
Abstract
Background
Hepatitis C virus (HCV) is considered a worldwide health problem since it affects over 3% of the population and causes 300,000 fatalities per year. Chronic infection causes high morbidity and mortality in patients, leading to liver cirrhosis, hepatocellular carcinoma, fibrosis, liver cancer, and other HCV-related illnesses. Finding novel and better HCV treatments is a top international health goal right now. As a result, the pressing need for new HCV antiviral drugs has fueled research into the structural requirements of NS5B polymerase inhibitors at a molecular basis.
Results
In this study, an in silico technique was applied to study 79 compounds with HCV inhibitory bioactivity, with the best statistical results (\(R^{2}\)= 0.7051, \(Q^{2}\) = 0.6455, \(R_{{{\text{pred}}}}^{2}\) = 0.6992, \(^{{\text{c}}} R_{{\text{r}}}^{2}\) = 0.6570, SEE = 0.2694).
Conclusions
This QSAR investigation allowed the research team to evaluate the influence of straightforward descriptors, shedding insight into the critical elements that guide the design of innovative effective molecules. Most of the innovative effective molecules exhibited better binding affinity (− 195.6 kcal/mol) than dasabuvir the reference drug (− 171.0 kcal/mol) with the target receptor (hepatitis C virus NS5B RNA polymerase). ADMET prediction disclosed enhanced pharmacokinetic properties with lower MRTD (maximum tolerated dose) of some new derivatives.
Similar content being viewed by others
Background
Hepatitis C Virus (HCV) is considered a worldwide health problem since it affects over 3% of the population and causes 300,000 fatalities per year [1]. Chronic infection causes high morbidity and mortality in patients, leading to liver cirrhosis, hepatocellular carcinoma, fibrosis, liver cancer, and other HCV-related illnesses [2]. HCV is an encapsulated single-strand linear positively oriented RNA virus belonging to the species Hepacivirus, which belongs to the Flaviviridae family. It has six genotypes and various subtypes, each with a different geographic distribution [1].
The host ribosomes translate HCV-RNA into a polyprotein, which is then processed by host and viral proteases into ten proteins: three structural proteins (core, E1, and E2), an ion channel (p7), and six non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [2]. The virus nucleocapsid is made up of structural proteins, while non-structural proteins are involved in virus development, maturation, and reproduction. The HCV NS5B protein is a multifunctional RNA binding protein that is critical for virus duplication. NS5B protein inhibition has been shown in multiple studies to successfully suppress viral replication [3]. As a result, the NS5B protein is a well-established therapeutic target for which several medicines have been licensed and released.
Anti-HCV vaccination is currently unavailable; nonetheless, in the 1990s, interferon-alpha was used to treat HCV, and in the next decade, pegylated interferon-alpha in a mixture with ribavirin was used to treat HCV. Interferon-alpha inhibitors, DNA and RNA polymerase inhibitors, NS3/4A RNA protease inhibitors, NS5 RNA serine protease inhibitors, and NS5B RNA polymerase inhibitors have all been licensed for use in clinical trials since 2011 [1, 4].
Current HCV medication has a low continued viral success rate, quick onset of drug resistance, especially in genotype 1 HCV patients, and considerable adverse effects, all of which lead to therapy cessation [5]. As a result, better anti-HCV medications with fewer problems are urgently needed. Finding the most essential molecular descriptors can aid in the discovery of new biologically active chemicals that can be used as anti-HCV medications and provide clinically successful treatment [6].
With advances in computational algorithms and simulation software, computer drug design has become widely used for drug discovery and development because of the advantages of being less time-consuming, cost-effective, and high effectiveness in silico screening and prediction of candidate medications [7]. Throughout the previous few decades, QSAR procedures have been developed and widely employed in a diversity of sectors, including chemical/biological, chemistry, and associated fields [4]. In this study, a QSAR model was proposed based on the structure of drugs having significant NS5B polymerase inhibitor bioactivity, and the model was externally validated to evaluate its genuine predictive capabilities. Based on the good efficacy of QSAR and molecular docking, the proposed model was efficient in understanding the relationship between structure and bioactivity and making suitable structure modifications for more promising suppression. Drug-likeness and ADMET pharmacokinetic characteristics were also tested on the novel compounds.
