
Abstract
Background
Isolation of post-partum umbilical cord Wharton’s jelly stem cells has gained attention as an alternative source of the bone marrow. Because easy isolation, lack of ethical concerns, and the presence of both embryonic and adult stem cells have made them a valuable source for use in therapeutic applications and regenerative medicine. The study utilized a modified protocol using in-house human pooled cord blood serum for isolation and expansion of the mesenchymal stem cells obtained from the human umbilical cord Wharton’s jelly. Cell proliferation and population doubling time and tri-lineage differentiation were assessed, and the expressions of mesenchymal cell surface markers CD44, CD90, CD105, and CD34 were assessed by flow cytometry and RT-PCR. The genetic stability of the isolated cells was assessed by chromosomal karyotype.
Results
The isolated cells displayed fibroblastic-like morphology and tri-lineage differentiation into adipocyte, chondrocyte, and osteocyte. The isolated cells maintained the proliferative competence with a doubling time ranged from 38 to 42h and corresponded well with the standard positive and negative molecular markers (CD44+, CD90+, CD 105+, and CD34−). Cell senescence occurred at the later passage of the cells (P15) affecting, about 25% of the population. Metaphases spread of the cells showed normal diploid karyotypes, with typical chromosomal plates indicating genetic stability of the isolated cells.
Conclusion
The primary cultures exhibited success in isolating the umbilical cord Wharton’s jelly mesenchymal stem cells, which maintained their tri-lineage differentiation potential, phenotypes and karyotype characteristics on further passage and expansion.
Similar content being viewed by others

Background
Stem cells are a unique type of cells that possess a regenerative potential to maintain and repair cellular damage throughout the life span of these tissues. Interest in adult stem cells leads to the isolation of various cells from different origins, such as mesenchymal stem cells (MSCs), hematopoietic stem cells, and other progenitors [1, 2]. The number of retrieved stem cells is limited, as isolation generally involves an invasive and technically complicated process, which compromises the quality and the biological activity of the isolated cells and limits their proliferation and differentiation potential during in vitro culture [3,4,5].
During embryonic development, hematopoietic and MSCs migrate from the umbilical cord to the placenta, and hence some colonies are trapped in the Wharton jelly [6,7,8]. The trapped MSCs produce microenvironment constituents, such as platelet-derived growth factor, insulin-like growth factor, and a specific stream, ensuring the preservation of cell population and maintenance of their function [9, 10]. Such an environment makes the human umbilical cord is an excellent source of MSCs, with more primitive cells and good regenerative properties than other tissues [11,12,13].
Various established protocols to isolate MSCs from the umbilical cord tissue apply either enzymatic digestion of the umbilical cord tissues or the direct explant methods that mainly rely on the migration ability of these MSCs from tissue explants without using any digestive enzymes [14, 15]. Supplements of human origin are necessary to supply growth factors to avoid the antigens of non-human sources present in fetal bovine serum and, therefore, are more suitable for cell therapy which requires strict quality control, safety, and maintenance during the harvesting and expansion process to minimize the risk of genetic alterations of the isolated cells [16,17,18,19,20]. In the study, a simple protocol used pooled cord blood serum (CBS) to isolate and grow Wharton’s jelly mesenchymal stem cells (WJ-MSCs). The proliferative, the differentiation potential, and the expression of different cell surface markers CD44, CD90, CD105, and CD34 were evaluated. Chromosomal karyotype analysis by G-banding was carried out to ensure the genetic stability of the isolated stem cells.
Methods
The study took place from March 2018 to July 2019 in the Department of Biochemistry and Molecular Diagnostics, National Liver Institute, Menouffia University, Egypt. The research ethics committees of approved the research proposal and the protocols to comply with national research guidelines. Patients provided informed written consent for the use of tissue for research purposes. The human umbilical cord tissue and venous cord blood were obtained immediately under sterile aseptic condition after placental delivery of a full-term third-trimester birth by either vaginal or cesarean section.
