
Abstract
Background
Lung cancer remains the leading and deadly type of cancer worldwide. It was estimated to account for about 25% of the 7 million people that died as a result of cancer-related issues/mortality every year in the world. Non-small cell lung cancer (NSCLC) is the lethal/deadly class of lung cancer with nearly 1.5 million reported cases and less than 20% survival rate. Therefore, it becomes necessary to explore more effective NSCLC drugs.
Result
A computational approach was employed here to design ten new EGFRWT inhibitors using compound 18 as a template for the design identified with the best binding affinity and good pharmacokinetic properties previously reported in our work. The modeled inhibitory activities of these newly designed EGFRWT inhibitors (range from 7.746966 to 11.09261) were better than that of the hit compound with pIC50 of 7.5639 and gefitinib the positive control with pIC50 of 5.879426. The ligand-binding interaction between these newly designed EGFRWT inhibitors and the EGFR tyrosine kinase receptor as shown in Table 3 was investigated and elucidated using molecular docking protocol. Based on the molecular docking results, the binding affinities of these newly designed EGFRWT inhibitors were found to be between − 8.8 and − 9.5 kcal/mol. The designed compound SFD10 has the highest binding affinity of − 9.5 kcal/mol followed by compound SFD8 (with a binding affinity of − 9.3 kcal/mol), then by compound SFD9 and 4 (each with a binding affinity of − 9.3 kcal/mol). None of them was found to have more than one violation of the filtering criterion used in this study thereby showing good ADMET properties.
Conclusion
The modeled inhibitory activities and binding affinities of these newly designed EGFRWT inhibitors were found to be higher than that of the template compound and the control (gefitinib) used in this research. They were also seen to be non-toxic with good pharmacokinetic properties.
Similar content being viewed by others


Background
Lung cancer is still the leading and deadly type of all cancers worldwide. Lung cancer was estimated to account for about 25% of the 7 million people that died as a result of cancer-related issues/mortality every year in the world [1,2,3]. The lung is classified traditionally into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [4]. NSCLC is the lethal/deadly class of lung cancer with nearly 1.5 million reported cases and less than 20% survival rate [5].
Receptor kinases (RKs) belong to the ErbB family, located on cell membranes of living organisms, and played a significant role in the management of the neoplasms’/malignant tumors’ physiological cycle [6]. EGFR is a member of RKs which was recognized to be the most significant target for the management of neoplasms/malignant tumors, which plays a vital role in the control of cancer cell growth, proliferation, and differentiation [7, 8].
Most of the EGFR inhibitors/NSCLC therapeutic agents share a common structure of quinazoline and acrylamide. Many EGFR inhibitors/NSCLC therapeutic agents have been designed and developed starting from the first, the second, and up to the third generations. The first-generation EGFR inhibitors/NSCLC therapeutic agents were developed to treat patients with EGFR mutation caused by the L858R mutation [9]. The first-generation EGFR inhibitors were reported to have a remarkable therapeutic effect in the early stage of clinical treatment; in the first year of treatment with these drugs, more than 50% of the patients developed resistance to these drugs by the T790M mutation [10]. As such, many second-generation EGFR inhibitors/NSCLC therapeutic agents such as afatinib and docatininb were designed and developed to manage the resistance caused by the T790M mutation. The stated objective was not achieved due to serious side effects caused by the so-called second-generation EGFR inhibitors/NSCLC therapeutic agents such as diarrhea and skin rashes. Many third-generation EGFR inhibitors/NSCLC therapeutic agents such as AZD9291, CO1686, and WZ4002 were designed and synthesized to manage the developed resistance caused by the EGFRT790M/L858R mutations [11].
This work targets the computational design of new EGFRWT inhibitors with improved inhibitory activities, as well as docking investigation and pharmacokinetic property prediction of these new EGFRWT inhibitors.
Method
Compound selection
From our previous study, virtual screening via molecular docking was performed which identified molecule 18 (Fig. 1) with the most promising affinity of − 8.6 kcal/mol toward the receptor and a pIC50 of 7.5639 as the template hit compound [12, 13].

