
Abstract
Background
Vildagliptin is a dipeptidyl peptidase-4 inhibitor used to treat diabetes mellitus. No bioequivalence study data have been published for the Indian population comparing bioequivalence of vildagliptin brands Galvus, Zomelis, and Jalra. This study aimed to evaluate the bioequivalence between three brands of vildagliptin 50 mg tablet (test 1, Zomelis; test 2, Jalra; and reference, Galvus) and to compare these test formulations with the reference formulation to meet the regulatory requirements of bioequivalence of CDSCO, India. The study was conducted in the clinical research center of the college after enrolling 12 healthy volunteers. This study was a single-dose, randomized, open-label, balanced, three treatment, three period, under fasting condition in 12 adult healthy volunteers. After overnight fasting, the subjects received a single dose of either of any three brands of the vildagliptin tablet (T1—test 1; T2—test 2; and R—reference). The washout period was 7 days. Randomization was in the way of T1T2R in the first period, T2T1R in the second period, and RT1T2 in the third period. Blood samples were collected, after that drug concentration in the plasma was measured with the help of HPLC. Outcome measures 90% confidence interval of the geometric mean ratios (test/reference) for the LnCmax, LnAUC0-t, and LnAUC0-∞ was calculated.
Results
The AUC0-t was 1390.03, 1401.50, and 1409.37 ng h/ml for the T1, T2, and R, respectively. Cmax was 287.89, 287.41, and 285.17 ng/ml for the T1, T2, and R, respectively. AUC0-∞ was 1452.03, 1467.59, and 1473.53 ng h/ml for the T1, T2, and R, respectively. No significant difference was observed in the pharmacokinetic parameters between the T1, T2, and R. The geometric mean ratios for T1/R for LnCmax, LnAUC0-t, and LnAUC0-∞ were 1.0014 (90% CI, 1.0002–1.0026), 0.9992 (90% CI, 0.9971–1.0013), and 0.9994 (90% CI, 0.9973–1.0016), respectively. For the T2/R, geometric mean ratios for LnCmax, LnAUC0-t, and LnAUC0-∞ were 1.0003 (90% CI, 0.9992–1.0013), 0.9988 (90% CI, 0.9969–1.0008), and 0.9985 (90% CI, 0.9961–1.0010), respectively.
Conclusion
In this single-dose study involving Indian healthy volunteers under fasting conditions, the three brands of vildagliptin (Zomelis, Jalra, and Galvus) were bioequivalent as per the bioequivalence criterion of CDSCO, India.
Similar content being viewed by others

Background
Diabetes mellitus is a metabolic disorder which is characterized by hyperglycemia with disturbances in the metabolism of carbohydrates, fats, and proteins which results because of the defects in secretion of insulin, action of insulin, or both. Thirst, polyuria, blurring of vision, and weight loss are the characteristic symptoms of diabetes [1]. Gliptins are prominent medications in type 2 diabetes management as five different molecules have commercialized, and they are also in combination with metformin [2]. Vildagliptin is one of gliptin drugs. Dipeptidyl peptidase 4 (DPP-4, DPP-IV) is found in the plasma, kidney, and brush-border membranes of the intestine, hepatocytes, on capillary endothelial cells’ surface, and a subset of T lymphocytes [3]. DPP-4 rapidly inactivate incretin glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide. GLP-1 stimulates postprandial-induced insulin secretion and leads to glucose homeostasis [4, 5]. GLP-1, which is in circulation, degraded rapidly, and its degradation is inhibited by DPP-4 [3, 6]. Inhibition of DPP-4 enzyme activity leads to GLP-1-increased activity; hence glycemic, control increased in human trials [7,8,9]. Therefore, augmentation of this GLP-1 activity in diabetic patients leads to lowering of glycemia [4, 10, 11]. Vildagliptin (1-[[3-hydroxy-1-adamantyl) amino] acetyl]-2-cyano-(S)-pyrrolidine) is orally active and highly selective inhibitor of DPP-4 [12]. After oral administration of vildagliptin, it is speedily absorbed, and it has eighty-fifth absolute bioavailability. Vildagliptin have low protein binding (9.3%); it equally distributes between the plasma and red blood cells. Eighty-five percent of the oral dose is ultimately excreted by the kidney as either metabolites or unchanged vildagliptin [13, 14]. Vildagliptin is a proprietary drug of Novartis, and it is sold under the brand name Galvus. Novartis licensed the drug to Abbott, USV, and Emcure [15]. Abbott sells vildagliptin as Zomelis, USV sells it as Jalra, and Emcure sells it as Vysov. These brands of vildagliptin recorded the sale of 822 crore out of 10,000 crore market of antidiabetic drugs in the year 2016 [16]. In our study, we decided to take three brands of vildagliptin Zomelis, Jalra, and Galvus to check their bioequivalence. Here, Zomelis was taken as test 1 drug (T1), Jalra was taken as test 2 (T2) drug, and Galvus was taken as the reference drug (R). The prices of these formulations are nearly the same.
