
Abstract
Background
Inborn errors of metabolism (IEMs) represent a special challenge in pediatric practice. Despite the unquestionable clinical significance of newborn screening, it just offers a snapshot of an extremely minor subgroup of metabolic disorders. So, it is crucial to use multiple techniques for accurate diagnosis of a wider spectrum of IEMs early in infancy to prevent overwhelming irreversible neurological complications in a cohort of high-risk Egyptian pediatrics. This study included four thousand and eighty suspected IEMs patients. They were referred to the Chromatography Unit, Clinical Biochemistry and Molecular Diagnostics Laboratories, National Liver Institute (NLI) for laboratory assessment in the period from March 2016 to November 2020. Separation of amino acids and acylcarnitines using tandem mass spectrometry (LC/MS) and organic acids using gas chromatography mass spectrometry (GC/MS) was done.
Results
Three hundred and twenty (320/4080, 7.8%) patients were diagnosed with IEMs. The following disorders were identified: organic acidopathies—200 (62.5%) including methylmalonic acidemia (MMA) (48/320, 15%), glutaric academia (GA) (40/320, 12, 5%), propionic acidemia (PA), (32/320, 10%), isovaleric acidemia (IVA) (40/320, 12.5%), methylcrotonyl glyceinuria (16/320, 5%), and orotic acidemia (24/320, 7.5%); amino acidopathies—80 (25%) including maple syrup urine disease (MSUD) (32/320, 10%), phenylketonuria (24/320, 7.5%), homocystinuria (16/320, 5%), and nonketotic hyperglycinemia (8/320, 2.5%) in addition to fatty acid disorders (FAO): 24 (7.5%) and lactic academia (LA), 16 (5%).
Conclusion
Early detection of IEMs by rapid non-invasive techniques. LC/MS and GC/MS. is a crucial process for early diagnosis of different types of IEMs to install therapeutic clue in a group of high-risk Egyptian pediatrics for proper treatment and better outcome
Similar content being viewed by others


Background
Inborn errors of metabolism (IEMs) are a diverse group of metabolic disorders caused by enzymatic defect in a metabolic pathway, its cofactor, or a transporter, leading to an accumulation of a substrate or lack of the product [1]. The increase of these toxic elements or their metabolites and lack of products of the defective pathway led to dysfunction of metabolism resulting in the pathophysiology of these disorders. Clinical manifestations generally happen in early infancy and childhood, with a diverse clinical spectrum [2].
IEMs have been categorized into different classes; one of them is characterized by the physiological disturbances of amino acids, known as aminoacidopathies [2]. Another class of IEMs is organic acidemias that relates to a set of disorders characterized by the excretion of non-amino organic acids in urine. Some organic acidemias are due to dysfunction in a definite stage of amino acid catabolism, commonly from inadequate enzyme activity. The majority of the classic organic acid disorders are the result of abnormal branched-chain amino acids or lysine catabolism [3]. There are also urea cycle disorders (UCDs) which are inborn errors of nitrogen detoxification/arginine synthesis due to defects in the urea cycle enzymes [4]. Mitochondrial fatty acid oxidation disorders (FAODs) are a varied group caused by the changed activity of the enzymes of the fatty acid transport and their oxidation in the mitochondria [5].
IEMs are rare diseases when taken individually, but totally these disorders are relatively frequent in distinct populations [6]. The cumulative incidence is about 1:800 in live births [7]. They are common throughout the Middle East, most probably because of the relatively high degrees of consanguinity (38.5%) [8]. Most IEMs are found in many families in Egypt causing high morbidity and mortality rates due to neurological damages which happen at a very young age [9].
Despite the unquestionable clinical significance of newborn screening and its principal role in the community health care, it just offers a snapshot of an extremely minor subgroup of metabolic disorders by the screening markers in dried blood spot (DBS) cases via the use of tandem mass spectrometry (MS/MS); moreover, several IEMs are not detected by routine newborn screening protocols [10]. Even when targeted screening programs focused on a single disease or a group of related disorders, up to 79% false-positive results were registered in a study in Taiwan [11]. There are also many causes for false-negative and false-positive results as a single biomarker or set of two metabolites may be a biomarker for more than one IEM [12]. Metabolomics gives the chance to chart metabolic pathways disorders, together with the network of metabolites indicating the origin of the metabolic disorder [13]. So, an effective screening program using multiple metabolomic techniques, for early detection of these disorders has long been documented as a critical, life-saving, and real preventive community health facility, not only for early treatment but also for genetic counseling and prenatal diagnosis in future gestations [14].
In this regard, this prospective study may be the first time that multiple metabolomic approaches have been utilized to widen the scope of the diagnosis of IEMs in Egypt to diminish morbidity and mortality in high-risk children.
