
Abstract
Background
Erdheim-Chester disease (ECD) is a rare histiocytic disorder recently recognized as a neoplasm due to the discovery of activating MAPK pathway mutations. Hepatic involvement by ECD is extremely rare.
Case presentation
We describe a case of a 64-year-old male who presented with pruritis, weight loss, and cholestatic liver function tests. Magnetic resonance imaging of the abdomen showed beaded appearance of the intrahepatic biliary tree. A liver biopsy was suggestive of primary or secondary sclerosing cholangitis. Computerized tomography (CT) of the abdomen showed perinephric and periaortic soft tissue stranding suggestive of ECD. 18F-fluorodeoxyglucose positron emission/computerized tomography scan showed a mediastinal hilar mass which turned out to be follicular lymphoma. Histopathology of molluscum-like skin lesions showed CD68 + , Factor XIIIa + , and CD1a-foamy histiocytes with multiple giant cells suggestive of ECD. The patient developed recurrent episodes of ascending cholangitis and his hyperbilirubinemia continued to worsen despite stenting of a common hepatic duct stricture found on endoscopic retrograde cholangiopancreatography.
Conclusions
The absence of associated inflammatory bowel disease and anti-neutrophil cytoplasmic antibody, as well as the rapidity of disease progression, makes us consider the possibility of hepatic involvement by ECD or an overlap syndrome. We want to highlight that negative histopathology should not delay the diagnosis of ECD as effective and potentially lifesaving therapies with BRAF or MEK pathway inhibitors are now available for these patients.
Similar content being viewed by others


Background
Erdheim-Chester disease (ECD) is a rare histiocytic disorder that has been recently recognized as a neoplasm. Over the last few years, many advances have been made in the understanding of the disease pathology such as the discovery of BRAF-V600E and activating MAPK pathway mutations [1]. This has led to the availability of promising therapeutic tools like BRAF inhibitors which have been outlined expertly in the 2020 consensus recommendations on ECD [2]. Hepatic involvement is extremely rare with only 5 previously reported cases. We present a case of a 64-year-old male who was referred to us for management of presumed primary sclerosing cholangitis however this diagnosis is challenged by several atypical features.
Case presentation
A 64-year-old Caucasian male was referred to our hepatology clinic for liver biopsy suggestive of primary sclerosing cholangitis (PSC). His past medical history included hypertension and hyperlipidemia. He was a non-smoker and drank 1–2 glasses of wine a day. He developed fatigue, pruritis, and a 15-lb weight loss over one year. Liver function tests revealed a bilirubin of 1.8 mg/dL (0.3–1) and alkaline phosphatase (ALP) 1671 IU/L (44–147). Acute and chronic viral hepatitis panels, anti-nuclear antibody (ANA), anti-smooth muscle antibody, anti-mitochondrial antibody, and anti-neutrophil cytoplasmic antibody (ANCA) were negative.
Triple phase magnetic resonance imaging (MRI) of the abdomen revealed subtle multifocal short segment intrahepatic biliary dilatation and multifocal signal abnormality suggestive of a mild fibrotic process in the liver with a normal extrahepatic biliary tree. Liver biopsy showed periportal and focal bridging fibrosis suggestive of primary or secondary sclerosing cholangitis.
A few weeks before his visit, he had developed progressive weakness, fatigue, and emesis and was admitted to his local hospital. He had leukocytosis and was empirically treated for ascending cholangitis with intravenous antibiotics. He was discharged home after his symptoms improved. On follow-up to our liver clinic, he had redeveloped the symptoms a few days after his antibiotics were stopped. He was noted to have leukocytosis again and was readmitted for intravenous antibiotics. CT abdomen revealed heterogenous perinephric soft-tissue stranding and diffuse large vessel stranding which first raised the possibility of ECD. Differentials included IgG4-related disease or lymphoma. IgG subclasses including IgG4 were normal. The absence of significant lymphadenopathy made lymphoma less likely. The presence of a left external iliac pseudoaneurysm raised suspicion of vasculitis. As ANA and ANCA were negative, vasculitis was unlikely per rheumatology.
