
Abstract
Background
Non-alcoholic fatty liver disease (NAFLD) is a metabolic disorder characterised by enhanced hepatic fat deposition and inflammation. Efforts to manage NAFLD are limited by the poorly characterised pathological processes and the lack of precise non-invasive markers, thus, proving the need to further study the involved cytokines, which, in turn, may represent novel molecular targets with possible diagnostic and therapeutic applications. Hence, we aimed to assess the diagnostic utility of serum interleukin 32 (IL-32) in NAFLD cases. This case-control study included 40 NAFLD patients and 40 healthy controls. The serum IL-32 concentrations were assessed by the enzyme-linked immunosorbent assay (ELISA).
Results
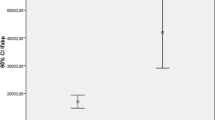
The serum IL-32 concentrations were significantly higher in NAFLD cases than controls (76 [45.5–111.125] vs. 13 [8–15] pg/mL, P < 0.001, respectively). IL-32 at a cut-off point > 22.5 pg/mL had 100% sensitivity, 87.50% specificity, 88.9% positive predictive value, 100% negative predictive value, and 98.2% accuracy in detecting the NAFLD cases.
Conclusion
Serum IL-32 could be considered a novel non-invasive marker for NAFLD. Further investigations are warranted to verify the potential utility of IL-32 in the clinical setting.
Similar content being viewed by others


Background
Non-alcoholic fatty liver disease (NAFLD) is currently a major driver of progressive hepatic disease globally, with a 15–30% prevalence rate. It is considered to be the hepatic component of metabolic syndrome [1, 2]. NAFLD has a broad spectrum of diseases. Simple steatosis is defined as the hepatic accumulation of triglycerides; it accounts for approximately 80% of NAFLD cases and is associated with a good prognosis. Approximately 25% of cases with simple steatosis develop into the more severe stage of non-alcoholic steatohepatitis (NASH), in which liver steatosis coexists with hepatic inflammation that can be accompanied by the ballooning of hepatocytes and liver fibrosis [3, 4]. Eventually, 20% of NASH cases progress to cirrhosis or hepatocellular carcinoma over 20–30 years [5].
The pathogenesis of NAFLD is not yet fully understood. Its existing pathophysiology proposes the multifactorial interaction of various metabolic, genetic and environmental influences, such as the gut microbiota and innate immunity interaction, mitochondrial dysfunction, abnormalities of iron metabolism, and increased fructose consumption [6,7,8,9], with the proliferation, dysfunction, and inflammation of adipose tissue [10, 11].
The infiltration of adipose tissue with immune cells results in altered adipokines and promotes metabolic diseases [10, 12]. Moreover, chronic inflammation and insulin resistance stimulate the release of free fatty acids (FFAs) from the adipose tissue, causing hepatocellular fat deposition. Subsequently, inflammatory adipokines promote the deposition of fibrous tissue, which is the hallmark of the disease progressing to cirrhosis [10, 12]. However, the inflammatory response is also essential for tissue repair during the early phases of hepatic damage [13]. This dual aspect of the inflammatory system may be an interesting target in the management of NAFLD. For example, interleukins such as IL-6, IL-8, IL-12, IL-18, and IL-34 have been shown to play a part in NAFLD disease [14,15,16,17,18].
Among several inflammatory biomarkers linked to obesity and NAFLD, IL-32 is evolving as a main regulator of obesity-driven inflammation and lipotoxicity. IL-32 was initially discovered in 2005 [19]. Formerly, it was recognised as NK4, a transcript produced after activating natural killer (NK) and T cells by IL-2 and mitogens, respectively [20, 21]. The IL-32 gene exists only in humans on chromosome 16p13.3. It has a full length of approximately 1.2 kbp and comprises eight small exons. IL-32 has nine isoforms which have different and sometimes opposing functions [22, 23].
There is still controversy regarding IL-32 localisation in and secretion from cells [22]. It is found mainly intracellularly [24]. It has been observed that IL-32 is localised in the cytoplasm and nucleus of Leishmania-infected macrophages, the endoplasmic reticulum of endothelial cells [24, 25], the Golgi compartment [26], and in the mitochondria of breast cancer cells [27]. IL-32 has also been found to colocalise with lysosomes [28]. IL-32 has also been observed in the supernatant of activated cells, suggesting that IL-32 is secreted or released from cells [29]. Owing to its localisation in the Golgi compartment, IL-32 may also be secreted across the endoplasmic reticulum-Golgi pathway [26]. However, the secretory path of IL-32 has not been defined in detail nor has a cognate receptor of IL-32 been discovered; the most closely related receptor is the proteinase 3-proteinase activated receptor 2 axis [30]. Consequently, thorough research should be performed to identify the localisation and secretion of IL-32.