Methods
Ethics and study population
The protocol of this study was approved by the ethics committee of the National Liver Institute, Menoufia University (NLI IRB protocol N 00193/2020). The study was carried out at Chromatography Unit, Clinical Biochemistry and Molecular Diagnostics Laboratories, NLI. Suspected cases of IEMs were recruited from March 2016 to November 2020.
The cases were referred to the unit for laboratory assessment based on the clinical suspicion by the pediatricians to have IEMs. The patients were subjected to complete history taking regarding nutritional history and growth charts with stress on birth date, sex, gestational age, antenatal, prenatal history, and consanguinity. Pedigree construction and recording of previous neonatal deaths and similar affected cases within the family were also recorded. Full clinical examination (general and neurological) by the pediatric clinicians was also performed.
Basic laboratory investigations
For all cases, routine laboratory investigations were done including liver function tests, kidney function tests, blood sugar, and lactate using Clinical Auto analyzer (Beckman Instruments, Fullerton, CA, USA), plasma homocysteine using (The ARCHITECT i1000SR immunoassay analyzer) and arterial blood gases (ABG) using an automated MedicaEasyLyte® Analyzer microprocessor-controlled electrolyte system that uses Ion Selective Electrode (ISE).
Sample collection and special metabolic investigations
Blood spots were obtained from infants by heel puncture or from the big finger of older children and spotted on filter paper (Guthrie card made of Whatman 903, purchased from GE Healthcare, NJ, USA), left to dry on a clean surface, and then stored at – 80 °C until analysis of amino acids and acylcarnitine using tandem mass spectrometry (MS/MS). Urine samples were collected from all subjects in special sterile plastic bags then evacuated in a plastic laboratory container without the addition of any preservatives or diet restriction before the sample collection. Urine samples were divided into two samples: one for testing reducing sugars and ketones immediately by dipstick tests and the other was stored immediately at – 80 °C till analysis of organic acids using gas chromatography/mass spectrometry (GC/MS).
Chemicals and reagents
Component of MassChrom® amino acids and acylcarnitines from dried blood/non-derivatized (Chromsystems Instruments & Chemicals GmbH, München, Germany) were purchased.
Pentadecanoic acid (PDA) was obtained from Across organics (NJ, USA). N, O-bis-(trimethylsilyl)–trifluoroacetamide (BSTFA) plus 1% trimethylchlorosilane (TMCS), purchased from SUPELCO, Bellefonte PA, USA, were used as derivatizing reagents. The solvent was of HPLC Grade; methanol was purchased from Fisher Scientific (Loughborough, UK). Distilled water was produced by Thermo Scientific Barnstead LabTower RO Water Purification System. Ethyl acetate was from Fisher Scientific (Loughborough, UK) used as an extraction reagent. Acetonitrile was from Fisher Scientific (Loughborough, UK). Anhydrous sodium bicarbonate was obtained from Merck (Darmstadt, Germany) and sodium chloride was from SIGMA-ALDRICH (Fluka) St. Louis, Mo, USA; both were of analytical grade. All other chemicals and reagents were purchased from SIGMA-ALDRICH (Fluka) St. Louis, Mo, USA.
Amino acids and acylcarnitine assay
Previously reported method [15] was used after some modifications; 3 mm of the dried blood spot disk punched into a well of v-bottomed plate, containing 100 μl of lyophilized internal standard that is reconstituted with exactly 25 ml extraction buffer according to the manufacturing recommendation) (Chromsystems Instruments & Chemicals GmbH, München, Germany). Then, the plate was sealed with a protective sheet and agitated at 600 rpm for 20 min at ambient temperature. The supernatant was transmitted to a new v-bottomed well plate and covered by aluminum foil protective sheet. Then samples were prepared for injection. Ten microliters of the elute was inserted into the MS/MS system (ACQUITY UPLC H-Class. Water Corporation, MA, USA) at 2 min interval in a flowing stream of 80% acetonitrile. The flow rate was 200 μl/min and reduced to 20 μl/min in 0.25 min. Then, the flow rate was increased to 600 μl/min in 1.25 min then decreased again to 200 μl/min. The scan time of the MS/MS system was 1.25 min. The obtained spectra of all analytes were analyzed with multiple reaction monitoring (MRM) mode. The mass to charge ratio of all the amino acids and acylcarnitines was recorded (Supplementary Table S1). The data were calculated using Neolynx software (Neolynx Inc., Glendale, CA, USA).