The 2D structure of the selected hit compound
Design, optimum conformation search, and preparations of designed EGFRWT inhibitors
According to the results of molecular docking virtual screening and pharmacokinetic study investigated in our previous research on some EGFRWT inhibitors, molecule 18 with a binding affinity of − 8.6 kcal/mol and good pharmacokinetic properties was retained as a template to be modified for designing new EGFRWT inhibitors. Ten new EGFRWT inhibitors were designed by the addition of halogens, halo substituted phenyl ring, phenyl methanone, and amino rings on the meta and para positions of the phenyl ring on the sulfinyl group of the template compound.
The Chemdraw software developed by Cambridge University was then utilized to draw the two-dimensional (2D) structures of these newly designed EGFRWT inhibitors (Table 1) [14, 15]. The transformation of two-dimensional (2D) to three-dimensional (3D) structures of these newly designed EGFRWT inhibitors was carried out by direct importation of the structures onto the interface of Spartan. The search for the optimum conformation of all the newly designed EGFRWT inhibitors was performed using Merck molecular force field (MMFF) with density functional theory (DFT) at Becke’s three-parameter hybrid function utilizing LYP correlation functional using 6-311G* basis set. Then, the stable conformations of the anilinopyrimidine analogs were then saved in a file format that is going to be used in the next investigation (Protein Data Bank file format) [12, 16].
Retrieval and preparation of EGFR kinase receptor and molecular docking execution
The crystal structure of EGFR kinase domain T790M mutation in complex with AEE788 with pdb entry code 4ZAU was successfully retrieved from the RCSB Protein Data Bank database, prepared and used in this study [17].
Vina of the Pyrex-virtual screening tool was utilize for the docking execution of these newly designed EGFRWT inhibitors with the active site of EGFR kinase receptor [18]. The UCSF chimera software was then used to re-couple the docked newly designed EGFRWT inhibitors and the EGFR kinase receptor and saved in pdb format for the investigation of the respective amino acids the newly designed EGFRWT inhibitors interacted with in the active site of the EGFR kinase receptor. Discovery studio was used for the investigation of the respective amino acids the newly designed EGFRWT inhibitors interacted with in the active site of the EGFR kinase receptor [19].
Pharmacokinetic properties prediction of newly designed EGFRWT inhibitors
SWISSADME (http://www.swissadme.ch/index.php), an online web tool, was utilized in predicting the drug-likeness of these newly designed EGFRWT inhibitors, while the ADMET properties of these newly designed EGFRWT inhibitors under investigation were predicted using the pkCSM, also an online web server (http://structure.bioc.cam.ac.uk/pkcsm) which uses graph-based signatures to generate predictive models of central ADMET properties for drug discovery. Many rules were developed to guide the choice of compounds in the early phases of drug discovery. Among the famous rules applied for the selection of compounds based on the drug-likeness properties in the early phase of drug discovery was Lipinski’s rule of five (RO5). The studied compounds would be evaluated for their drug-likeness properties based on the RO5 criteria [20,21,22].
Results
Activity modeling of newly designed EGFRWT inhibitors
The result of the activity modeling is shown in Table 2.
Molecular docking investigation of newly designed EGFRWT inhibitors
The results of the molecular docking are presented in Table 3 and Figs. 2, 3, 4, and 5.

2D and 3-dimensional structure of the SFD10 receptor

2D and 3-dimensional structure of the SCD8 receptor

2D and 3-dimensional structure of the SCD9 receptor

2D and 3-dimensional structure of the SCD4 receptor
Pharmacokinetic properties of newly designed EGFRWT inhibitors
The results of the pharmacokinetic properties are presented in Tables 4 and 5.
Discussion
Activity modeling of newly designed EGFRWT inhibitors
The predictive power of the model developed in our previous study (Eq. 1) was further confirmed in this study by modeling/predicting the pIC50 of these newly designed EGFRWT inhibitors (Table 2). The model was not just chosen but because of its significance with R2trng = 0.9459, R2adj = 0.9311, Q2cv = 0.8947, R2test = 0.7008, and LOF = 0.1195 [23]. From the table, it can be inferred that the predicted pIC50 of these newly designed EGFRWT inhibitors (range from 7.746966 to 11.09261) were better than that of the hit compound with pIC50 of 7.563837 and gefitinib the positive control used in this study with pIC50 of 5.879426. This further reaffirmed the high predicting performance of the model used in modeling the pIC50 of these newly designed EGFRWT inhibitors.