Methods
Dataset
79 compounds of HCV NS5B polymerase inhibitors were employed, which were sourced from online resources (https://pubchem.ncbi.nlm.nih.gov/). The EC50 (µM) values of the identified compounds' bioactivity were provided. The bioactivity in EC50 was transformed to Log (1/EC50), which gives pEC50 [8]. Additional file 1: Table S1 in the Additional file 1 contains the datasets.
Optimization and descriptors calculation
The optimization was carried out using the quantum mechanical approach in Spartan 14 and the density function theory B3LYP/631G** [9]. PaDEL-Descriptors were used to create roughly 2000 molecular descriptors for every species using the preprocessing molecules.
Dataset treatment, division, and Selection of optimal descriptors
The Drug Theoretics and Cheminformatics Laboratory software (DTC-QSAR v1.0.5) freely available to the public at http://dtclab.webs.com/software-tools was used in performing the dataset pre-treatment, division, and selection of optimal descriptors for Model building [10]. The total dataset was split into two parts: 70% model building data and 30% model test data [8]. The DTC-QSAR tool v1.0.5’s Genetic Function Algorithm (GFA) was used to choose a set of descriptors that best explained the diversity in bioactivity values of the investigated compounds. GFA is a heuristic search approach for locating exact or approximate answers to optimization and search issues. It has the advantage of constructing many models, use the lack of fit function to avoid overfitting, and allowing the user to regulate the length of the equation [11, 12].
Model applicability domain (AD)
The length of projection methodology was used to set up the AD for QSAR models in the research, which is established on leverage principles of the research compounds and standardized residual (SR) of the model. The horizontal component of the hat state vector was used to create compound leverages (hi) as given in Eq. (1):
XT is the transpose of X, and X is the descriptor array. SR is calculated using the model's error function with Eq. (2)
p represents the actual bioactivity value for the trained or test data, \(\overline{p}\). represents the model's estimated bioactivity value and n stands for the number of molecules in the trained or test data.
The borders > − 3, < 3 and < h* were used to design the AD models, where h* stands for caution leverage, which is calculated using Eq. (3)
t is the proportion of structural properties in the model and n has the same meaning as in Eq. (2) [13].
Design of new chemical entities
New chemical entities are designed by a careful selection of template molecule (T) with high bioactivity, low standardized residual and within the model AD from the dataset and making modifications based on the description of the descriptor selected for optimal model (see Additional file 1: Table S3).
Accessing the design entities
To determine the reliability of these new design entities, they were evaluated using the online tools SwissADME (www.swissadme.ch/) and pkCSM (http://structure.bioc.cam.ac.uk/pkcsm) to estimate the drug-likeness and pharmacokinetics ADMET profile.
Docking process
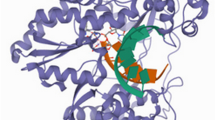
The target receptor was accessed from a protein data bank deposited in 2013 by Schoenfeld and co-worker [14] with the following information: PDB ID of 4MKA, the resolution of 2.05 Å, and the R value free of 0.217. The co-crystallized molecule was removed after which the target receptor was imported into the Molegro Virtual Docker tools (MVD) and the Preparation tab was selected on the dialog and Assign All was set to Always. This ensures that all preparation will be done by MVD. The detailed docking process was applied as described in our previous work [13]. The target receptor is presented in Fig. 1

Structure of the target receptor (hepatitis C virus NS5B RNA polymerase) in complex with dasabuvir
Results
Three mathematical models relating the bioactivity of hepatitis C virus NS5B RNA polymerase inhibitors with its structure were built in the present research, and the QSAR models are presented in Table 1 with their coefficient of determination (R2), standard error of estimate (SEE), and mean average error (MAE) values.