Qualitative urinary organic acid assay
Frozen urine samples were liquefied by incubation at 37 °C for 15 min and vortexed for 15 s. Urine creatinine levels were measured by the Beckman Coulter (Synchron CX 9 ALX) Clinical Autoanalyzer, USA, and adjusted to 1 μMol creatinine [16]. Extraction and derivatization of the urine samples were done according to the previously reported method [17]. Internal standard (PDA) stock solution was prepared (48.8mg/100mL) in absolute methanol. One microliter aliquot of derivatized sample was injected in splitless mode into an Agilent 7890 GC system supported by a 30.0 m× 0.25 mm i.d. fused-silica capillary column with 0.25 μm HP-5MS stationary phase (Agilent, USA). The injector temperature was rest at 250 °C. Helium was used as carrier gas at a flow rate of 1 mL/min through the column. The column temperature was primarily kept at 80 °C for 2 min and then increased to 280 °C by 4 °C/min, where it was held for 3 min. Run time was 55 min. The column effluent was introduced into the ion source of an Agilent 5975 mass selective detector (Agilent Technologies). The MS quadruple temperature was fixed at 150 °C and the ion source temperature at 230 °C. The acceleration voltage was turned on after a solvent delay of 3 min. Masses were attained from m/z 50 to 550. The total ion current (TIC) chromatograms were studied to identify the peaks of the trimethylsilyl (TMS) derivatives of the organic acids. Retention time and mass spectrum were recorded for each derivative (see Supplementary Table S2). GC/MSD ChemStation Software (Agilent, USA) was used for auto-acquisition of GC total ion chromatograms (TICs) and fragmentation patterns.
Results
This study was conducted on 4080 cases that were referred to the unit for laboratory assessment based on the clinical suspicion by the pediatricians of IEMs. The study included 2360 males (57.8%) and 1720 females (42.2%), and their ages ranged from 15 days to 3 years.
Table 1 represented the symptoms and signs of the different types of disorders detected among the studied patients according to age groups. The most shared and frequent presenting features were sepsis-like symptoms (in 75% of diagnosed cases) (poor feeding, reduced activity, and poor crying) and convulsions. However, hyperammonemia (52.5%) and metabolic acidosis (47.5%) were among the obvious laboratory abnormalities.
Table 2 represented the characteristics of IEMs, patients detected in the current study. A total of 320/4080 (7.8%) patients were found to have IEMs; they came from 280 families. Consanguinity was detected in 192/320 (60%) patients and there were history of previous siblings’ deaths and similarly affected cases in the family (up to three similar cases in the same family) in 96/320 (30%) and 88/320 (27.5%) patients, respectively.
Organic acidemia was the most frequent disorder (200/320, 62.5%) detected among the studied patients, followed by aminoacidpathies (80/320, 25%), FAO defects (24/320, 7.5%), and finally lactic academia (LA) was detected in 16 patients (5%).
The detected types of organic acidemias were methylmalonic acidemia (MMA) (48/320, 15 %), Glutaric academia (GA) (40/320, 12, 5%), Propionic acidemia (PA), (32/320, 10%), isovaleric acidemia (IVA) (40/320, 12.5%), methylcrotonyl glyceinuria (16/320, 5%), and orotic acidemia (24/320, 7.5%).
The detected types of aminoacidopathies were maple syrup urine disease (MSUD) (32/320, 10%), phenylketonuria (24/320, 7.5%), homocystinuria (16/320, 5%), and nonketotic hyperglycinemia (8/320, 2.5%).
Lactic acidemia was detected in (16/320, 5%) and fatty acids oxidation defects were detected in (24/320, 7.5%).
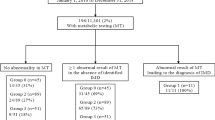
Tables 3 and 4 represented the laboratory and metabolomics findings in the diagnosed cases of IEMs. The preliminary diagnosis was outlined in the workflow chart (Fig. 1) built on the detected abnormalities in the amino acids and acylcarnitines (for normal ranges see Supplementary Table S3) profiles in dried blood spot samples measured by LC (MS/MS) and confirmed by the identification of elevated peaks of diagnostic urinary metabolites by GC/MS (Supplementary Figs. S1–S9).

Workflow chart
Discussion
The introduction of MS/MS and GC/MS allowed the detection of IEMs that supposed to be rare conditions; however, together they are not that rare. These technologies have considerably enhanced the diagnostic value for such disorders, and hence early management can be assumed before permanent clinical squeal takes place [6].
In such scenario, extended metabolic screening by MS/MS showed particular findings in 288 patients out of 4080; so, the second alternate test, such as urine organic acid analysis, was furthermore necessary as a confirmatory follow-up diagnostic test. Furthermore, this study revealed that IEMs were diagnosed in 320/2080 (7.8%) of the cases by the application of two metabolome analyses (MS/MS δ GC/MS).
In Egypt, a lack of awareness of such inherited diseases by the physicians particularly in the rural community leads to delayed diagnosis or misdiagnosis and recurrent metabolic crises with irreversible damage or death of the affected neonates before starting the diagnosis [18]. This agreed with the findings of the present study as there was a delayed diagnosis of the patients (presenting age was around the end of the first year of life), which may be the cause of those severe and irreversible clinical complications. These results were consistent with reports by other studies [6, 19].