IL-32 is expressed in several human tissues, but it is more prominent in immune cells (such as NK cells, macrophages, monocytes, and T cells) than in non-immune cells (such as epithelial cells, endothelial cells, mesenchymal stromal cells, fibroblasts, and hepatocytes) [21, 29, 31]. IL-32 expression is also seen in various cancer cell lines [21, 23, 26, 32]. Although IL-32 can be expressed in normal circumstances, IL-32 expression is robustly stimulated in reaction to the proinflammatory cytokines [28, 33], infections [28, 34], oxidative stress responses [35], and various disorders such as type 2 diabetes [36], allergic rhinitis [37], chronic obstructive pulmonary disease [38], atopic dermatitis [25], systemic lupus erythematosus [39], and rheumatoid arthritis [40]. Regarding hepatic diseases, IL-32 is upregulated in hepatitis C and B viral infections and NAFLD and is also associated with the severity of hepatic disease [41,42,43,44,45].
IL-32 exhibits several biological activities including monocyte differentiation and stimulation of pro/anti-inflammatory cytokines as tumour necrosis factor-α (TNFα) and the activation of nuclear factor kappa (NF-κB) signalling pathways [19, 22]. Conversely, TNFα and interferon γ (IFNγ) have been found to stimulate IL-32 secretion in various pathological conditions [46, 47]. Notably, the mutual positive transcription between IL-32 and TNFα triggered a low-grade inflammation in lipid-storing organs through the enhanced expression of proinflammatory cytokines such as IL-1b, IL-6, and IL-10, whereas IL-32 silencing yielded opposite effects [48]. However, other reports have shown IL-32 to have the opposite function by stimulating anti-inflammatory cytokines such as IL-10 and immunosuppressive molecules such as Indoleamine 2, 3-dioxygenase in immune cells [49].
The function of IL-32 in metabolic conditions is unclear. It may be initially upregulated as a protective response against the accumulation of FFAs. Additionally, the overexpression of IL-32 in HepG2 cells resulted in reduced intracellular fat accumulation, whereas shRNA knockdown of IL-32 enhanced fat accumulation. However, over time, the proinflammatory impacts of IL-32 and its influences on insulin resistance could play an essential part in the pathogenesis of metabolic syndrome [45, 50, 51].
A variety of mechanisms have been proposed to ascertain the role of IL-32 in lipid metabolism. IL-32 promoters have binding locations for fatty acid-responsive transcription factors, which are upregulated during lipotoxicity [44]. IL-32 has been linked to endothelial inflammation in response to the postprandial increase in FFAs [52]. In addition, it has a role in regulating the intracellular content of lipids by modulating cholesterol efflux [53, 54]. Moreover, a positive association between IL-32 mRNA and the lipid regulatory receptor liver X receptor α (LXRα), the lipid transporters ATP-binding cassette subfamily A member 1 (ABCA1), ATP-binding cassette subfamily G member 1 (ABCG1), and the fatty acid carrier ApoA1 in primary liver cells has been detected [31]. Hence, we aimed to estimate the serum IL-32 concentrations in NAFLD cases and assess its diagnostic performance for the detection of NAFLD.
Methods
This case-control study included 40 NAFLD cases and 40 healthy controls, who were recruited from the Internal Medicine and Hepatology outpatient clinics at Ain Shams University Hospitals, Cairo, Egypt, from March 2020 to March 2021. Exclusion criteria were viral hepatitis, autoimmune liver diseases, metabolic liver diseases (hemochromatosis, Wilson’s disease, alpha-1-antitrypsin deficiency, and cystic fibrosis), alcoholic liver disease (quantity of ethanol consumption > 20 g/day for females and > 30 g/day for males), concurrent infections, and concomitant conditions causing secondary steatohepatitis, such as endocrine disorders, primary dyslipidaemia, or malnutrition.
All the participants underwent a detailed medical history, physical examination, laboratory investigations, and ultrasonography [55]. Ultrasonography was performed for the diagnosis of NAFLD [55]. The indices of the fatty liver index (FLI) [56] and NAFLD fibrosis score were calculated as reported previously [57]. According to Bedogni et al., the FLI ranges from 0 to 100; NAFLD is ruled out by an FLI <30 and confirmed with an FLI ≥60 [56]. According to Angulo P et al., NAFLD score < − 1.455 = F0–F2, NAFLD score − 1.455–0.675 = indeterminate score, NAFLD score > 0.675 = F3–F4 [57].
Serum IL-32 was measured by the human IL-32 enzyme-linked immunosorbent assay (ELISA) kit (MyBioSource, CA, USA, Cat. No: MBS029816). The detection range = 12.5–400 pg/mL, sensitivity = 2.0 pg/mL, and intra- and inter-assay precision were less than 15%.
The study protocol followed the Declaration of Helsinki ethical guidelines. The study was certified by the Ethical Committee of the Faculty of Medicine, Ain Shams University (FWA 000017585). An informed written approval was gained from all the participants prior to their inclusion in the research.