The R2 and the error measurement (SEE and MAE) were used to assess the performance of QSAR models, resulting in model 2 being chosen for further investigation. The better the model, the higher the R2 scores or the lower the error values. In addition, the Tropsha parameters [8] were utilized to assess the top models' reliability. All the statistical tools and necessary predictive capability as a benchmark for a good QSAR model are listed in Table 2.
The descriptors included in the selected model, as well as their descriptions, are listed in the Additional file 1: Table S2, and the numerical values of the descriptor used in the selected model are presented in Additional file 1: Table S3, while Table 3 presents observed EC50, pEC50, predicted pEC50, and residual of the entire datasets.
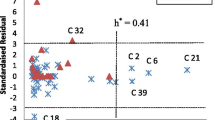
William’s plots were generated and are presented in Fig. 2 for the selected QSAR model, based on the plot of standardized residual (SR) versus leverage values (hi), to get an immediate and straightforward graphical detection of both the Y outliers and the X outliers of the model, and Fig. 3 depicts a display of estimated vs actual bioactivity over the whole data, revealing a strong link between actual and estimated bioactivity levels in the models.

The model applicable domain

Plot of the model predicted against experimental anti-hepatitis C bioactivity values
Based on the proven and verified QSAR model, molecule 16 in dataset Table (Additional file 1: Table S1), pictured in Fig. 4, was utilized as a standard to characterize the molecule. Molecule 16 was utilized as a foundation for developing new molecules because this was chosen cautiously from Fig. 2, recognizing the data point as more active, low SR, and detected inside the existing model’s AD.

(E)-3-[6-[[2-[(3-cyclopentyl-1-methyl-2-pyridin-2-ylindole-6-carbonyl)amino]-2-methylpropanoyl]amino]-2-ethoxypyridin-3-yl]prop-2-enoic acid (Template Molecule)
The bioactivity of the precursor drug, newly created molecules, and reference molecule (dasabuvir) was predictable using the previously proved QSAR model (model 2). All modified variants, as well as dasabuvir, had a greater pEC50 value than the prototype, according to the data except N5 (see Additional file 1: Table S4).
The interaction of molecules N15, R, and T with the target receptor (hepatitis C virus NS5B RNA polymerase, PDB ID: 4MKA) was visualized using the Discovery Studio software and the Molecular Operating Environment (MOE) software, as shown in Figs. 5, 6, and 7, respectively.

A 3D diagram for the interaction of molecule 15 (N15) in complex with the target receptor (hepatitis C virus NS5B RNA polymerase, PDB ID: 4MKA), B 2D diagram for the interaction of molecule 15 (N15) in complex with the target receptor (hepatitis C virus NS5B RNA polymerase),

A 3D diagram for the interaction of reference molecule R (dasabuvir) in complex with the target receptor (hepatitis C virus NS5B RNA polymerase, PDB ID: 4MKA), B 2D diagram for the interaction of reference molecule R (dasabuvir) in complex with the target receptor (hepatitis C virus NS5B RNA polymerase)

A 3D diagram for the interaction of template molecule T in complex with the target receptor (hepatitis C virus NS5B RNA polymerase, PDB ID: 4MKA), B 2D diagram for the interaction of template molecule T in complex with the target receptor (hepatitis C virus NS5B RNA polymerase)
A molecular docking assessment utilizing Molegro Virtual Docker version 6.0 verified that molecule N15 had the most energetically favorable ligand conformation with the MolDock scoring function value of − 195.6 kcal/mol which is better than that of the reference drug of − 171 kcal/mol. The results of the ligand–receptor interactions of the novel molecule (N15) in Fig. 5, the reference molecule (R) in Fig. 6, and the template molecule (T) in Fig. 7 are summarized in Table 4.