Furthermore, many older children exhibited poor response to particular therapies when started, while younger patients belonging to the same families displayed a considerably improved response. This indicates the absolute necessity for early recognition and intervention for promising results in such disorders [6].
Variable ratios of IEMs were found in diverse studies according to the type of selection of patients. In a study done by Shawky and his colleagues, IEMs were diagnosed in 20/50 (40%) infants and children who were supposed to have IEMs [20]. In another study done by Selim and his colleagues, IEMs were confirmed in 6% (203/3380) of children suspected to have IEMs [6]. Also, high incidence of IEMs (32.5%) was reported in the study of Shawky and his colleagues [21] compared to other studies that was due to the selection criteria of neonates with suspected IEMs, not all that admitted to the Neonatal Intensive Care Unit (NICU).
In the current study, out of 320 patients with IEMs, 184 patients (57.5%) had consanguineous parents. This was in line with other studies by El-Mesellamy et al. and Selim et al. [6, 22] in addition, there were a history of previous siblings’ death and similar cases in the family (up to 3 children in some cases) in 96/320 (30 %) and 88/320 (27.5%) patients respectively (Table 2). This might not be clarified only by the high consanguinity in Egypt that extends up to 35.3% [23], but also by the fact that most diagnosed IEMs are autosomal recessive which rise in occurrence by consanguineous coupling because relatives more often share abnormal genes innate from a shared ancestor [21]. Hence, a history of parent consanguinity and/or sibling deaths would increase the suspicion of IEMs [24]. This reflected the important influence of consanguinity in this specific health problem in Egypt due to lack of early detection, and absence of genetic follow-up and counseling when having a confirmed case [6].
In the current study, the most common IEMs were organic acidemias detected in 62.5%, aminoacidopathies in 25%, fatty acid oxidation disorders in 7.5%, and lactic acidemia in 4%. Methylmalonic acidemia (MMA) and maple syrup urine disease (MSUD) were the most frequently diagnosed types in the current study, while Phenylketonuria (PKU) and MMA, were the most commonly diagnosed disorders in a study done by Karam and his colleagues [25] similar to that reported from other Mediterranean countries like Tunisia [26]. Furthermore, Selim and her colleagues [6] demonstrated that aminoacidopathies were in 127/203 (62.6%), mainly PKU in 100/203 (49.3%), followed by organic acidemias in 69/203 (34%).
The variation in the commonest types of IEMs in the current study is due to variation in performed investigations, the inconsistent diagnostic strategies (ammonia and lactate were measured in our study), and age variation of the patients.
In the current study, the shared presenting symptoms of the cases were sepsis-like symptoms (poor feeding, reduced activity, and poor crying) which were in agreement with the study of Shawky et al. [21] who recommended to take in mind inborn errors of metabolism at the same time as common developed disorders, for example, sepsis. The further common presenting symptom among the detected cases was convulsions. This came in agreement with the study of Shawky et al. [21] who reported that the major symptoms in neonates with the risk of metabolic disorders were convulsions and lethargy.
In the current study, there was a high frequency of hyperammonemia in 168 patients (52.5%) among the diagnosed cases of IEMs; 136 had organic acidemias, 16 had Maple syrup urine disease (MSUD) and 16 had orotic aciduria (data not shown), this was in line with the study of Shawky et al. [21]. They were mainly presented with lethargy, poor feeding, suckling, crying, convulsions, persistent vomiting, and acidosis. This means that blood ammonia has to be measured for each neonate/infant presented by these symptoms, or supposed to have IEMs [21].
Methylmalonic acidemia (MMA) was detected in 48/320 (15%), by elevations of propionylcarnitine (C3) and the ratio of acetyl carnitine: propionylcarnitine (C3:C2) in dried blood spot samples. Diagnosis was confirmed by the elevated peaks of urinary methylmalonic acid, methylcitric acid, moderate elevation of 3-hydroxy propionic acid, and propionyl glycine detected by GC/MS. The diagnosis was according to the findings of El-Mesellamy et al. and Shennar et al. [22, 27]. It results from either deficiency of the methylmalonyl CoA mutase enzyme or the synthesis of its cofactor Vit B12 [22].
Glutaric acidemia (GA) was detected in 40/320 (12.5%): by elevation of glutarylcarnitine (C5DC) in dried blood spot samples. The diagnosis was confirmed by the detection of elevated peaks of urinary glutaric and 3-OH glutaric acids by GC/MS, which are the diagnostic metabolites according to the findings of the researchers [22, 27, 28].
Glutaric acidemia type 1 (GA-1) is caused by the defect in functional glutaryl-CoA dehydrogenase (GCDH) activity, an important enzyme in the catabolism of l-tryptophan, l-lysine, and l-hydroxylysine, leads to the accumulation of glutaric acid, 3-hydroxyglutaric acid (3-OH-GA), and occasionally glutaconic acid in the different body fluids [28].