Statistical analysis
All the results were analysed using SPSS Statistics for Windows, v. 25.0 (Armonk, NY: IBM Corp.). Variables were shown as frequency and percentages for categorical variables, mean and standard deviation for parametric numerical variables, and median (interquartile range) for non-parametric numerical variables. The Student t-test, Mann-Whitney test, chi-square test, and Fisher’s exact test were used when appropriate. Correlation analyses between serum IL-32 levels and other variables were performed using Spearman’s rho. A receiver-operating characteristics (ROC) curve analysis was applied to evaluate the diagnostic accuracy of IL-32 for the differentiation of NAFLD patients. A two-tailed P < 0.05 was considered significant.
Results
This study included 40 NAFLD cases with a mean age of 49.40 ± 12.01 years, 67.50% of whom were females, and 40 healthy controls with a mean age of 45.66 ± 10.33 years, 47.50% of whom were females (Table 1).
Serum IL-32 concentrations were significantly higher in the NAFLD cases than controls (76 [45.5–111.125] vs. 13 [8–15] pg/mL, P < 0.001, respectively; Table 1 and Fig. 1). Serum IL-32 levels did not differ between NAFLD patients with regard to gender, smoking, co-existing hypertension, FLI scores, or NAFLD fibrosis scores (P ≥ 0.05; Table 2). Serum IL-32 levels correlated only with albumin levels (P = 0.029; Table 3). Fatty liver index and NAFLD fibrosis score of all participants are shown in Tables 4 and 5. IL-32 at a cut-off point > 22.5 pg/mL had 100% sensitivity, 87.50% specificity, 88.9% positive predictive value (PPV), 100% negative predictive value (NPV), and 98.2% accuracy for the differentiation of NAFLD cases (Table 6, Fig. 2).

Serum interleukin 32 levels in NAFLD cases and controls

ROC curve analysis for the diagnostic performance of IL-32 for NAFLD detection
There were statistically significant differences between the NAFLD cases and controls with regard to co-existing hypertension, weight, body mass index, waist circumference, aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin, triglycerides, cholesterol, high-density lipoprotein, low-density lipoprotein, FLI scores, and NAFLD fibrosis scores (P < 0.001; Table 1).
Discussion
At present, liver biopsy remains the favoured method for evaluating the degree of hepatic necro-inflammation and fibrosis in NAFLD cases. However, this invasive procedure has multiple drawbacks including the high cost, the limited representation of the total liver mass, intra- and interobserver inconsistency, and post-procedural complications [58]. These drawbacks prevent the liver biopsy from being a tool for successive disease evaluation in clinical practice. Alternatively, numerous non-invasive laboratory and imaging methods have been proposed as more appropriate for first-line investigations [59,60,61]. However, the present non-invasive methods lack sufficient sensitivity and specificity. Therefore, innovative strategies are being considered for biomarker discovery and therapeutic target identification, focussing not only on the extent of hepatic fibrosis but also on hepatic inflammation, which represents one other critical part of hepatic disease pathology [62, 63].
According to the current literature, there is an association between IL-32 and obesity-linked inflammation [44, 46]. Furthermore, IL-32 expression increased in visceral adipocytes subjected to inflammatory stimuli as hypoxia, lipopolysaccharide, and TNF-α. In addition, IL-32 triggered the expression of inflammatory biomarkers in these cells [46]. Serum IL-32 concentrations were also increased in type 2 diabetes and correlated with body mass and fasting blood glucose [36] and decreased after bariatric surgery [48]. Therefore, further investigations are essential to determine whether IL-32 is a possible biomarker for NAFLD.
In the current study, we observed significantly higher serum IL-32 concentrations in the NAFLD cases than in the controls. Moreover, serum IL-32 had a reliable diagnostic accuracy in differentiating NAFLD cases. Similarly, an earlier study showed that serum IL32 concentrations were higher in NAFLD cases compared to controls (P < 0.01) [44]. Furthermore, higher serum IL-32 levels were observed in patients with severe NAFLD than those without severe disease (P < 0.01) [44]. In the same study, the inclusion of IL-32 in the ALT-AST model caused a 24% increase in AUC for the differentiation of NAFLD (AUC = 0.92 vs. 0.81) [44].
In accordance with the current findings, Dali-Youcef et al. reported that IL-32 protein levels were increased in hepatic samples of NAFLD cases. Additionally, compared to controls, IL-32 expression was raised 2.5-fold in the NAFLD cases (P < 0.001), while a statistically non-significant increase in IL-32 expression was detected in obese patients with normal livers (1.6-fold). Dali-Youcef et al. also noticed a positive association between IL-32 and waist circumference, body mass index, aminotransferases, and NAFLD score, suggesting that this gene contributes to liver steatosis and metabolic syndrome [45].