To establish the viability of these molecules as medications, the research team looked at the drug-like and pharmacokinetic properties (ADMET) of these novel compounds, using dasabuvir as a reference. The ADMET properties given in Table 6 were estimated using the pkCSM software, while the drug-likeness attributes presented in Table 5 were originally assessed using the web tool SwissADME. Drug-likeness characteristics are an important criterion for drug candidates in the early stages of development [15]. The practice of correlating a molecule's physicochemical characteristics with its biopharmaceutical characteristics in the human body, notably its impact on oral bioavailability, is also known as the drug-likeness approach [16].
Discussion
The selected QSAR model is statistically sound because it met the conditions listed in Table 2 as a threshold, and hence, has an appropriate predictive capability. Model 2 met Tropsha's and the Organization for Economic Cooperation and Development's (OECD) requirements [12, 17] as it explains 71% and predicts 70% of the variances of the HCV NS5B polymerase inhibitors with their bioactivity as presented in Table 2. It indicates that the models precisely regressed the data and that it can predict the fitting training set for the model, as it predicted around 70% of the data and so met the minimum criteria of 60% [11]. The result of the y-randomization test demonstrates that the model's random R2 (cR2p = 0.6570) is significantly greater than the recommended value of 0.50, indicating that the model is not the result of pure chance [12].
In this study, no Y outlier was detected and one X outlier was detected (molecule 51) accounting for 1% of the entire dataset (see Fig. 2). The majority of the compounds fall within the application domain, indicating that the chemical molecules adhere to a well-defined applicability region. In summary, the optimization techniques have a great deal of potential and are extremely successful. As a consequence, it can be utilized to enhance the bioactivity of any of the chemicals being studied.
Figure 3 shows the estimated vs. actual bioactivity levels across the entire dataset, indicating a close relationship between actual and predicted bioactivity levels in the models which are further confirmed by low residual values presented in Table 3. Because the models have the internal and external statistical capacity and are structurally error-free, they can be used to infer whether or not a known molecule is active if it falls inside the models' AD. As indicated in Additional file 1: Table S4, N15 has the highest predicted bioactivity of all the molecules, much exceeding dasabuvir. It shows the template structure, newly improved molecules, and licensed molecule (dasabuvir), as well as their expected responses, leverages, and MolDock score (binding affinity). Except for molecule 51, the majority of the molecules' leveraging findings were logical and below the leveraging cutoff point (h* = 0.43), showing that the improved molecules, as well as dasabuvir, met the requirements and were within the model's applicability restrictions.
GGI10 is a topological descriptor that measures atom-to-atom charge transfer. It has the largest positive coefficient in the calculated linear equation in the chosen model. This means that increasing the charge transfer between the pair of atoms will increase the pEC50 values, and the more active the molecule will be. Conversely, decreasing the charge transfer between the pair of atoms will result in a decrease in the pEC50 values, and the less active the molecule will be [18]. GATS2p and VR2Dzi are the other positive correlation descriptors in the chosen model, in order of positive contribution as measured by their coefficients. On the other hand, the ATSC5e is the most negative descriptor contributor in the chosen model, and it is defined as a based Broto–Moreau autocorrelation of lag 5 measured by electronegativities that explains how electronegativities effects are spread along with the topological structures of the molecule [19], and its presence in the model linked the electronegativities of pairs atoms that are bonded together. The built model revealed that increasing ATSC5e reduces the activity of the compounds and that other descriptors such as GATS6p, nX, and MDEC-24 have a significant negative impact on reported anti-HCV bioactivity in decreasing order.