Bilateral knee X-rays revealed patchy medullary sclerosis of tibial metaphysis which could be seen in ECD but could be nonspecific findings. The patient underwent a head-to-toe 18F-fluorodeoxyglucose positron emission/computerized tomography (18FDG PET/CT) scan to identify a target for biopsy which showed a 2.1 × 2.9-cm highly FDG avid left hilar mass in the chest concerning for malignancy. There were no findings to suggest vasculitis. Endobronchial ultrasound-guided biopsy of the left hilar mass revealed a low-grade follicular lymphoma. Bone marrow biopsy and flow cytometry were negative for lymphoma involvement. Due to the low disease burden and lack of symptoms, the decision was made to manage the lymphoma with surveillance. His symptoms of fatigue and pruritis were attributed to PSC. Of note, no target could be identified on 18FDG PET/CT scan to confirm the diagnosis of ECD on histopathology.
Two months later, he presented with deepening jaundice, bilirubin 10.7 mg/dL, and ALP 1277 IU/L. His creatinine was elevated at 3 mg/dL (0.7–1.3). CT abdomen revealed hepatomegaly and extensive soft tissue encasing the kidneys, renal hilum, ureters, pelvis, aorta, and mesenteric arteries which had progressed since the prior scan (Fig. 1). On endoscopic retrograde cholangiopancreatography (ERCP), a common hepatic duct (CHD) stricture was identified with non-filling of the right hepatic duct. He underwent balloon dilatation of the CHD followed by stent placement. Brush cytology and biopsy from the CHD stricture showed reactive columnar epithelium and no malignant cells. Post-ERCP his bilirubin continued to rise.

Perinephric soft tissue stranding and encasement on CT abdomen pelvis. CT: computer tomography
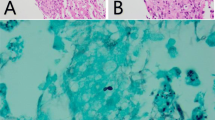
He had now also developed molluscum-like dome-shaped yellow-pink firm papules located on the hands, posterior leg, and neck (Fig. 2). Biopsy of skin lesion showed dermal proliferation of histiocytic cells which were CD68 positive, Factor XIIIa positive, and CD1a negative confirming the diagnosis of ECD; along with lipidized giant cells with glassy pink cytoplasm displaying a weak BRAF signal (Fig. 3).

Molluscum-like dome-shaped papule on right wrist

Dermal proliferation of histiocytic cells (majority of cells) and multiple giant cells (arrows)H&E × 20
Targeted therapy with BRAF or MEK inhibitors versus Interferon-alpha treatment was considered; however, there was concern that he may be too sick to receive these treatments. He and his family eventually decided to pursue comfort care and unfortunately, the patient was discharged to hospice and passed away shortly afterward.
Conclusions
ECD is an exceedingly rare condition with about 800 patients described in literature according to the 2020 ECD consensus guidelines [2]. It is a histiocytic disorder that has recently been recognized as a neoplasm due to the high prevalence of activating MAPK pathway and BRAF-V600E mutations on molecular analysis [1]. The mean age at diagnosis in the USA is 46 years with a 70–75% male preponderance [1].
ECD is a multisystem disease with varied organ involvement. Osseous involvement is most common with symmetric osteosclerosis of long bones seen in more than 95% of patients [1]. Other common findings are perinephric/periaortic soft-tissue stranding and encasement on imaging, atrioventricular tumors, diabetes insipidus, cardiac, and pulmonary involvement which are reported in 50–60% of cases [1]. Cutaneous involvement is seen in 25–30% of patients, most commonly with a xanthelasma-like lesion [3] To our knowledge, molluscum-like lesions such as those seen in our patient have not previously been reported. Associated myeloid neoplasms can be found in 10% of patients [4]. To the best of our knowledge, there are only 5 previously reported cases with hepatic involvement of ECD and there is variation in the nature of hepatic involvement among these as well. Previously reported presentations include liver fibrosis presenting with decompensated cirrhosis [5], xanthogranulomatous lesions of the bile duct masquerading as Klatskin’s tumor [6], hepatomegaly with histiocytic infiltration of the liver [7], or hepatic nodules with histiocytic infiltrates on biopsy [8, 9].
A skull-to-toe 18FDG PET scan including long bones is recommended at diagnosis to assess the extent of the disease and identify a target for biopsy [2]. However, it may not be the best initial diagnostic modality [10] and was unhelpful in reaching the diagnosis of ECD in our case. Technitium-99 m bone scan may be more sensitive for detecting osseous lesions. Brain and Cardiac MRI are recommended in all patients at diagnosis [2].