Propionic acidemia (PA) was detected in 32/320 (10%) by elevations of C3 and the ratio of C3:C2 in dried blood spot samples. Diagnosis was confirmed by the highly elevated peaks of urinary 3-hydroxypropionic acid, propionylglycine, methylcitric acid, and the absence of methylmalonic acid by GC/MS (Fig. S3). The diagnosis was according to the findings of other studies [22, 27].
Isovaleric acidemia (IVA) was detected in 40/320 (12.5%) by elevations of isovalerylcarnitine (C5) in dried blood spot samples. It was confirmed by elevated peaks of urinary isovalerylglycine (mono and di-derivatives) and 3-hydroxyisovaleric acid by GC/MS as it was reported by other studies [27, 29]. It is caused by a defect in the function of the Apo enzyme of the mitochondrial enzyme isovaleryl-CoA dehydrogenase [29].
Methylcrotonyl glycinuria (MCG) was detected in twelve males and four female patients 16/320 (5%) by elevation of hydroxy isovalerylcarnitine (C5-OH) in dried blood spot samples. Diagnosis was confirmed by the detection of 3 hydroxyisovaleric acid, methylcrotonyl glycine, and methyl citric acid by GC/MS. These results were in line with other studies [6, 30]. It is caused by deficiency of 3-methyl crotonyl CoA carboxylase (3-MCC) enzyme; one of the four biotin-containing carboxylases known in human beings; hence, leucine catabolism is blocked [30].
Orotic acid was detected in 24/320 (7.5%). Low citrulline and high arginine levels were identified in four patients, associated with the detection of elevated peak of urinary orotic acid on GC/MS analysis that implies primary orotic acidemia/aciduria which is a disorder of pyrimidine metabolism or urea cycle disorder; however, genetic testing is the method of the first choice to confirm the diagnosis [4] that was consistent with the findings of El-Mesellamy and his colleagues [31]. Likewise, family pedigree construction revealed a similar case (she was a female carrier) as it is x-linked disease that implies ornithine transcarboxylase OTC deficiency [32]. The other cases were diagnosed by elevated peak of urinary orotic acid. These findings were in line with El-Mesellamy and his colleagues [31] who depended on the detection of orotic acid in urine for diagnosis of urea cycle defects.
MSUD was detected in 32/320 (10%); by elevated the ratio of Leucine/Isoleucine (Leu/Ile) and valine in dried blood spot samples and by confirmed by the detection of elevated peaks of alloisoleucine, 2-oxoisocaproate, and 2-hydroxyisovalerate in urine by GC/MS. In a study done by Fateen and his colleagues [18], MSUD, caused by a deficiency in the branched chain a-ketoacid dehydrogenase complex, was diagnosed in 12 cases [(12/1041 (1.2%)] which represent 4% out of 302 patients supposed to be aminoacidopathy; however, they were diagnosed by performing thin layer chromatographic separation of amino acids in plasma and urine that showed abnormal quantities of branched chain amino acids (valine, leucine, and isoleucine).
Phenylketonuria (PKU) was detected in 24/320 (7.5%) by quantitative elevation of phenylalanine level and the ratio of phenylalanine to tyrosine in dried blood spot samples according to the findings of the study of Fateen and his colleagues [18]. It is caused by mutations in the phenylalanine hydroxylase (PAH) gene causing the accumulation of excessive quantities of phenylalanine which are harmful to brain development [18].
Around 1/3 of diagnosed cases with aminoacidopathies were identified as MSUD or PKU which is reasonably a great number, this was in line with the study done by Fateen and his colleagues [18].
Non-ketotic hyperglycinemia (NKH); was detected in eight male cases 8/320 (2.5%) by elevated glycine level in dried blood spot samples. It is a hereditary disorder characterized by unusual great levels of glycine produced by a defective glycine cleavage system, causing too much glycine concentrations in both blood and brain [33]. In a study done by Fateen and his colleagues [18], only 3 patients (3/1041 0.3%) gave a band of glycine in thin layer chromatography (TLC) separation of free amino acids in urine and blood and this signifies 1% out of 302 cases diagnosed with aminoacidopathy.
Homocytinuria (HCU) was detected in 16/320 (5%) by increased level of blood methionine concentrations in filter paper. Quantitative analysis of plasma homocysteine was done for conformation of the diagnosis. It is a genetic deficiency of cystathionine β-synthase (CBS). This enzyme is involved in the transsulfuration of homocysteine to form cystathionine using pyridoxal-phosphate as a cofactor [34]. In another study by Selim and her colleagues [6], Homocytinuria was detected in 4/203 (2%).
Fatty acid oxidation disorders (FAODs) are inborn errors of metabolism due to disruption of either mitochondrial β-oxidation or the fatty acid transport using the carnitine transport pathway. The presentation of a FAOD will depend upon the specific disorder, but common elements may be seen, and ultimately require a similar treatment. Furthermore, they are an important group to consider in hypoketotic hypoglycaemia conditions due to decreased ability to use stored fat as a source of energy during fasting periods [5].