Methods
Aim of the study
This study was done to evaluate the bioequivalence of three different brands of vildagliptin tablet (50 mg) formulations following single-dose administration in healthy volunteers after an overnight fasting of 10 h in order to compare the bioequivalence of these preparations.
Compliance with ethical standards
For the compounds to be bioequivalent, the compounds should have the calculated 90% confidence interval for AUC and Cmax within the bioequivalent range, usually 80–125% [17]. Informed consent was taken from all the subjects as per the specifications of the Central Drug Standard Control Organization. The Institutional Review Board of our university approved this study protocol and informed consent. This study was conducted as per the Declaration of Helsinki and Good Clinical Practices as per the schedule Y of Drug and Cosmetic Rule 1945 of India and Central Drug Standard Control Organization (CDSCO) [18, 19]. This study was not funded by any organization, and no conflict of interest was reported.
Subjects
Twelve healthy Indian male volunteers between the age group of 18 and 25 years were enrolled in the study, and their body mass index was in the range of 18.50–24.90 kg/m2 (both inclusive) [20]. Written informed consent was taken from all volunteers before the start of the study. For the subjects, some exclusion criteria were there.
Study design and procedures
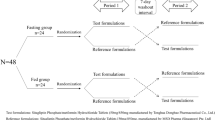
This study was single-dose, open-label, randomized, 3-treatment, 3-period. After a night fasting, the subjects took a single dose of either of any three brands of the vildagliptin tablet {T1 (Batch No. BF488), T2 (Batch no. BE448), R (Batch no.BL218)}. There were 7 days of washout period between each period of the study. The randomization schedule was like T1T2R in the first period, T2T1R in the second period, and RT1T2 in the third period (T1 = Zomelis, T2 = Jalra, and R = Galvus). The study was explained to each subject, and a written informed consent was taken from each subject according to the Schedule Y of the Drug and Cosmetic Act and Rules 1945 of India. After taking the informed consent, a clinical examination was done for the subjects. Routine clinical tests were performed at the DIPSAR Clinical Research Laboratory 1 week before the start of the study. Before the dose administration, the subjects were enrolled in the clinical research center of DIPSAR and were kept confined to the center for the duration of the study. A peripheral venous catheter was placed in the antecubital vein of the subjects, and it was flushed with 0.5 ml of heparin in normal saline (NS) solution (1:20). To get samples free from NS solution and heparin, first, 1 ml of blood was discarded. For the determination of the amount of vildagliptin in the plasma, blood samples (4 ml) were collected in K2EDTA vacutainers at the following given times: predose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 h. These blood samples were centrifuged at 2500 rpm for 15 min at 4 °C. The resulting plasma samples were separated and stored at − 75 °C until analysis. For the adverse events, subjects were monitored for 24 h during the study using clinical measurements like blood pressure (BP), pulse rate, and body temperature and by orally asking any complaint felt by the subjects.