Molecular docking of newly designed EGFRWT inhibitors
The ligand-binding interaction between these newly designed EGFRWT inhibitors and the EGFR tyrosine kinase receptor as shown in Table 3 was investigated and elucidated using the molecular docking protocol. Based on the molecular docking results, the binding affinities of these newly designed EGFRWT inhibitors were found to be between − 8.8 and − 9.5 kcal/mol. The designed compound SFD10 has the highest binding affinity of − 9.5 kcal/mol followed by compound SFD8 (with a binding affinity of − 9.3 kcal/mol), then by compound SFD9 and 4 (each with a binding affinity of − 9.3 kcal/mol).
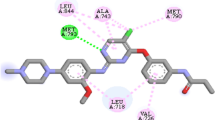
SFD10 (− 9.5 kcal/mol) formed conventional and carbon-hydrogen bonds with THR790 (2.48 Å) and PRO794 (3.31 Å) amino acids backbone of the receptor. Besides hydrogen bonds, it formed halogen with ALA743, ILE744, and LEU788 amino acids, electrostatics bond with LYS745 amino acid, and hydrophobic bond with LYS745, LEU792, LYS728, PRO794, VAL726, and LEU718 amino acid residues of the receptor.
The second best designed compound SFD8 (− 9.3 kcal/mol) formed a conventional hydrogen bond with PRO794 (2.61178 Å) and LYS745 (2.23017 Å) amino acid residues. It also formed a carbon-hydrogen bond with PHE795 (3.35205 Å) and TYR801 (3.6175 Å). On the other hand, it also formed a halogen bond with LYS745 amino acids. In addition to the halogen bond, it formed a hydrophobic bond with PHE795, GLY796, LEU844 (2), LEU718, VAL726, ALA743 (2), LYS745, and HIS805 amino acid residues of the target protein.
Only one conventional carbon-hydrogen bond interaction was observed between the designed compound SFD9 (− 9.2 kcal/mol) and the binding site of the target protein with PRO794 (2.43) amino acid residue. Also, two carbon-hydrogen bond interactions were observed between the designed compound SFD9 and the binding site of the target protein with GLN791 (3.52) and PHE795 (3.42) amino acid residues, respectively. The following amino acid residues PHE795, GLY796, ALA743, MET793, LEU788, LEU718, LEU844, VAL726, and LYS745 were also observed between the designed compound SFD9 and the binding site of the target protein employing hydrophobic interaction.
SFD4 among the designed compounds with higher affinity (− 9.2 kcal/mol) toward the target interacted in the binding site of the target receptor with PRO794 (2.46) and THR790 (2.32) residues employing conventional carbon-hydrogen bond. Two carbon-hydrogen bond interactions were observed between the designed compound SFD4 and the binding site of the target protein with GLN791 (3.33) and MET793 (3.79) amino acid residues, respectively. The binding site of the receptor was seen to interact with SFD4 employing hydrophobic interaction with PHE795, GLY796, MET793, LEU844, MET766, LEU718, LEU844, and VAL726. Not only that, ALA743 and LEU788 amino acid residues were observed to interact with SFD4 employing a halogen bond. LYS745 amino acid in the binding site of the receptor interacted with SFD4 employing electrostatic interaction.
The following amino acid residues, PRO794, LEU718, ALA743, VAL726, and LYS745 are common to the best designed compounds which might be the reason why they have a higher binding affinity. On comparing the designed compounds with the template and the control gefitinib, the designed compounds possessed better binding energy than the template and the control gefitinib. Furthermore, the 3D and 2D structures of the best four discussed designed compounds are shown in Figs. 2, 3, 4 and 5.