In this study, fatty acid oxidation defects were detected only in 24 cases (7.5%); that was similar to other reports in some Arab populations like Lebanon (4%) [25] while Bahrain [35] have considerably higher prevalence rates for fatty acid oxidation defects (20%). The diagnosis was established according to Fingerhut et al. [36] by increased Hexanoylcarnitine (C6)-Decanoylcarnitine (C10) and low level of Hexadecanoylcarnitine (C16) and Linoleoylcarnitine (C18:1) and high free carnitines in eight cases and elevated dicarboxylic acids in urine such as adipic, suberic, dehydrosuberic, sebacic, dehydrosebacic, and modest amounts of 3-OH dicarboxylics-hexanoyl glycine in the other cases.
Lactic acid (di- and tri-TMS derivatives) were detected in (16/320, 5%) associated by elevated peaks of urinary 4-hydroxyphenyllactic acid by GC/MS. Lactic acidemia is a physiological condition that can occur due to a number of causes [37]. It is also may be due to enzyme defect in krebs cycle leading to respiratory chain deficiencies such as pyruvate dehydrogenase (PDH), pyruvate carboxylase (PC), and electron transport chain (ETC) deficiencies as well as 4-hydroxyphenyllactic acid that is actually a tyrosine metabolite and may be elevated secondary to disorders of tyrosine catabolism, liver disease, or lactic acidemia so more enzyme studies and molecular genetic studies may be required [38]. Interestingly, determining the acid-base status is important in the assessment of a patient with a potential inherited metabolic defect, because high anion gap metabolic acidosis is usually caused by the accumulation of organic acids including lactic acid, ketone bodies, or unusual acids and their derivatives. In contrast, diarrhea and renal tubular acidosis are the main causes of metabolic acidosis with a normal anion gap [20].
The only limitations to metabolome coverage result from the available volume of sample, restrictions of metabolite extraction, and the sensitivity and specificity of the analytical techniques in addition to the pretreatment of urine samples analyzed via GC-MS by urease to eliminate the significant amount of urea, as it might affect the chemical derivatization, causing overloading of column, peak alterations, and co-elution of metabolites peaks [39].
Conclusion
The use of such rapid non-invasive robust coupled metabolomics techniques (MS/MS and GC/MS) should be taken into consideration as a novel mean to enhance the scale of IEMs disease classes correlated with pathophysiology, therapy modalities, and follow-up strategies in addition to genetic counseling and prenatal diagnosis in future gestations.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- IEMs:
-
Inborn errors of metabolism
- MS/MS:
-
Tandem mass spectrometry
- GC/MS:
-
Gas chromatography mass spectrometry
- FAODs:
-
Fatty acid oxidation disorders
- LA:
-
Lactic academia
- UCDs:
-
Urea cycle disorders
- DBS:
-
Dried blood spot
- BSTFA:
-
Bis-(trimethylsilyl)–trifluoroacetamide
- TMS:
-
Trimethylsilyl
- TIC:
-
Total ion current
- MSUD:
-
Maple syrup urine disease
- MMA:
-
Methylmalonic acidemia
- PA:
-
Propionic acidemia
- GCDH:
-
Glutaryl-CoA dehydrogenase
- IVA:
-
Isovaleric acidemia
- MCG:
-
Methylcrotonyl glycinuria
- 3-MCC:
-
3-Methyl crotonyl CoA carboxylase
- 3-OH-GA:
-
3-Mydroxyglutaric acid
- MCG:
-
Methylcrotonyl glycinuria
- OTC:
-
Ornithine transcarboxylase
- OA:
-
Orotic acidemia
- PKU:
-
Phenylketonuria
- PAH:
-
Phenylalanine hydroxylase
- NKH:
-
Non-ketotic hyperglycinemia
- TLC:
-
Thin layer chromatography
- HCU:
-
Homocytinuria
- CBS:
-
Cystathionine β-synthase
- PDH:
-
Pyruvate dehydrogenase
- PC:
-
Pyruvate carboxylase
- ETC:
-
Electron transport chain
- Phe:tyr:
-
Phenylalanine:tyrosine ratio
- Leu/Ile:
-
Leucine:isoleucine
- Gly:
-
Glycine
- CSF:
-
Cereprospinal fluid
- NR:
-
Not remarkable
- C0:
-
Free carnitine
- C6:
-
Hexanoylcarnitine
- C10:
-
Decanoylcarnitine
- C16:
-
Hexadecanoylcarnitine
- C18:1:
-
Linoleoylcarnitine
- C2:
-
Acetyl carnitine
- C3:
-
Propionylcarnitine
- C5:
-
Isovalerylcarnitine
- C5DC:
-
Glutarylcarnitine dicarboxylate
- C5OH:
-
Hydroxy isovalerylcarnitine
References
Agana M et al (2018) Common metabolic disorder (inborn errors of metabolism) concerns in primary care practice. Ann Transl Med 6(24):469–469
Wajner M, Sitta A, Kayser A, Deon M, Groehs AC, Coelho DM, Vargas CR (2019) Screening for organic acidurias and aminoacidopathies in high-risk Brazilian patients: eleven-year experience of a reference center. Genet Mol Biol 42(1 Suppl 1):178–185