The molecular docking method determines the most thermodynamically favorable ligand conformation using an energy-based scoring function. Lower energy scores, in comparison with higher energy values, are thought to imply better protein–ligand bindings (binding affinity). As a result, molecular docking can be thought of as an optimization problem in which the goal is to discover the lowest-energy ligand-binding configuration. The results presented in Additional file 1: Table S4 show that N15 has the highest predicted pEC50 value of 12.05 and Molegro Virtual Docker version 6.0 was used to further confirm the result by performing molecular docking analysis which revealed that molecule N15 had the lowest-energy ligand-binding configuration (see Additional file 1: Table S4). The predicted chemical N15, according to the data, outperforms both the reference and template molecules. It had the most favorable MolDock score (binding affinity) of -195.6 kcal/mol, and total interaction energy of − 13.0 kcal/mol resulting from six H-bonds and one ionic bond as reported in Table 4, wherewith NH of the piperidine, the OH of the carboxyl group of the acetic acid moiety attached to benzene ring formed H—bond donor with GLU493, HIS293, MET485 and SER297 of the target residue, respectively, while the oxygen atom (O) of the carboxyl group of the acetic acid moiety attached to pyrrolidine and piperidine formed H—bond acceptor with ARG481 and TRP501, respectively, and finally the NH of piperidine moiety formed ionic interaction with GLU493 of the target receptor residue. All these interactions stabilize the N15 in the binding pocket of the receptor, making N15 the most potent novel inhibitor of the target receptor as observed in Table 4 and Fig. 5, and these could be attributed to the presence of pyrrolidine and piperidine groups. The dasabuvir (reference molecule) had a lower MolDock score (binding affinity) of − 171.0 kcal/mol, and total interaction energy of -8.0 kcal/mol resulting from four H-bonds. The methylsulfonyl group attached to dimethylamine connected to the naphthalene ring formed two H—bond acceptors with ARG467 residue, while –NH attached to pyridine ring and –OCH3 attached to benzene ring formed H—bond donor each with ASP454 and THR416 target residue, respectively, as reported in Table 4.
Discovery Studio and Molecular Operating Environment (MOE) software were used for virtual evaluation which shed more light on the interaction of molecules N15, R, and T with the target receptor (hepatitis C virus NS5B RNA polymerase, PDB ID: 4MKA). When compared to the reference molecule, molecule N15 has a higher degree of interaction with the target receptor.
The Lipinski idea of five, which argues that if a molecule violates more than two of the qualities given in Table 5, it would be poorly absorbed [15], is one of the most popular and useful directions in the early clinical phase of drug development. The selected compounds, except for N6, N7, and N11, are regarded to meet Lipinski's criterion because the majority of them did not breach more than two, and so maybe categorized as drug-like molecules. Furthermore, an evaluation was carried out using ABS standards, with the template and all generated molecules, including the standard molecule, having a value in the range of 0.11 to 0.56. These criteria were established based on the probability value of a molecule with an optimal permeability and bioavailability outline, where this range represents total compliance of the Lipinski concept of five, which is a characteristic value of more than 10% [20]. On a scale of 1 to 10, the synthetic accessibility of the chemicals in question was also graded (extremely easy to synthesize to complicated to synthesize) [20]. Except for N5, N6, N7, N11, and N13, which have synthetic accessibility of 5.01–5.26, making them easy to synthesize, the majority of the compounds studied have synthetic accessibility of ≤ 5 (see Table 5).
The ADMET profiles of identified compounds and the reference molecule were assessed and are displayed in Table 6 using web-based pkCSM tools. BBB penetrations values help determine whether or not a chemical will pass across the blood–brain barrier (BBB). A Log BB > 0.3 indicates that a molecule can easily pass the blood–brain barrier, whereas a Log BB < − 1 indicates that the molecule is poorly distributed [21]. Except for N7 all of the compounds studied easily crossed the blood–brain barrier in this investigation (see Table 6).
A molecule with a Papp > 8 × 10−6 cm/s is considered to have a high Caco-2 permeability as a credible in vitro model for estimating oral drug permeability [21]. The findings of this study revealed that all of the compounds studied have only modest cell uptake in Caco-2 cells.
The HIA forecasts the percentage of a substance that will be absorbed through the human gut. Poorly absorbed molecules are those that have absorption of less than 30% [15]. The study findings revealed that all of the compounds studied have excellent HIA scores.