The current study is limited by the small sample size and the lack of a paired histological evaluation of NAFLD due to the invasiveness of liver biopsy. Finally, IL-32 has emerged as an adipokine with a substantial influence on a range of human diseases including NAFLD; this influence suggests IL-32 and its signalling pathway could be a potential therapeutic target [45]. Further large-scale studies should be performed to determine the molecular function of IL-32 and recognition of IL-32 interacting molecules will help achieve this goal [64].
Conclusions
Serum IL-32 could be considered a novel non-invasive marker for NAFLD. Further investigations are warranted to verify the potential utility of IL-32 in the clinical setting.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABCA1:
-
ATP-binding cassette subfamily A member 1
- ABCG1:
-
ATP-binding cassette subfamily G member 1
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- AUC:
-
Area under the curve
- ELISA:
-
Enzyme-linked immunosorbent assay
- FFAs:
-
Free fatty acids
- IFNγ:
-
Interferon γ
- IL:
-
Interleukin
- LXRα:
-
Lipid regulatory receptor liver X receptor α
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- NK cells:
-
Natural killer cells
- NPP:
-
Negative predictive value
- NF-κB:
-
Nuclear factor kappa B
- PPV:
-
Positive predictive value
- ROC:
-
Receiver-operating characteristics curve
- TNF-α:
-
Tumour necrosis factor-α
References
Eslam M, Sanyal AJ, George J (2020) International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 158(7):1999–2014.e1. https://doi.org/10.1053/j.gastro.2019.11.312 PMID: 32044314
Valenti L, Pelusi S (2020) Redefining fatty liver disease classification in 2020. Liver Int. 40(5):1016–1017. https://doi.org/10.1111/liv.14430 PMID: 32352234
Romeo S, Sanyal A, Valenti L (2020) Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab. 31(1):35–45. https://doi.org/10.1016/j.cmet.2019.12.002 PMID: 31914377
Pelusi S, Cespiati A, Rametta R, Pennisi G, Mannisto V, Rosso C, Baselli G, Dongiovanni P, Fracanzani AL, Badiali S, Maggioni M, Craxi A, Fargion S, Prati D, Nobili V, Bugianesi E, Romeo S, Pihlajamaki J, Petta S, Valenti L (2019) Prevalence and risk factors of significant fibrosis in patients with nonalcoholic fatty liver without steatohepatitis. Clin Gastroenterol Hepatol. 17(11):2310–2319.e6. https://doi.org/10.1016/j.cgh.2019.01.027 PMID: 30708111
Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, Colombo M, Craxi A, Crespo J, Day CP, Eguchi Y, Geier A, Kondili LA, Kroy DC, Lazarus JV, Loomba R, Manns MP, Marchesini G, Nakajima A, Negro F, Petta S, Ratziu V, Romero-Gomez M, Sanyal A, Schattenberg JM, Tacke F, Tanaka J, Trautwein C, Wei L, Zeuzem S, Razavi H (2018) Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 69(4):896–904. https://doi.org/10.1016/j.jhep.2018.05.036 PMID: 29886156
Banini BA, Kumar DP, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK, Wang L, Guan HP, Oseini AM, Alonso C, Bedossa P, Koduru SV, Min HK, Sanyal AJ (2021) Identification of a metabolic, transcriptomic, and molecular signature of patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatology. 73(4):1290–1306. https://doi.org/10.1002/hep.31609 PMID: 33131062; PMCID: PMC8046714
Lang S, Demir M, Martin A, Jiang L, Zhang X, Duan Y, Gao B, Wisplinghoff H, Kasper P, Roderburg C, Tacke F, Steffen HM, Goeser T, Abraldes JG, Tu XM, Loomba R, Stärkel P, Pride D, Fouts DE, Schnabl B (2020) Intestinal virome signature associated with severity of nonalcoholic fatty liver disease. Gastroenterology. 159(5):1839–1852. https://doi.org/10.1053/j.gastro.2020.07.005 PMID: 32652145; PMCID: PMC8404510.