Determination of vildagliptin plasma concentration
Plasma concentrations of vildagliptin were determined in the Clinical Research Center Laboratory of DIPSAR, using a validated HPLC-UV method. HPLC system used was of Shimadzu Corporation (LC-2010C HT) model. The HPLC components consist of the following equipment: System Controller (LC-2010HT), Sample Cooler (LC-2010CHT), Degasser (5 line degasser), Column Oven, Autosampler injector, Pump (4 pump system), UV-visual detector, and reservoir tray. Vildagliptin was extracted from the plasma by using the protein precipitation extraction method. Plasma samples which were stored at − 75 °C were taken out from the deep freezer. The method which was followed was a slightly modified version of Santhakumari et al.’s method [20]. An aliquot of 210 μl plasma was taken into the Eppendorf tubes and added 50 μl of internal standard dilution (tolbutamide 1000 ng/ml) and then it was vortexed to mix the contents; 1200 μl methanol was used as a precipitating solvent to precipitate vildagliptin. After that, it was vortexed for 1 min, and after that, it was centrifuged at 4 °C, 7000 rpm for 10 min. The resulting supernatant was taken out and transferred to HPLC vials. The mobile phases which were used in this method were 50 mM ammonium bicarbonate (pH 7.8) (phase A) and 100% acetonitrile (phase B). One milliliter per minute was the flow rate of mobile phases. The chromatographic system consisted of a C-18 column, and the UV detector was set at 210 nm (Table 1). The total run time was 20 min. The injection volume was 20 μl.
Statistical methods and data analysis
Pharmacokinetic analysis
There was no dropout of subjects in the study, and all the subjects participated till the last period of the study. To determine the pharmacokinetic parameters of vildagliptin, the non-compartmental pharmacokinetic method was employed. By visual inspection of each subject’s plasma, concentration-time profile maximum plasma concentration (Cmax, ng/ml) and time to reach peak plasma concentration (Tmax, h) were obtained. The area under the curve (AUC) from time 0 to the last measurable concentration time (AUC0-t) was calculated by the trapezoidal method. Microsoft Excel was used as the software to calculate various parameters in this bioequivalence study. The t½ (h) was calculated as 0.693/kel where kel is terminal elimination rate constant. The AUC from time 0 to infinite time (AUC0-∞) was calculated as AUC0-t + Ct/kel; here, Ct represents the last quantifiable concentration, and kel represents the terminal elimination rate constant. kel was calculated by least-squares regression analysis during the terminal log-linear phase of the concentration-time curve [21, 22].
Statistical analysis
To determine the bioequivalence between the products, analysis of variance (ANOVA) at α = 0.05 was performed to determine the statistical differences of Cmax, AUC0-t, and AUC0-∞ which represented the rate and extent of drug absorption. ANOVA was performed on the logarithmically (Ln) transformed data of Cmax, AUC0-t, and AUC0-∞. The bioequivalence between the three formulations was assessed by calculation of the 90% confidence interval for the ratios of Cmax, AUC0-t, and AUC0-∞ obtained after the administration of the three formulations using logarithmically transformed data. For the products to be bioequivalent, the requirement is the 90% CI of the Cmax, AUC0-t, and AUC0-∞ to be within the acceptance criteria of 0.8–1.25 (CDSCO guidelines, 2005). Any statistical difference at P < 0.05 was considered as significant.
Results
Bioanalytical method
The calibration curve of vildagliptin was linear ranging from 10 to 1000 ng/ml, and the linear regression of the drug concentration versus peak height ratios (vildagliptin/IS) gave coefficients of determination (r2) = 0.9992. The lower limit of quantitation was 10.0 ng/ml with the accuracy (%) and precision (CV %) of 97.37and 1.48, respectively.