On histopathology, ECD is characterized by xanthogranulomatous infiltrates with foamy histiocytes that are CD68 + , CD163 + , Factor XIIIa + , and CD1a- and multinucleated Touton giant cells [11]. Tissue is most commonly obtained from osseous, perinephric, or cutaneous lesions. However, the diagnosis should be considered even if histopathology is inconclusive if clinical presentation and imaging are suggestive as histopathology can often be obscured by nonspecific fibrosis [2].
The recent discovery of the MAPK pathway and BRAF-V600E mutations have opened avenues for targeted therapy with good outcomes. BRAF inhibitors such as vemurafenib are recommended as first-line treatment for patients with BRAF-V600E mutations [2, 12]. MEK inhibitors are strongly recommended for BRAF-wild-type ECD cases [2]. Interferon-α was the traditional treatment before the advent of targeted therapy and can be used when targeted treatment is not available, as can systemic chemotherapy such as cladribine [2].
Diagnosis of ECD is challenging due to the rarity of the disease, varied nature of its presentation, and inconsistent results of histopathology. The mean duration from presentation to diagnosis was 4.2 years in one study [1].
Our patient presented with symptoms of pruritis and weight loss. Liver function showed a cholestatic pattern. MRI showed a beaded appearance of the intrahepatic biliary tree and liver biopsy showed periportal and focal bridging fibrosis. These features would suggest a diagnosis of PSC. However, some elements make us doubt this diagnosis. Associated inflammatory bowel disease and positive ANCA which are seen in 60–80% of patients with PSC were both absent in our patient [13]. PSC usually affects both the intrahepatic and extrahepatic biliary tree. Isolated small duct involvement can be seen in 5% of patients and usually runs a more benign course. Patients with small duct PSC can later develop involvement of the extrahepatic ducts [14, 15]. Eventually, this patient did develop a large duct stricture. PSC in general runs a more indolent course with an average survival from time of diagnosis to death or liver transplantation being 10–12 years [16]. Our patient was transitioned to hospice within 6 months. Even though a review of the liver and CHD biopsies did not show evidence of ECD, as discussed above, it is not uncommon for findings of ECD to be obscured by non-specific fibrosis on histopathology. One case of ECD associated with PSC has been previously described. The patient had associated ulcerative colitis and succumbed to a hepatobiliary malignancy 3 years after diagnosis of PSC [17].
Although we cannot rule out PSC completely, the above evidence makes us suspect hepatic involvement by ECD or an overlap syndrome; or that PSC associated with ECD may have a much more aggressive course. We recommend a full skin exam as minor lesions may not be reported due to being overshadowed by other issues. It was the molluscum-like skin lesions, which have not been previously described in ECD, that eventually clinched the diagnosis for our patient. However, we want to stress that negative histopathology should not delay the diagnosis of ECD if imaging is suggestive, especially since effective, and potentially lifesaving therapy is now available.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ECD:
-
Erdheim-Chester disease
- CT:
-
Computerized tomography
- PSC:
-
Primary sclerosing cholangitis
- ALP:
-
Alkaline phosphatase
- ANA:
-
Anti-nuclear antibody
- ANCA:
-
Anti-neutrophil cytoplasmic antibody
- MRI:
-
Magnetic resonance imaging
- 18FDG PET/CT:
-
18F-fluorodeoxyglucose positron emission/computerized tomography
- ERCP:
-
Endoscopic retrograde cholangiopancreatography
- CHD:
-
Common hepatic duct
References
Estrada-Veras JI, O’Brien KJ, Boyd LC, Dave RH, Durham B, Xi L et al (2017) The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 1(6):357–366. https://doi.org/10.1182/bloodadvances.2016001784
Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH et al (2020) Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 135(22):1929–1945. https://doi.org/10.1182/blood.2019003507
Kobic A, Shah KK, Schmitt AR, Goyal G, Go RS, Guo R et al (2020) Erdheim-Chester disease: expanding the spectrum of cutaneous manifestations. Br J Dermatol 182(2):405–409. https://doi.org/10.1111/bjd.18153