Lang S, Schnabl B (2020) Microbiota and fatty liver disease-the known, the unknown, and the future. Cell Host Microbe 28(2):233–244. https://doi.org/10.1016/j.chom.2020.07.007 PMID: 32791115; PMCID: PMC7467841
Trépo E, Valenti L (2020) Update on NAFLD genetics: from new variants to the clinic. J Hepatol. 72(6):1196–1209. https://doi.org/10.1016/j.jhep.2020.02.020 PMID: 32145256
Cordeiro A, Costa R, Andrade N, Silva C, Canabrava N, Pena MJ, Rodrigues I, Andrade S, Ramalho A (2020) Does adipose tissue inflammation drive the development of non-alcoholic fatty liver disease in obesity? Clin Res Hepatol Gastroenterol. 44(4):394–402. https://doi.org/10.1016/j.clinre.2019.10.001 PMID: 32044284
Gastaldelli A, Cusi K (2019) From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 1(4):312–328. https://doi.org/10.1016/j.jhepr.2019.07.002 PMID: 32039382; PMCID: PMC7001557
Lefere S, Tacke F (2019) Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep. 1(1):30–43. https://doi.org/10.1016/j.jhepr.2019.02.004 PMID: 32149275; PMCID: PMC7052781
Hossain M, Kubes P (2019) Innate immune cells orchestrate the repair of sterile injury in the liver and beyond. Eur J Immunol. 49(6):831–841. https://doi.org/10.1002/eji.201847485 PMID: 31001813
Fricker ZP, Pedley A, Massaro JM, Vasan RS, Hoffmann U, Benjamin EJ, Long MT (2019) Liver fat is associated with markers of inflammation and oxidative stress in analysis of data from the framingham heart study. Clin Gastroenterol Hepatol. 17(6):1157–1164.e4. https://doi.org/10.1016/j.cgh.2018.11.037 PMID: 30476583; PMCID: PMC6475462
Auguet T, Bertran L, Binetti J, Aguilar C, Martínez S, Sabench F, Lopez-Dupla JM, Porras JA, Riesco D, Del Castillo D, Richart C (2020) Relationship between IL-8 circulating levels and TLR2 hepatic expression in women with morbid obesity and nonalcoholic steatohepatitis. Int J Mol Sci. 21(11):4189. https://doi.org/10.3390/ijms21114189 PMID: 32545403; PMCID: PMC7312372
Darmadi D, Ruslie RH (2021) Association between serum interleukin (IL)-12 level and severity of non-alcoholic fatty liver disease (NAFLD). Rom J Intern Med. 59(1):66–72. https://doi.org/10.2478/rjim-2020-0029 PMID: 33055315
Flisiak-Jackiewicz M, Bobrus-Chociej A, Tarasów E, Wojtkowska M, Białokoz-Kalinowska I, Lebensztejn DM (2018) Predictive role of interleukin-18 in liver steatosis in obese children. Can J Gastroenterol Hepatol. 2018:3870454. https://doi.org/10.1155/2018/3870454 PMID: 29854715; PMCID: PMC5944203
Shoji H, Yoshio S, Mano Y, Kumagai E, Sugiyama M, Korenaga M, Arai T, Itokawa N, Atsukawa M, Aikata H, Hyogo H, Chayama K, Ohashi T, Ito K, Yoneda M, Nozaki Y, Kawaguchi T, Torimura T, Abe M, Hiasa Y, Fukai M, Kamiyama T, Taketomi A, Mizokami M, Kanto T (2016) Interleukin-34 as a fibroblast-derived marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. 6:28814. https://doi.org/10.1038/srep28814 PMID: 27363523; PMCID: PMC4929441
Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA (2005) Interleukin-32: a cytokine and inducer of TNF alpha. Immunity. 22(1):131–142. https://doi.org/10.1016/j.immuni.2004.12.003 PMID: 15664165
Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis-Pistofidis R, Cai S, Reidy M, Rahmat M, Flaifel A, Ferland B, Su NK, Agius MP, Park J, Manier S, Bustoros M, Huynh D, Capelletti M, Berrios B, Liu CJ, He MX, Braggio E, Fonseca R, Maruvka Y, Guerriero JL, Goldman M, van Allen E, McCarroll SA, Azzi J, Getz G, Ghobrial IM (2020) Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer. 1(5):493–506. https://doi.org/10.1038/s43018-020-0053-3 PMID: 33409501; PMCID: PMC7785110
Kallionpää H, Somani J, Tuomela S, Ullah U, de Albuquerque R, Lönnberg T, Komsi E, Siljander H, Honkanen J, Härkönen T, Peet A, Tillmann V, Chandra V, Anagandula MK, Frisk G, Otonkoski T, Rasool O, Lund R, Lähdesmäki H, Knip M, Lahesmaa R (2019) Early detection of peripheral blood cell signature in children developing β-cell autoimmunity at a young age. Diabetes. 68(10):2024–2034. https://doi.org/10.2337/db19-0287 PMID: 31311800
Hong JT, Son DJ, Lee CK, Yoon DY, Lee DH, Park MH (2017) Interleukin 32, inflammation and cancer. Pharmacol Ther. 174:127–137. https://doi.org/10.1016/j.pharmthera.2017.02.025 PMID: 28223235
