentin JP (2019) Today’s challenges to de-risk and predict drug safety in human “mind-the-gap.” Toxicol Sci 167:307–321. https://doi.org/10.1093/toxsci/kfy270
Bddeilmann M, Boonen H, Czich A, Dear G, Hewitt P, Mow T, Newham P, Oinonen T, Pognan F, Roth A, Valentin JP (2019) Optimizing drug discovery by investigative toxicology: current and future trends. ALTEX-Alternatives to animal experimentation 36:289–313. https://doi.org/10.14573/altex.1808181
International Conference of Harmonisation. ICH (2003) Q1A(R2), Stability testing of new drug substances and products. International Conference on Harmonization, Geneva
International Conference of Harmonisation. ICH (1996) Q1B, Stability testing: photostability testing of new drug substances and products. International Conference on Harmonization, Geneva
International Conference of Harmonisation. ICH (1994) Q2(R1), Validation of analytical procedures: text and methodology. International Conference on Harmonization, Geneva
Lisbeth P, Vincent H, Kristof M, Bernard S, Manuel M, Hugo N (2016) Genotype and co-medication dependent CYP2D6 metabolic activity: effects on serum concentrations of aripiprazole, haloperidol, risperidone, paliperidone and zuclopenthixol. Eur J Clin Pharmacol 72(2):175–184. https://doi.org/10.1007/s00228-015-1965-1
Fond G, Macgregor A, Tamouza R, Hamdani N, Meary A, Leboyer M, Dubremetz JF (2014) Comparative analysis of anti-toxoplasmic activity of antipsychotic drugs and valproate. Eur Arch Psychiatry Clin Neurosci 264:179–183. https://doi.org/10.1007/s00406-013-0413-4
Bryan EJ, Purcell MA, Kumar A (2017) Zuclopenthixol dihydrochloride for schizophrenia. Cochrane Database of Syst Rev 11:5474. https://doi.org/10.1002/14651858.CD005474.pub2
Viala A, Ba B, Durand A et al (1988) Comparative study of the pharmacokinetics of zuclopenthixol decanoate and fluphenazine decanoate. Psychopharmacology 94:293–297. https://doi.org/10.1007/BF00174677
Tracqui A, Kintz P, Cirimele V, Berthault F, Mangin P, Ludes B (1997) HPLC-DAD and HPLC-MS findings in fatality involving (Z)-cis-clopenthixol (zuclopenthixol). J Anal Toxico 21:314–318. https://doi.org/10.1093/jat/21.4.314
Kollroser M, Henning G, Gatternig R, Schober C (2001) HPLC-ESI-MS/MS determination of zuclopenthixol in a fatal intoxication during psychiatric therapy. Forensic Sci Int 123:243–247. https://doi.org/10.1016/S0379-0738(01)00540-0
Bodil BH, Steen HH (1944) Determination of zuclopenthixol and its main N-dealkylated metabolite in biological fluids using high-performance liquid chromatography with post-column photochemical derivatization and fluorescence detection. J Chromatogr B: Biomed Sci Appl 658:319–325. https://doi.org/10.1016/0378-4347(94)00245-2
Bikshal BK, Useni RM, Venkateswara RA, Maheshwara RL (2018) Intended high-performance liquid chromatography procedure for the quantification of norfloxacin and its potential impurities in active pharmaceutical ingredient and tablet dosage forms. Thai J Pharm Sci 42:27–36
Mallu UR, Anna VR, Kasimala BB (2019) Rapid stability indicating HPLC method for the analysis of Leflunomide and its related impurities in bulk drug and formulations. Turk J Pharm Sci 16:457–465. https://doi.org/10.4274/tjps.galenos.2018.34635
Bikshal BK, Venkateswara RA, Useni RM (2018) Stability-indicating reversed-phase HPLC method for the separation and estimation of related impurities of Cilnidipine in pharmaceutical formulations. Indian Drugs 55:41–49. https://doi.org/10.53879/id.55.12.11185
Sri Girija K, Bikshal BK, Venkateswara RA (2021) A new high-performance liquid chromatography method for the separation and simultaneous quantification of eptifibatide and its impurities in pharmaceutical injection formulation. Int J Appl Pharm 13:165–172. https://doi.org/10.22159/ijap.2021v13i2.39895
Cherukula SR, Chilakabattina NNB, Bhagya KT, Nalleboyina V, Venkateswara RA (2023) Development of a stability-indicating HPLC method for Lasmiditan and its process related impurities with characterization of degradation products by LC-MS/MS. J Chem Metrol 17:79–92. https://doi.org/10.25135/jcm.89.2304.2767
Varma RB, Rao BS (2022) Gas Chromatography-Head Space-Flame Ionization Sensor based assessment of four residuary solvents in rivaroxaban bulk medication. Res J Pharm Technol 15:5158–5163. https://doi.org/10.52711/0974-360X.2022.00868
Varma BHR, Rao BS (2023) Gas chromatography-head space-mass spectrometry sensor based quality control of dobutamine hydrochloride bulk material for a mutagenic impurity, 2-bromopropane. Res J Chem Environ 27:54–61. https://doi.org/10.25303/2702rjce054061
Rajesh VB, Battula SR, Kapavarapu MVNR, Mandapati VR (2022) A novel Rivaroxaban degradation impurity detection by RP-HPLC extraction by preparative chromatography, and characterization by LC-MS, NMR and FT-IR: analysis of novel impurity in batch samples and tablets of Rivaroxaban. Rasayan J Chem 15:2373–2381. https://doi.org/10.31788/RJC.2022.1547008
Rajesh VB, Sreenivasa RB, Maruthi VNRK, Varaprasad RM (2023) Assessment of gas chromatography methodology approach for the trace evaluation of carcinogenic impurity, methyl chloride, in trimetazidine dihydrochloride. Ann Pharm Fr 81:64–73. https://doi.org/10.1016/j.pharma.2022.06.012
Acknowledgements
The corresponding author wish to acknowledge department of Engineering Chemistry, College of Engineering, Koneru Lakshmaiah Education Foundation, Vaddeswaram, Guntur—522302, A.P., India for support to carry out this research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
We have assured that “all authors have read and approved the manuscript.” All the authors have equal contribution and participation in this research work. BS and JSK had completed his work under the supervision of Dr. VRA, IVKV and GSR helped to carry research work and guides to resolve the complications.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
There is no living organism involved to carry out this research work and hence ethical approval not required.
Consent for publication
The corresponding author on behalf of all author approved consent for publication of submitted paper in the journal.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sesharamsingh, B., Kumar, J.S., Viswanath, I.V.K. et al. Development of stability-indicating HPLC method for quantification of pharmacopeia impurities of Zuclopenthixol and characterization of its stress degradation products by LCMS/MS. Futur J Pharm Sci 9, 101 (2023). https://doi.org/10.1186/s43094-023-00554-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-023-00554-5