Inter-formulation variations between T1/T2 and R were analyzed by significance testing (ANOVA) for each logarithmically transformed data of AUC0-t, AUC0-∞, and Cmax, with the results of F values and P values were as given below:
ANOVA of Cmax | ||||||
|---|---|---|---|---|---|---|
Source of variation | SS | df | MS | F | P value | F crit |
Rows | 0.000894 | 11 | 8.13E−05 | 4.628591 | 0.001101 | 2.258518 |
Columns | 0.000118 | 2 | 5.92E−05 | 3.374427 | 0.052702 | 3.443357 |
Error | 0.000386 | 22 | 1.76E−05 | |||
Total | 0.001399 | 35 | ||||
ANOVA of AUC0-t | ||||||
|---|---|---|---|---|---|---|
Source of variation | SS | df | MS | F | P value | F crit |
Rows | 0.000723 | 11 | 6.57E−05 | 0.767611 | 0.667168 | 2.258518 |
Columns | 0.000216 | 2 | 0.000108 | 1.261262 | 0.302993 | 3.443357 |
Error | 0.001883 | 22 | 8.56E−05 | |||
Total | 0.002822 | 35 | ||||
ANOVA of AUC0-∞ | ||||||
|---|---|---|---|---|---|---|
Source of variation | SS | df | MS | F | P value | F crit |
Rows | 0.001064 | 11 | 9.67E−05 | 0.859377 | 0.588844 | 2.258518 |
Columns | 0.000257 | 2 | 0.000128 | 1.141905 | 0.337415 | 3.443357 |
Error | 0.002475 | 22 | 0.000113 | |||
Total | 0.003796 | 35 | ||||
Pharmacokinetic analysis
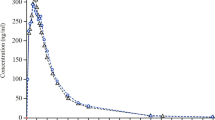
The mean plasma concentration vs. the time profile of all the three formulations in the 12 treated healthy subjects is shown in Fig. 1. After oral administration, the drug was absorbed rapidly of all three formulations, and it was available to the systemic circulation. Peak plasma concentrations (averaged) were 287.89, 287.41, and 285.17 ng/ml after the oral administration of the test 1, test 2, and reference formulations, respectively. The concentrations were reached at a mean time of 1.8 h after drug administration for all three formulations. The AUC0-t and AUC0-∞ values averaged 1390.03 and 1452.03 after the administration of the test 1 formulation, 1401.50 and 1467.59 ng h/ml after the administration of test 2 formulation, and 1409.37 and 1473.35 ng h/ml after the administration of the reference formulation, respectively. The data are shown in Table 2.

Mean plasma concentrations of vildagliptin after the administration of test 1, test 2, and reference formulations. Here, series 1 = test 1, series 2 = test 2, and series 3 = reference
Bioequivalence analysis
The mean ratio and the 90% confidence interval for Ln-transformed Cmax, AUC0-t, and AUC0-∞ are presented in Table 3. The lower and upper limits of the 90% confidence intervals were 1.0002–1.0026 for LnCmax, 0.9971–1.0013 for Ln AUC0-t, and 0.9973–1.0016 for LnAUC0–∞ of the test 1 formulation. The lower and upper limits of the 90% confidence intervals were 0.9992–1.0013 for LnCmax, 0.9969–1.0008 for LnAUC0-t, and 0.9961–1.0010 for LnAUC0–∞ of the test 2 formulation. It can be seen that these values lie in the range of bioequivalence (0.80–1.25). The Cmax values of vildagliptin after the administration of the three formulations did not differ significantly.
Discussion
Vildagliptin is a new orally effective antihyperglycaemic drug used in the treatment of type 2 diabetes. It is a specific inhibitor of dipeptidyl peptidase-IV (DPP-IV) inhibitor [23]. This inhibition prevents the degradation of the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). It leads to improve glycaemic control which is determined by glycated hemoglobin (HbA(1c)) and fasting plasma glucose (FPG) levels, and pancreatic alpha and beta cell functions also enhanced by the use of vildagliptin [24]. There were no adverse events encountered in this study. The objective of this study was to evaluate the bioequivalence of three brands of vildagliptin 50 mg tablet as test 1, test 2, and reference tablet which were administered as a single dose orally. As per the protocol, primary pharmacokinetic parameters AUC0-t, AUC0-∞, and Cmax were evaluated. According to the protocol, any concentration lower than the lower limit of quantification was considered as 0. Extrapolated AUC of vildagliptin was low having a mean value of 4.5% after the administration of the test 1 formulation, 4.7% of the test 2 formulation, and 4.6% of the reference formulation. It was less than 20% of AUC0-t which indicates that the time used for checking the bioequivalence between the products was good enough to calculate the plasma concentration-time profile of the drug. The 90% confidence intervals were calculated for test 1/reference tablet and test 2/reference tablet of LnCmax, LnAUC0-t, and LnAUC0-∞ of vildagliptin. After this, these confidence intervals were compared with the acceptance range of bioequivalence which is 0.80–1.25 as per the CDSCO criterion for bioequivalence [19]. In our study, we found that the 90% confidence interval of the test 1 and test 2 formulations of vildagliptin was within the bioequivalence criterion set for bioequivalence by CDSCO, India. For the Cmax values, no statistical difference was observed after the oral administration of vildagliptin tablet formulations of the test 1, test 2, and reference formulations. Based on this study, it can be stated that these three formulations of vildagliptin test 1, test 2, and reference (Zomelis, Jalra, and Galvus) are bioequivalent. The findings of this study can be used in the future for the purpose of bioequivalence and pharmacokinetic profiling of the vildagliptin; however, further studies can be done on it by taking more numbers of healthy subjects, more sensitive analytical methods, and more advanced software for the calculation of the pharmacokinetic profile.