Sloot YJE, Smit JW, Joosten LAB, Netea-Maier RT (2018) Insights into the role of IL-32 in cancer. Semin Immunol. 38:24–32. https://doi.org/10.1016/j.smim.2018.03.004 PMID: 29747940
Kobayashi H, Lin PC (2009) Molecular characterization of IL-32 in human endothelial cells. Cytokine. 46(3):351–358. https://doi.org/10.1016/j.cyto.2009.03.007 PMID: 19364659; PMCID: PMC2704890
Meyer N, Zimmermann M, Bürgler S, Bassin C, Woehrl S, Moritz K, Rhyner C, Indermitte P, Schmid-Grendelmeier P, Akdis M, Menz G, Akdis CA (2010) IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol. 125(4):858–865.e10. https://doi.org/10.1016/j.jaci.2010.01.016 PMID: 20227751
Zahoor M, Westhrin M, Aass KR, Moen SH, Misund K, Psonka-Antonczyk KM, Giliberto M, Buene G, Sundan A, Waage A, Sponaas AM, Standal T (2017) Hypoxia promotes IL-32 expression in myeloma cells, and high expression is associated with poor survival and bone loss. Blood Adv. 1(27):2656–2666. https://doi.org/10.1182/bloodadvances.2017010801 PMID: 29296919; PMCID: PMC5745138
Park JS, Lee S, Jeong AL, Han S, Ka HI, Lim JS, Lee MS, Yoon DY, Lee JH, Yang Y (2015) Hypoxia-induced IL-32β increases glycolysis in breast cancer cells. Cancer Lett. 356(2 Pt B):800–808. https://doi.org/10.1016/j.canlet.2014.10.030 PMID: 25449783
Dos Santos JC, Heinhuis B, Gomes RS, Damen MS, Real F, Mortara RA, Keating ST, Dinarello CA, Joosten LA, Ribeiro-Dias F (2017) Cytokines and microbicidal molecules regulated by IL-32 in THP-1-derived human macrophages infected with New World Leishmania species. PLoS Negl Trop Dis. 11(2):e0005413. https://doi.org/10.1371/journal.pntd.0005413 PMID: 28241012; PMCID: PMC5344527
Wen S, Hou Y, Fu L, Xi L, Yang D, Zhao M, Qin Y, Sun K, Teng Y, Liu M (2019) Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 442:320–332. https://doi.org/10.1016/j.canlet.2018.10.015 PMID: 30391782
Nakayama M, Niki Y, Kawasaki T, Takeda Y, Ikegami H, Toyama Y, Miyamoto T (2013) IL-32-PAR2 axis is an innate immunity sensor providing alternative signaling for LPS-TRIF axis. Sci Rep. 3:2960. https://doi.org/10.1038/srep02960 PMID: 24129891; PMCID: PMC3797434
Damen MSMA, Dos Santos JC, Hermsen R, Adam van der Vliet J, Netea MG, Riksen NP, Dinarello CA, Joosten LAB, Heinhuis B (2018) Interleukin-32 upregulates the expression of ABCA1 and ABCG1 resulting in reduced intracellular lipid concentrations in primary human hepatocytes. Atherosclerosis. 271:193–202. https://doi.org/10.1016/j.atherosclerosis.2018.02.027 PMID: 29524862
Paz H, Tsoi J, Kalbasi A, Grasso CS, McBride WH, Schaue D, Butterfield LH, Maurer DM, Ribas A, Graeber TG, Economou JS (2019) Interleukin 32 expression in human melanoma. J Transl Med. 17(1):113. https://doi.org/10.1186/s12967-019-1862-y PMID: 30953519; PMCID: PMC6449995
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, Spinelli L, Cervera-Marzal I, Ebbo M, Girard-Madoux M, Jaeger S, Bollon E, Hamed S, Hardwigsen J, Ugolini S, Vély F, Narni-Mancinelli E, Vivier E (2018) High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 49(5):971–986.e5. https://doi.org/10.1016/j.immuni.2018.09.009 PMID: 30413361; PMCID: PMC6269138
Ribeiro-Dias F, Saar Gomes R, de Lima Silva LL, Dos Santos JC, Joosten LA (2017) Interleukin 32: a novel player in the control of infectious diseases. J Leukoc Biol. 101(1):39–52. https://doi.org/10.1189/jlb.4RU0416-175RR PMID: 27793959
Masson N, Keeley TP, Giuntoli B, White MD, Puerta ML, Perata P, Hopkinson RJ, Flashman E, Licausi F, Ratcliffe PJ (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science. 365(6448):65–69. https://doi.org/10.1126/science.aaw0112 PMID: 31273118; PMCID: PMC6715447
Fadaei R, Bagheri N, Heidarian E, Nouri A, Hesari Z, Moradi N, Ahmadi A, Ahmadi R (2020) Serum levels of IL-32 in patients with type 2 diabetes mellitus and its relationship with TNF-α and IL-6. Cytokine. 125:154832. https://doi.org/10.1016/j.cyto.2019.154832 PMID: 31479874
Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS, Kim HM (2011) IL-32 up-regulation is associated with inflammatory cytokine production in allergic rhinitis. J Pathol. 224(4):553–563. https://doi.org/10.1002/path.2899 PMID: 21598250