Papo M, Diamond EL, Cohen-Aubart F, Emile JF, Roos-Weil D, Gupta N et al (2017) High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood 130(8):1007–1013. https://doi.org/10.1182/blood-2017-01-761718
Balasubramanian G, Modiri A, Affi M, Hagen CE, Batdorf B, Oshima K et al (2017) a fatal case of Erdheim-chester disease with hepatic involvement. ACG Case Rep J. 4:e95. https://doi.org/10.14309/crj.2017.95
Gundling F, Nerlich A, Heitland WU, Schepp W (2007) Biliary manifestation of Erdheim-Chester disease mimicking Klatskin’s carcinoma. Am J Gastroenterol 102(2):452–454. https://doi.org/10.1111/j.1572-0241.2006.00893.x
Pan A, Doyle T, Schlup M, Lubcke R, Schultz M (2011) Unusual manifestation of Erdheim-Chester disease. BMC Gastroenterol 11:77. https://doi.org/10.1186/1471-230X-11-77
Ivan D, Neto A, Lemos L, Gupta A (2003) Erdheim-Chester disease: a unique presentation with liver involvement and vertebral osteolytic lesions. Arch Pathol Lab Med 127(8):e337–e339. https://doi.org/10.5858/2003-127-e337-EDAUPW
Gupta A, Aman K, Al-Babtain M, Al-Wazzan H, Morouf R (2007) Multisystem Erdheim-Chester disease; a unique presentation with liver and axial skeletal involvement. Br J Haematol 138(3):280. https://doi.org/10.1111/j.1365-2141.2007.06642.x
Kirchner J, Hatzoglou V, Buthorn JB, Bossert D, Sigler AM, Reiner AS et al (2021) (18)F-FDG PET/CT versus anatomic imaging for evaluating disease extent and clinical trial eligibility in Erdheim-Chester disease: results from 50 patients in a registry study. Eur J Nucl Med Mol Imaging 48(4):1154–1165. https://doi.org/10.1007/s00259-020-05047-8
Ozkaya N, Rosenblum MK, Durham BH, Pichardo JD, Abdel-Wahab O, Hameed MR et al (2018) The histopathology of Erdheim-Chester disease: a comprehensive review of a molecularly characterized cohort. Mod Pathol 31(4):581–597. https://doi.org/10.1038/modpathol.2017.160
Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I et al (2018) Vemurafenib for BRAF V600-mutant Erdheim-Chester disease and langerhans cell histiocytosis: analysis of data from the histology-independent, phase 2, open-label VE-BASKET Study. JAMA Oncol 4(3):384–388. https://doi.org/10.1001/jamaoncol.2017.5029
Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B et al (2010) Diagnosis and management of primary sclerosing cholangitis. Hepatology 51(2):660–678. https://doi.org/10.1002/hep.23294
Bjornsson E, Olsson R, Bergquist A, Lindgren S, Braden B, Chapman RW et al (2008) The natural history of small-duct primary sclerosing cholangitis. Gastroenterology 134(4):975–980. https://doi.org/10.1053/j.gastro.2008.01.042
Lindor KD, Kowdley KV, Harrison ME, American College of G (2015) ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 110(5):646–59. https://doi.org/10.1038/ajg.2015.112. (quiz 60)
Wiesner RH, Grambsch PM, Dickson ER, Ludwig J, MacCarty RL, Hunter EB et al (1989) Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology 10(4):430–436. https://doi.org/10.1002/hep.1840100406
Spoerl D, Andre R, Bornand A, Seebach JD (2021) Three cases of BRAF mutation negative Erdheim-Chester disease with a challenging distinction from IgG4-related disease. Allergy Asthma Clin Immunol 17(1):6. https://doi.org/10.1186/s13223-020-00505-2
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
RS wrote the manuscript, edited the final version with contributions and suggestions from all other authors, formatted and edited the images. MS provided histopathology slides and contributed to manuscript. DM provided oncological input to the manuscript. AB reviewed liver and common biopsies to ensure ECD was not seen on them. GK critically reviewed, edited, and approved the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient has unfortunately passed away. His wife has provided informed verbal consent over the phone for publication of the details of the case. She does not live in the same city, but should a written informed consent be required after the manuscript has been accepted, we should be able to provide it.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, R., Stone, M.S., Macfarlane, D.E. et al. Erdheim-Chester disease associated with an aggressive form of sclerosing cholangitis. Egypt Liver Journal 13, 4 (2023). https://doi.org/10.1186/s43066-023-00242-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43066-023-00242-2