Conclusion
In this single-dose study involving healthy male volunteers under fasting conditions, the three brands of vildagliptin ((1) Zomelis, (2) Jalra, and (3) Galvus) were found to be bioequivalent as per the bioequivalence criterion of CDSCO, India. Further studies can be done to compare vildagliptin bioequivalence by taking more numbers of healthy subjects, more sensitive analytical methods, and more advanced software for the calculation of the pharmacokinetic profile.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- T1:
-
Test 1 (Zomelis)
- T2:
-
Test 2 (Jalra)
- R:
-
Reference (Galvus)
- DPP-4:
-
Dipeptidyl peptidase-4
- GLP-1:
-
Glucagon-like peptide-1
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- HbA(1c):
-
Glycated hemoglobin
- FPG:
-
Fasting plasma glucose
- CDSCO:
-
Central Drug Standard Control Organization
- NS:
-
Normal saline
- BP:
-
Blood pressure
- C max :
-
Maximum plasma concentration
- T max :
-
Time to reach peak plasma concentration
- AUC:
-
Area under the curve
- ANOVA:
-
Analysis of variance
References
Alberti KGMM, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553 doi: 10.1002/(SICI)1096-9136(199807)15:7 < 539::AID-DIA668 > 3.0.CO;2-S
Scheen AJ (2015) How I treat… the role of dpp-4 inhibitors (gliptins) in the treatment of type 2 diabetes. Rev Med Liege 70(12):593–599
Deacon CF, Johnsen AH, Holst JJ (1995) Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 80:952–957
Gutniak M, Orskov C, Holst JJ, Ahre’n B, Efendic S (1992) Antidiabetogenic effect of glucagon-like peptide-1 (7–36) amide in normal subjects and patients with diabetes mellitus. N Engl J Med 326(20):1316–1322. https://doi.org/10.1056/NEJM199205143262003
Kieffer TJ, Habener JF (1999) The glucagons-like peptides. Endocr Rev 20(6):876–913. https://doi.org/10.1210/edrv.20.6.0385
Mentlein R (1999) Dipeptidyl-peptidase IV (CD26)—role in the inactivation of regulatory peptides. Regul Pept 85:9–24
Balkan B, Kwasnik L, Miserendino R, Holst JJ, Li X (1999) Inhibition of dipeptidyl peptidase IV with NVP-DPP728 increases plasma GLP-1 (7–36 amide) concentrations and improves oral glucose tolerance in obese Zucker rats. Diabetologia 42(11):1324–1331. https://doi.org/10.1007/s001250051445
Ahre’n B, Landin-Olsson M, Jansson P-A, Svensson M, Holmes D, Schweizer A (2004) Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab 85:2078–2084
Reimer MK, Holst JJ, Ahre’n B (2002) Long-term inhibition of dipeptidyl peptidase IV improves glucose tolerance and preserves islet function in mice. Eur J Endocrinol 146:717–727
Drucker DJ (2003) Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. Expert Opin Investig Drugs 12(1):87–100. https://doi.org/10.1517/13543784.12.1.87
Mest HJ, Mentlein R (2005) Dipeptidyl peptidase inhibitors as new drugs for the treatment of type 2 diabetes. Diabetologia 48(4):616–620. https://doi.org/10.1007/s00125-005-1707-5
Villhauer EB, Brinkman JA, Naderi GB, Burkey BF, Dunning BE, Prasad K, Mangold BL, Russell ME, Hughes TE (2003) 1-[[(3-Hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J Med Chem 46(13):2774–2789. https://doi.org/10.1021/jm030091l
He YL (2012) Clinical pharmacokinetics and pharmacodynamics of vildagliptin. Clin Pharmacokinet 51(3):147–162. https://doi.org/10.2165/11598080-000000000-00000
Galvus, INN- Vildagliptin. Summary of product characteristics. Available via Galvus summary of product characteristics: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000771/WC500020327.pdf Accessed 30 May 2017.