Gasiuniene E, Lavinskiene S, Sakalauskas R, Sitkauskiene B (2016) Levels of IL-32 in serum, induced sputum supernatant, and bronchial lavage fluid of patients with chronic obstructive pulmonary disease. COPD. 13(5):569–575. https://doi.org/10.3109/15412555.2016.1145201 PMID: 27018873
Kwon OC, Ghang B, Lee EJ, Hong S, Lee CK, Yoo B, Kim S, Kim YG (2019) Interleukin-32γ: Possible association with the activity and development of nephritis in patients with systemic lupus erythematosus. Int J Rheum Dis. 22(7):1305–1311. https://doi.org/10.1111/1756-185X.13550 PMID: 30941928
Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB (2006) IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 103(9):3298–3303. https://doi.org/10.1073/pnas.0511233103 PMID: 16492735; PMCID: PMC1413916
Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, Barrie HD, Powell EE, Kim SH, Dinarello CA, Bartenschlager R, Jonsson JR, Tilg H (2011) Interleukin-32: a new proinflammatory cytokine involved in hepatitis C virus-related liver inflammation and fibrosis. Hepatology. 53(6):1819–1829. https://doi.org/10.1002/hep.24285 PMID: 21381070
Kim DH, Park ES, Lee AR, Park S, Park YK, Ahn SH, Kang HS, Won JH, Ha YN, Jae B, Kim DS, Chung WC, Song MJ, Kim KH, Park SH, Kim SH, Kim KH (2018) Intracellular interleukin-32γ mediates antiviral activity of cytokines against hepatitis B virus. Nat Commun. 9(1):3284. https://doi.org/10.1038/s41467-018-05782-5 PMID: 30115930; PMCID: PMC6095909
Liu B, Ma X, Wang Q, Luo S, Zhang L, Wang W, Fu Y, Allain JP, Li C, Li T (2020) Marmoset viral hepatic inflammation induced by hepatitis c virus core protein via IL-32. Front Cell Infect Microbiol. 10:135. https://doi.org/10.3389/fcimb.2020.00135 PMID: 32373543; PMCID: PMC7186372
Baselli GA, Dongiovanni P, Rametta R, Meroni M, Pelusi S, Maggioni M, Badiali S, Pingitore P, Maurotti S, Montalcini T, Taliento AE, Prati D, Rossi G, Fracanzani AL, Mancina RM, Romeo S, Valenti L (2020) Liver transcriptomics highlights interleukin-32 as novel NAFLD-related cytokine and candidate biomarker. Gut. 69(10):1855–1866. https://doi.org/10.1136/gutjnl-2019-319226 PMID: 32001554; PMCID: PMC7497582
Dali-Youcef N, Vix M, Costantino F, El-Saghire H, Lhermitte B, Callari C, D'Agostino J, Perretta S, Paveliu S, Gualtierotti M, Dumeny E, Oudot MA, Jaulin A, Dembélé D, Zeisel MB, Tomasetto C, Baumert TF, Doffoël M (2019) Interleukin-32 contributes to human nonalcoholic fatty liver disease and insulin resistance. Hepatol Commun. 3(9):1205–1220. https://doi.org/10.1002/hep4.1396 PMID: 31497742; PMCID: PMC6719754
Suga H, Sugaya M, Miyagaki T, Kawaguchi M, Fujita H, Asano Y, Tada Y, Kadono T, Sato S (2014) The role of IL-32 in cutaneous T-cell lymphoma. J Invest Dermatol. 134(5):1428–1435. https://doi.org/10.1038/jid.2013.488 PMID: 24226419
Heinhuis B, Koenders MI, van Riel PL, van de Loo FA, Dinarello CA, Netea MG, van den Berg WB, Joosten LA (2011) Tumour necrosis factor alpha-driven IL-32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann Rheum Dis. 70(4):660–667. https://doi.org/10.1136/ard.2010.139196 Epub 2010 Dec 27. PMID: 21187297
Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Valentí V, Moncada R, Landecho MF, Silva C, Salvador J, Frühbeck G (2016) Increased interleukin-32 levels in obesity promote adipose tissue inflammation and extracellular matrix remodeling: effect of weight loss. Diabetes. 65(12):3636–3648. https://doi.org/10.2337/db16-0287 PMID: 27630206
Smith AJ, Toledo CM, Wietgrefe SW, Duan L, Schacker TW, Reilly CS, Haase AT (2011) The immunosuppressive role of IL-32 in lymphatic tissue during HIV-1 infection. J Immunol. 186(11):6576–6584. https://doi.org/10.4049/jimmunol.1100277 PMID: 21525393; PMCID: PMC3098930
Damen MSMA, Ballak D, Sapinsley Z, Bai X, Chan ED, Seals DR, Popa CD, Joosten LAB (2020) Transgenic mice expressing human IL-32 develop adipokine profiles resembling those of obesity-induced metabolic changes. Cytokine. 125:154793. https://doi.org/10.1016/j.cyto.2019.154793 PMID: 31398626
Lee DH, Hong JE, Yun HM, Hwang CJ, Park JH, Han SB, Yoon DY, Song MJ, Hong JT (2015) Interleukin-32β ameliorates metabolic disorder and liver damage in mice fed high-fat diet. Obesity (Silver Spring). 23(3):615–622. https://doi.org/10.1002/oby.21001 PMID: 25645248