The maximum tolerated dose (MRDT) is a measurement of a chemical's hazardous dosage threshold in humans. An MRTD of less than or equal to 0.477 log(mg/kg/day) is considered low for a specific chemical, while significantly greater than 0.477 log(mg/kg/day) is considered high [15, 20]. Except for N14, all of the compounds in this investigation had a low toxicity.
Conclusions
The statistically tested QSAR models produced justified describing the antiviral properties of the compounds investigated. The models are statistically valid, with R2 values of 0.7051 and 0.6992 for internal and external validation, respectively, and match the criteria for a good QSAR model, as advocated by many organizations. According to the molecular docking simulation, most of the modified compounds had a higher binding affinity for the target receptor (hepatitis C virus NS5B RNA polymerase) than dasabuvir, the reference medication. The enhanced therapeutic features of some novel derivatives were revealed by ADMET prediction, with lowered MRTD (maximum tolerable dosage). This inquiry can be useful to comprehend the physicochemical and biological bioactivities of hepatitis C virus NS5B RNA polymerase inhibitors and their modified derivatives using a QSAR model, molecular docking, drug-likeness, and ADMET analysis to find new and improved HCV antiviral medicines.
Availability of data and materials
All data and materials are available upon request.
Abbreviations
- HCV:
-
Hepatitis C virus
- NS:
-
Non-structural proteins
- QSAR:
-
Quantitative structure–activity relationship
- PDB:
-
Protein data bank
- AD:
-
Applicability domain
- SR:
-
Standardized residual
- IC50 :
-
Half maximal inhibitory concentration
- OECD:
-
Organization for economic co-operation and development
- MLR:
-
Multiple linear regression
References
Zając M, Muszalska I, Sobczak A, Dadej A, Tomczak S, Jelińska A (2019) Hepatitis C: new drugs and treatment prospects. Eur J Med Chem 165:225–249. https://doi.org/10.1016/j.ejmech.2019.01.025
Abuelizz HA, Marzouk M, Bakheit AH, Al-Salahi R (2020) Investigation of some benzoquinazoline and quinazoline derivatives as novel inhibitors of HCV-NS3/4A protease: biological, molecular docking and QSAR studies. RSC Adv 10(59):35820–35830. https://doi.org/10.1039/d0ra05604a
Qin Z, Yan A (2020) QSAR studies on hepatitis C virus NS5A protein tetracyclic inhibitors in wild type and mutants by CoMFA and CoMSIA, SAR QSAR. Environ Res 31(4):281–311. https://doi.org/10.1080/1062936X.2020.1740889
Li W, Si H, Li Y, Ge C, Song F, Ma X, Duan Y, Zhai H (2016) 3D-QSAR and molecular docking studies on designing inhibitors of the hepatitis C virus NS5B polymerase. J Mol Struct 1117:227–239. https://doi.org/10.1016/j.molstruc.2016.03.073
Therese PJ, Manvar D, Kondepudi S, Battu MB, Sriram D, Basu A, Yogeeswari P, Kaushik-Basu N (2014) Multiple e-pharmacophore modeling, 3D-QSAR, and high-throughput virtual screening of hepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model 54(2):539–552. https://doi.org/10.1021/ci400644r
Algamal ZY, Lee MH, Al-Fakih AM, Aziz M (2017) High-dimensional QSAR classification model for anti-hepatitis C virus activity of thiourea derivatives based on the sparse logistic regression model with a bridge penalty. J Chemom 31(6):1–8. https://doi.org/10.1002/cem.2889
Zheng M, Liu X, Xu Y, Li H, Luo C, Jiang H (2013) Computational methods for drug design and discovery: focus on China. Trends Pharmacol Sci 34(10):549–559. https://doi.org/10.1016/j.tips.2013.08.004
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inform 29(6–7):476–488. https://doi.org/10.1002/minf.201000061
Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O’Neill DP, DiStasio RA, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Yeh Lin C, Van Voorhis T, Hung Chien S et al (2006) Advances in methods and algorithms in a modern quantum chemistry program package. Phys Chem Chem Phys 8(27):3172–3191. https://doi.org/10.1039/b517914a