Pharmacokinetic properties of newly designed EGFRWT inhibitors
The drug-likeness properties of the designed compounds were also predicted following Lipinski’s rule of five (Table 4). All the newly designed compounds were found to have one violation for Lipinski’s rule of five (WM > 500). The number of hydrogen bond donors and acceptors for all was less than 5 and 10, respectively. The TPSA and the WLOGP values were less than 140 Å and 5, respectively. The synthetic accessibility scores of these newly designed compounds on the scale were in the easy portion (< 5). It means that there is high tendency these newly designed compounds can be easily synthesize in the laboratory. On that basis, the newly designed compounds predicted to be drug-like compounds, orally bioavailable, and active [21, 22].
The predicted ADMET properties of these newly designed compounds are represented in Table 5. The intestinal absorption values for these newly designed compounds were all above 90% but less than 100. Their intestinal absorption values have passed the threshold value of 30%, which clearly shows that these newly designed compounds have high human intestinal absorption properties. The BBB permeability (log BB) values of all newly designed series F compounds were all < − 1, which implies that all these newly designed compounds are poorly distributed through the brain. The CNS permeability (Log PS) values for all were > − 2 which are considered to penetrate the central nervous system. Moreover, they were found to be both substrate and inhibitors of CYP3A4, thereby affirming their metabolic properties. Furthermore, the total clearance for a drug molecule in the body for these newly designed compounds was within the accepted value. All the newly designed compounds were found to be non-toxic. Based on these predicted parameters, the newly designed compounds are said to have high absorption value, low toxicity level, and good permeability across the cell membrane. In general, all these newly designed NSCLC drugs were predicted to have good pharmacokinetic profiles [21, 22].
Conclusion
In the end, the modeled activities of these newly designed EGFRWT inhibitors were seen to be higher than that of the template compound and the control used in this research as well as their binding affinities toward their target (EGFR kinase enzyme). They were seen to possess drug-like properties by non-violating more than 1 of Lipinski’s rule of five, the filtering criterion used in this work, meaning they were predicted to be orally bioavailable. More so, they were also seen to have good ADMET properties, and none of them was found to be toxic. Furthermore, based on their synthetic accessibility, they can be easily synthesized in the laboratory. Therefore, this study recommends that these newly designed EGFRWT inhibitors should be synthesized.
Availability of data and materials
All data and materials are available upon request.
Abbreviations
- ADMET:
-
Absorption, distribution, metabolism, excretion, and toxicity
- BBB:
-
Blood-brain barrier
- B3LYP:
-
Becke’s three-parameter read-Yang-Parr hybrid
- CNS:
-
Central nervous system
- DFT:
-
Density functional theory
- EGFR:
-
Epidermal growth factor receptor
- G.I absorption:
-
Gastrointestinal absorption
- H-Bond:
-
Hydrogen bond
- MW:
-
Molecular weight
- NSCLC:
-
Non-small cell lung cancer agents
- PDB:
-
Protein Data Bank
- RK:
-
Receptor kinase
- RO5:
-
Rule of five
- TPSA:
-
Total polar surface area
References
Chico LK, Van Eldik LJ, Watterson DM (2009) Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov 8(11):892
Khan I, Garikapati KR, Setti A, Shaik AB, Makani VKK, Shareef MA, Rajpurohit H, Vangara N, Pal-Bhadra M, Kamal A (2019) Design, synthesis, in silico pharmacokinetics prediction and biological evaluation of 1, 4-dihydroindeno [1, 2-c] pyrazole chalcone as EGFR/Akt pathway inhibitors. Eur J Med Chem 163:636–648. https://doi.org/10.1016/j.ejmech.2018.12.011
Gschwind A, Fischer OM, Ullrich A (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4(5):361–370. https://doi.org/10.1038/nrc1360
Turker S, Sahinli H, Perkin P, Yazilitas D, Koklu NO, Imamoglu GI, Karacin C, Altinbas M (2018) “Squamos cell lung cancer” case applying with dyspepsia complaints. J Oncol Sci 4(3):147–148