Gorzelak-Pabiś P, Wozniak E, Wojdan K, Chalubinski M, Broncel M (2020) Single triglyceride-rich meal destabilizes barrier functions and initiates inflammatory processes of endothelial cells. J Interferon Cytokine Res. 40(1):43–53. https://doi.org/10.1089/jir.2018.0173 PMID: 31460824
Damen MS, Agca R, Holewijn S, de Graaf J, Dos Santos JC, van Riel PL, Fransen J, Coenen MJ, Nurmohamed MT, Netea MG, Dinarello CA, Joosten LA, Heinhuis B, Popa CD (2017) IL-32 promoter SNP rs4786370 predisposes to modified lipoprotein profiles in patients with rheumatoid arthritis. Sci Rep. 30(7):41629. https://doi.org/10.1038/srep41629 PMID: 28134327; PMCID: PMC5278556
Xu Z, Dong A, Feng Z, Li J (2017) Interleukin-32 promotes lipid accumulation through inhibition of cholesterol efflux. Exp Ther Med. 14(2):947–952. https://doi.org/10.3892/etm.2017.4596 PMID: 28781617; PMCID: PMC5526182
Sanyal AJ (2002) American Gastroenterological Association. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 123(5):1705–1725. https://doi.org/10.1053/gast.2002.36572 PMID: 12404245
Bedogni G, Bellentani S, Miglioli L, Masutti F, Passalacqua M, Castiglione A, Tiribelli C (2006) The fatty liver index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2(6):33. https://doi.org/10.1186/1471-230X-6-33 PMID: 17081293; PMCID: PMC1636651
Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, Saksena S, Burt AD, Bida JP, Lindor K, Sanderson SO, Lenzi M, Adams LA, Kench J, Therneau TM, Day CP (2007) The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 45(4):846–854. https://doi.org/10.1002/hep.21496 PMID: 17393509
Davison BA, Harrison SA, Cotter G, Alkhouri N, Sanyal A, Edwards C, Colca JR, Iwashita J, Koch GG, Dittrich HC (2020) Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. J Hepatol. 73(6):1322–1332. https://doi.org/10.1016/j.jhep.2020.06.025 PMID: 32610115
Piazzolla VA, Mangia A (2020) Noninvasive diagnosis of NAFLD and NASH. Cells. 9(4):1005. https://doi.org/10.3390/cells9041005 PMID: 32316690; PMCID: PMC7226476
Saleh SA, Salama MM, Alhusseini MM, Mohamed GA (2020) M2BPGi for assessing liver fibrosis in patients with hepatitis C treated with direct-acting antivirals. World J Gastroenterol. 26(21):2864–2876. https://doi.org/10.3748/wjg.v26.i21.2864 PMID: 32550761; PMCID: PMC7284180
Saleh SA, Abdelwahab KM, Mady AM, Mohamed GA (2020) The impact of achieving a sustained virological response with direct-acting antivirals on serum autotaxin levels in chronic hepatitis C patients. Egypt Liver J 10:52. https://doi.org/10.1186/s43066-020-00060-w
Drenth JPH, Schattenberg JM (2020) The nonalcoholic steatohepatitis (NASH) drug development graveyard: established hurdles and planning for future success. Expert Opin Investig Drugs. 29(12):1365–1375. https://doi.org/10.1080/13543784.2020.1839888 Epub 2020 Oct 27. PMID: 33074035
Lambrecht J, Tacke F (2021) Controversies and opportunities in the use of inflammatory markers for diagnosis or risk prediction in fatty liver disease. Front Immunol. 9(11):634409. https://doi.org/10.3389/fimmu.2020.634409 PMID: 33633748; PMCID: PMC7900147
Aass KR, Kastnes MH, Standal T (2021) Molecular interactions and functions of IL-32. J Leukoc Biol. 109(1):143–159. https://doi.org/10.1002/JLB.3MR0620-550R Epub 2020 Sep 1. PMID: 32869391
Acknowledgements
Not applicable
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MM, SG, GM designed the research; KA participated in the acquisition of data; MM, SG, KA, GM participated in the analysis and interpretation of the data; MM, SG, KA, GM revised the article critically for important intellectual content; GM wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Ethics Committee of the Faculty of Medicine, Ain Shams University (FWA 000017585). Informed written consent was obtained from each participant before enrollment in the study. This study was performed in accordance with the 1975 principles of the Declaration of Helsinki and its appendices.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohamed, M.S., Ghaly, S., Azmy, K.H. et al. Assessment of interleukin 32 as a novel biomarker for non-alcoholic fatty liver disease. Egypt Liver Journal 12, 26 (2022). https://doi.org/10.1186/s43066-022-00189-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43066-022-00189-w