Seashore MR (2020) Organic acid disorders. In: Rosenberg's molecular and genetic basis of neurological and psychiatric disease. Elsevier, pp 785–791
Häberle J et al (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 7(1):32
Djouadi F, Bastin J (2019) Mitochondrial genetic disorders: cell signaling and pharmacological therapies. Cells 8(4):289
Selim LA et al (2014) Selective screening for inborn errors of metabolism by tandem mass spectrometry in Egyptian children: a 5 year report. Clin Biochem 47(9):823–828
Pampols T (2010) Inherited metabolic rare disease. Adv Exp Med Biol 686:397
Rashed MS et al (1997) Screening blood spots for inborn errors of metabolism by electrospray tandem mass spectrometry with a microplate batch process and a computer algorithm for automated flagging of abnormal profiles. Clin Chem 43(7):1129–1141
Fateen E et al (2009) Inborn errors of metabolism revealed by organic acid profile analysis in high risk Egyptian patients: six years experience. Egypt J Med Human Genet 10(2)
Yuasa M et al (2019) Evaluation of metabolic defects in fatty acid oxidation using peripheral blood mononuclear cells loaded with deuterium-labeled fatty acids. Dis Markers 2019:2984747–2984747
Cheng K-H et al (2010) Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years' experience from two centers in Taiwan. J Chin Med Assoc 73(6):314–318
Ismail IT, Showalter MR, Fiehn O (2019) Inborn errors of metabolism in the era of untargeted metabolomics and lipidomics. Metabolites 9(10):242
Wangler MF et al (2018) A metabolomic map of Zellweger spectrum disorders reveals novel disease biomarkers. Genet Med 20(10):1274–1283
García-Cazorla A et al (2009) Mental retardation and inborn errors of metabolism. J Inherit Metab Dis 32(5):597–608
Oraby M (2009) Preliminary results of Egypt experience for use of tandem mass spectrometry for expanded metabolic screening. J Appl Sci Res 5(10):1425–1435
Rinaldo P, Cowan TM, Matern D (2008) Acylcarnitine profile analysis. Genet Med 10(2):151–156
Christou C et al (2014) GC-MS analysis of organic acids in human urine in clinical settings: a study of derivatization and other analytical parameters. J Chromatogr B 964:195–201
Fateen EM, Ibrahim M, Abdallah Z (2014) Fifteen years experience: Egyptian metabolic lab. Egypt J Med Human Genet 15(4):379–385
Narayanan M et al (2011) Diagnosis of major organic acidurias in children: two years experience at a tertiary care Centre. Indian J Clin Biochem 26(4):347–353
Shawky RM et al (2007) Study of amino acid disorders among a high risk group of Egyptian infants and children. Egypt J Med Human Genet 8(2):173–190
Shawky RM, Abd-El Khalek HS, Elakhdar SE (2015) Selective screening in neonates suspected to have inborn errors of metabolism. Egypt J Med Human Genet 16(2):165–171
El-Mesellamy H et al (2014) Disorders associated with abnormal acylcarnitine profile among high risk Egyptian children. Bratisl Lek Listy 115(5):300–306
Shawky RM, Sadik DI (2011) Congenital malformations prevalent among Egyptian children and associated risk factors. Egypt J Med Human Genet 12(1)
Ali YF et al (2017) Metabolic screening and its impact in children with nonsyndromic intellectual disability. Neuropsychiatr Dis Treat 13:1065
Karam PE et al (2013) Diagnostic challenges of aminoacidopathies and organic acidemias in a developing country: a twelve-year experience. Clin Biochem 46(18):1787–1792
Hadj-Taieb S et al (2012) Aminoacidopathies and organic acidurias in Tunisia: a retrospective survey over 23 years. La Tunisie Medicale 90(3):258–261
Shennar HK et al (2015) Diagnosis and clinical features of organic acidemias: a hospital-based study in a single center in Damascus. Syria Qatar Med J:9
Zaki OK et al (2014) Demographic and clinical features of glutaric acidemia type 1; a high frequency among isolates in upper Egypt. Egypt J Med Human Genet 15(2):187–192
Vockley J, Ensenauer R (2006) Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. In: American journal of medical genetics part C: seminars in medical genetics. Wiley Online Library
Sun W et al (2011) The screening of inborn errors of metabolism in sick Chinese infants by tandem mass spectrometry and gas chromatography/mass spectrometry. Clin Chim Acta 412(13):1270–1274
El-Mesallamy H, Gouda A, Fateen E, Elbaz A (2013) Detection of urinary organic acids in high risk Egyptian children by electrospray tandem mass spectrometry. J Appl Sci Res 9:1909–1916
Häberle J et al (2011) Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum Mutat 32(6):579–589