Ambure P, Gajewicz-Skretna A, Cordeiro MNDS, Roy K (2019) New workflow for QSAR model development from small data sets: small dataset curator and small dataset modeller. Integration of data curation, exhaustive double cross-validation, and a set of optimal model selection techniques. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00476
Arthur DE, Ejeh S, Uzairu A (2020) Quantitative structure-activity relationship (QSAR) and design of novel ligands that demonstrate high potency and target selectivity as protein tyrosine phosphatase 1B (PTP 1B) inhibitors as an effective strategy used to model anti-diabetic agents. J Recept Signal Transduct 40(6):501–520. https://doi.org/10.1080/10799893.2020.1759092
Ejeh S, Uzairu A, Shallangwa GA, Abechi SE (2021) Computer-aided identification of a series of novel ligands showing high potency as hepatitis C virus NS3/4A protease inhibitors. Bull Natl Res Cent. https://doi.org/10.1186/s42269-020-00467-w
Ejeh S, Uzairu A, Shallangwa GA, Abechi SE (2021) In silico design, drug-likeness and ADMET properties estimation of some substituted thienopyrimidines as HCV NS3/4A protease inhibitors. Chem Afr. https://doi.org/10.1007/s42250-021-00250-y
Schoenfeld RC, Bourdet DL, Brameld KA, Chin E, de Vicente J, Fung A, Harris SF, Lee EK, Le Pogam S, Leveque V, Li J, Lui A-T, Najera I, Rajyaguru S, Sangi M, Steiner S, Talamas FX, Taygerly JP, Zhao J (2013) Discovery of a novel series of potent non-nucleoside inhibitors of hepatitis C virus NS5B. J Med Chem 56(20):8163–8182. https://doi.org/10.1021/jm401266k
Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models: strategies and importance. Int J Drug Des Disocov 2(3):511–519
Yan F, Liu T, Jia Q, Wang Q (2019) Multiple toxicity endpoint–structure relationships for substituted phenols and anilines. Sci Total Environ 663:560–567. https://doi.org/10.1016/j.scitotenv.2019.01.362
Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2020) QSAR model for prediction of some non-nucleoside inhibitors of dengue virus serotype 4 NS5 using GFA-MLR approach. J Chem Lett 1:69–76
Todeschini R, Consonni V (2008) Handbook of molecular descriptors, vol 11. Wiley, New York
Umar AB, Uzairu A, Shallangwa GA, Uba S (2020) Docking-based strategy to design novel flavone-based arylamides as potent V600E-BRAF inhibitors with prediction of their drug-likeness and ADMET properties. Bull Natl Res Cent. https://doi.org/10.1186/s42269-020-00432-7
Danishuddin M, Khan SN, Khan AU (2010) Molecular interactions between mitochondrial membrane proteins and the C-terminal domain of PB1-F2: an in silico approach. J Mol Model 16(3):535–541. https://doi.org/10.1007/s00894-009-0555-5
Acknowledgements
Not applicable.
Funding
No fund received.
Author information
Authors and Affiliations
Contributions
SE and AU were in charge of selecting the research tool. SE, AU, GAS, and SEA were involved in the study design, paper writing, and subject selection and data collecting. The statistical analysis was carried out by SE, AU, GAS, and SEA, who also revised the final text. The completed paper was read and endorsed by all writers. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1. Chemical structures of the study compounds. Table S2. The descriptor used in the model and their descriptions. Table S3. Numerical values of the descriptor used in the selected model. Table S4. Chemical Structure, predicted pEC50, leverage values, and MolDock scores of template molecule (T), designed molecules (N), and reference Molecule (R).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ejeh, S., Uzairu, A., Shallangwa, G.A. et al. Computational insight to design new potential hepatitis C virus NS5B polymerase inhibitors with drug-likeness and pharmacokinetic ADMET parameters predictions. Futur J Pharm Sci 7, 219 (2021). https://doi.org/10.1186/s43094-021-00373-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00373-6