Hizal M, Sendur MA, Bilgin B, Akinci MB, Dede DS, Neselioglu S, Erel O, Yalcin B (2018) J Oncol Sci
Zhao B, Xiao Z, Qi J, Luo R, Lan Z, Zhang Y, Hu X, Tang Q, Zheng P, Xu S (2019) Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur J Med Chem 163:367–380. https://doi.org/10.1016/j.ejmech.2018.11.069
Chan BA, Hughes BG (2015) Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Translat Lung Cancer Res 4(1):36
Zhang Q, Wang Z, Guo J, Liu L, Han X, Li M, Fang S, Bi X, Tang N, Liu Y (2015) Comparison of single-agent chemotherapy and targeted therapy to first-line treatment in patients aged 80 years and older with advanced non-small-cell lung cancer. Onco Targets Ther 8:893
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I (2010) Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med 362(25):2380–2388. https://doi.org/10.1056/NEJMoa0909530
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B (2005) EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med 352(8):786–792. https://doi.org/10.1056/NEJMoa044238
Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462(7276):1070–1074. https://doi.org/10.1038/nature08622
Ibrahim MT, Uzairu A, Shallangwa GA, Uba S (2020) Computer-aided molecular modeling studies of some 2, 3-dihydro-[1, 4] dioxino [2, 3-f] quinazoline derivatives as EGFR WT inhibitors. Beni Suef Univ J Basic Appl Sci 9:1–10
Qin X, Li Z, Yang L, Liu P, Hu L, Zeng C, Pan Z (2016) Discovery of new [1, 4] dioxino [2, 3-f] quinazoline-based inhibitors of EGFR including the T790M/L858R mutant. Bioorg Med Chem 24(13):2871–2881. https://doi.org/10.1016/j.bmc.2016.01.003
Mills N (2006) ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www. cambridgesoft. com. Commercial Price: 1910fordownload, 2150 for CD-ROM; Academic Price: 710fordownload, 800 for CD-ROM. ACS Publications
Ibrahim MT, Uzairu A, Shallangwa GA, Uba S (2020) Structure-based design and activity modeling of novel epidermal growth factor receptor kinase inhibitors; an in silico approach. Sci Afr:e00503
Ghamali M, Chtita S, Hmamouchi R, Adad A, Bouachrine M, Lakhlifi T (2016) The inhibitory activity of aldose reductase of flavonoid compounds: combining DFT and QSAR calculations. J Taibah Univ Sci 10(4):534–542. https://doi.org/10.1016/j.jtusci.2015.09.006
Ibrahim MT, Uzairu A, Uba S, Shallangwa GA (2020) Computational virtual screening and structure-based design of some epidermal growth factor receptor inhibitors. Fut J Pharm Sci 6(1):1–16
Ibrahim MT, Uzairu A, Shallangwa GA, Uba S (2019) QSAR modelling and docking analysis of some thiazole analogues as⍺-glucosidase inhibitors. J Eng Exact Sci 5(3):0257–0270. https://doi.org/10.18540/jcecvl5iss3pp0257-0270
Ibrahim MT, Uzairu A, Uba S, Shallangwa GA (2020) Computational modeling of novel quinazoline derivatives as potent epidermal growth factor receptor inhibitors. Heliyon 6(2):e03289
Hadni H, Elhallaoui M (2020) 3D-QSAR, docking and ADMET properties of aurone analogues as antimalarial agents. Heliyon 6(4):e03580
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):42717. https://doi.org/10.1038/srep42717
Hosen S, Dash R, Khatun M, Akter R, Bhuiyan MHR, Rezaul M, Karim NJM, Ahamed F, Islam KS, Afrin S (2017) In silico ADME/T and 3D QSAR analysis of KDR inhibitors. J Appl Pharm Sci 7(01):120–128
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int J Drug Design Disc 3:511–519
Acknowledgements
The authors acknowledge the technical effort of Ahmadu Bello University, Zaria, Nigeria.
Funding
This research did not receive any funding from anybody.
Author information
Authors and Affiliations
Contributions
MTI contributed throughout the research work. AU gives directives and technical advices. SU also partake in technical activities. GAS also partake in technical activities. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ibrahim, M.T., Uzairu, A., Uba, S. et al. Design of more potent quinazoline derivatives as EGFRWT inhibitors for the treatment of NSCLC: a computational approach. Futur J Pharm Sci 7, 140 (2021). https://doi.org/10.1186/s43094-021-00279-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00279-3