Anti-diabetic drug cartel issue reaches CCI, Health Ministry. The Economic Times (2017). https://economictimes.indiatimes.com/industry/healthcare/biotech/pharmaceuticals/anti-diabetic-drug-cartel-issue-reaches-cci-health-ministry/articleshow/57800084.cms?from=mdr. Accessed 30 May 2017.
Rajagopal D (2017) Pricing of diabetes drug vildagliptin - drug pricing Cartel issue reaches CCI, Health Min. The Economic Times, Mumbai Accessed via Pricing Of Diabetes Drug Vildagliptin: http://epaperbeta.timesofindia.com/Article.aspx?eid=31818&articlexml=PRICING-OF-DIABETES-DRUG-VILDAGLIPTIN-Drug-Pricing-Cartel-24032017014005. Accessed 30 May 2017
Central Drugs Standard Control Organization. Guidelines for Bioavailability & Bioequivalence Studies (2005). Available via Guidelines for Bioavailability & Bioequivalence Studies CDSCO: http://cdsco.nic.in/html/be%20guidelines%20draft%20ver10%20march%2016,%2005.pdf Accessed 30 May 2017.
World Medical Association Declaration of Helsinki-Ethical Principles for Medical Research Involving Human Subject (2000). 52nd WMA General Assembly Edinburgh, Scotland. Available via World Medical Association Declaration of Helsinki-Ethical Principles for Medical Research Involving Human Subject: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ Accessed 30 May 2017
Good Clinical Practices for Clinical Research in India (2013) Law Relatings to Drugs & Cosmetics, Central Drugs Standard Control Organization. Dte.GHS, Ministry of Health and Family Welfare, Government of India Accessed via Good Clinical Practices For Clinical Research In India, CDSCO: http://www.sgpgi.ac.in/sop/GCP-%20Indian.pdf. Accessed 30 May 2017
Pharne AB, Santhakumari B, Ghemud AS, Jain HK, Kulkarni MJ (2012) Bioanalytical method development and validation of vildagliptin a novel dipeptidyl peptidase IV inhibitor by RP-HPLC method. Int J Pharm Pharmaceutic Sci 4(3):119–123
Guidance for Industry (2001) Statistical approaches to establishing bioequivalence. Center for Drug Evaluation and Research (CDER) https://www.fda.gov/media/70958/download. Accessed 30 May 2017
Tjandrawinata RR, Setiawati E, Yunaidi DA, Santoso ID, Setiawati A, Susanto LW (2012) Bioequivalence study of two formulations of bisoprolol fumarate film-coated tablets in healthy subjects. Drug Design Dev Ther. https://doi.org/10.2147/dddt.s36567
Mathieu C, Degrande E (2008) Vildagliptin: a new oral treatment for type 2 diabetes mellitus. Vasc Health Risk Manag 4(6):1349–1360. https://doi.org/10.2147/vhrm.s3005
Nauck MA, Juris JM (2018) Incretin hormones: their role in health and disease. Diabetes Obes Metab 20(Suppl. 1):5–21. https://doi.org/10.1111/dom.13129
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SS provided guidance during this study. MS performed the study and relevant work. MS was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board-DPSRU (ethics committee) registered with the DCGI as ECR/277/Indt/DL/2017 in its meeting. Informed consent form to participate in the study was also reviewed and approved by the ethics committee. The IRB-approved (ethics committee) informed consent to participate was taken from the participants in written form as per schedule Y of the Drug and Cosmetic Act 1945 (India). The informed consent was in English and vernacular language (Hindi). The study was described to each subject before their consent to participate in the study.
Consent for publication
Consent was taken from all participants to publish the research data of this study by concealing their personal identifiers.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, M., Agrawal, S.S. Bioequivalence study of different brands of vildagliptin in healthy human subjects. Futur J Pharm Sci 7, 155 (2021). https://doi.org/10.1186/s43094-021-00308-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00308-1