Baker PR 2nd et al (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137(Pt 2):366–379
van Vliet D et al (2014) Single amino acid supplementation in aminoacidopathies: a systematic review. Orphanet J Rare Dis 9:7–7
Golbahar J et al (2013) Selective newborn screening of inborn errors of amino acids, organic acids and fatty acids metabolism in the Kingdom of Bahrain. Mol Genet Metab 110(1-2):98–101
Fingerhut R et al (2001) Hepatic carnitine palmitoyltransferase I deficiency: acylcarnitine profiles in blood spots are highly specific. Clin Chem 47(10):1763–1768
Shetty PP et al (2020) Amino acid profile and lactate pyruvate ratio: potential adjunct markers for differentiating inborn errors of metabolism. Indian J Clin Biochem 35(4):430–435
Sandlers Y (2019) Amino acids profiling for the diagnosis of metabolic disorders. In: Biochemical testing-clinical correlation and diagnosis. IntechOpen
Roberts LD et al (2012) Targeted metabolomics. Curr Prot Mol Biol 98(1):30.2 1-30.2. 24
Acknowledgements
The authors would like to express their sincere appreciation to all the chemists of the Clinical Biochemistry and Molecular Diagnostics Department, National Liver Institute, for their contribution to the laboratory tests and to the guardians of the participant patients for their contribution to this work.
Funding
This research was performed using the instruments supplied by a grant from the Science and Technology Development Fund (STDF) (N2338).
Author information
Authors and Affiliations
Contributions
SA was the major contributor in performing the practical section of this work and the interpretation of the laboratory data. She also contributed to the writing, revision, and language polishing of the manuscript. ME and SA performed full clinical examination (general and neurological), complete history taking regarding nutrition and growth charts with stress on birth date, sex, gestational age, antenatal, prenatal history, consanguinity, pedigree construction, recording of previous neonatal deaths, and similar affected cases within the family. OZ contributed to the interpretation of the clinical data and supervision of the final diagnosis of the cases. MO contributed to the conception, design, and revision of the work. HE was the main coordinator of the study and contributed to the revision of the work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Ethics Committee of National Liver Institute, Menoufia University (NLI IRB protocol N 00193/2020). Informed written consents were obtained from the guardians of participants before enrollment in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The mass to charge ratio of the measured amino acids and acylcarnitines. Table S2. The m/z values for Q- and C-ions for all identified organic acids and their retention times. Table S3. The normal ranges of all reported metabolites by MS/MS.
Additional file 2: Figure S1.
A case of Methylmalonic aciduria (MMA) with elevated peaks of 1: methylmalonic acid, 2: Methyl citric acid, 3: internal standard. Figure S2. A case of Glutaric Acidemia (GA) with elevated peaks of 1:3 hydroxyglutaric acid, 2: glutaric acid, 3:internal standard. Figure S3. A case of Propionic Acidemia (PA) with elevated peaks of 1: 3 hydroxypropionic acid, 2: propionylglycine, 3: tiglylglycine 4: methylcitric acid, 5: internal standard. Figure S4. A case of Isovaleric Acidemia (IVA) with elevated peaks of 1: 3 hydroxyisovaleric acid, 2: isovalerylglycine, 3: internal standard. Figure S5. A case of methylcrotonyl glycinuria (MCG) with elevated peaks of 1: 3 hydroxyisovaleric acid, 2: methylcrotonylglycine, 3: internal standard. Figure S6. A case of Orotic Acidemia (OA) with elevated peaks of 1: orotic acid, 2: internal standard. Figure S7. A case of MSUD with elevated 1: 2-oxoisocaproic acid, 2: 2-hydroxy isovaleric acid, 3: internal standard. Figure S8. A case of fatty acid oxidation defects with elevated1:adipic acid ,2 :suberic acid, 3 :sebacic acid, 4 :internal standard, 5 :3-hydroxy sebacic acid. Figure S9. A case of lactic Acidemia (LA) with elevated peaks of 1: lactic acid, 2:3 hydroxy butyric acid, 3:4 hydroxyphenyllactic acid, 4:4 hydroxyphenylpyruvic acid, 5:internal standard. Figure S10. Total ion chromatogram for a normal OA profile.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdelsattar, S., Obada, M., El-Hawy, M.A. et al. Inherited metabolic disorders in a cohort of Egyptian children. Egypt Liver Journal 12, 20 (2022). https://doi.org/10.1186/s43066-022-00176-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43066-022-00176-1