Chemicals and reagents
DMEM high glucose and RPMI media from Lonza (Lonza, Bio Whittaker Germany). Fetal bovine serum (FBS), phosphate-based saline (PBS), trypsin, colcemid (10 μg/ml), and antibiotics (penicillin10,000 U/ml; streptomycin 10,000 μg/ml, gentamycine10,000 μg/ml), fungi zone, l-glutamine, trypan blue, Giemsa stain from, and Trizol (Life Technologies, Germany). Glacial acetic acid; methanol; potassium chloride; chloroform; isopropanol; ethanol; ultrapure water; dNTPs; and primers of the cluster differentiation markers CD34, CD44, CD90, CD105, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from (Thermo Fisher Scientific, Waltham, MA). Oil Red O, Alcian Blue, or 2% Alizarin Red from Sigma Aldrich, Germany). Fluorescein isothiocyanate (FITC) anti-CD34 antibody, FITC anti-CD45 antibody, anti-CD90, and anti-CD105 antibody from (BD Biosciences, USA).
Preparation of pooled cord blood serum
The cord blood was collected in a 50-ml sterile centrifuge tube without anticoagulant immediately after the birth of a full-term baby from the umbilical cord of the undelivered placenta. The collected blood, usually obtained from five donors at a time, was allowed to clot by incubation at room temperature for 3 h and then centrifuged at 10,000 rpm for 20 min. The supernatant sera was collected and pooled together, then heat-inactivated in a 60°C water bath for 30 min. The pooled serum was then filtered using 0.22-μm filters (Biotech, Germany). The serum was aliquot and stored at −20°C until further use.
Isolation of mesenchymal stem cell from umbilical cord Wharton’s jelly tissue
Five milligrams of the cord blood was collected in a sterile heparinized tube before placental delivery, then longitudinal pieces of the umbilical cord were collected in a sterile container and rinsed thoroughly with PBS to remove any blood. 10ml PBS was supplemented with 0.1% fungi zone, and penicillin/streptomycin (100 μg/ml) was added to the tube and transported on ice to the laboratory for further processing within 2 h of harvesting the cord.
In a clean and sterile laminar flow hood, the umbilical cord was washed with PBS and cut into small segments. The umbilical cord vessels were removed, and the Wharton’s jelly was scraped and cut with scissors into two mm2 pieces and placed in separate 10-cm dishes containing 5 ml of 0.05% trypsin-EDTA solution. The dishes were incubated at 37°C for 60 min for partial digestion of the Wharton’s jelly and release of the cells. The trypsin solution was neutralized by adding 10ml of the culture medium supplemented with 10% CBS. The contents of the dishes were transferred into 50 ml tubes and centrifuged, and then, the supernatant was discarded. A slurry and around 20 partially digested tissue pieces were transferred to a 75-cm2 culture flask containing 15 ml of the culture medium supplemented with 10% CBS and antibiotic solution (0.1% gentamicin, 0.2% streptomycin, 0.1% penicillin, and 0.1% fungi zone). The flask was put in a 5% CO2 incubator for 3 days. The culture medium was changed, and the flask was carefully examined by an inverted light microscope for attachment of the pieces to the plastic surface and to detect any bacterial or fungal contamination. The culture medium was changed every 3 days for days. The fibroblastic morphology of the outgrown cells was assessed by microscope and labeled as passage 0(P0). P0 cells were detached by 0.05 trypsin and sub-cultured in a new flask labeled passage number one (P1).
Proliferation assay of the isolated WJ-MSCs
Sets of WJ-MSCs were cultured in 6-cm dishes at a density of 20×104 cells per dish in triplicate and incubated at CO2 at 37°C. Cells were harvested every 24h by trypsinization and counted by the hemocytometer using the trypan blue exclusion to produce cell growth curves over a period of 7 days (Fig. 2A). The proliferation doubling time (PDT) was calculated from the growth curve by applying a nonlinear regression exponential growth equation [21].
Senescence β-galactosidase staining assay
The number of senescent cells was determined in WJ-MSC cultures at different passages during in vitro propagation by senescence β-galactosidase staining (Cell Signaling Technology, Danvers, MA, USA), according to the manufacturer’s instructions. Cells at different passages were seeded at a density of 500 × 10 4 /cm2 into 6-well plates and cultured for 72h. Cells were stained with β-galactosidase staining and examined by light microscopy. The percentage of senescent cells was determined by counting the number of cells with intracellular blue dye relative to the total number of cells in ten random fields.
Tri-lineage differentiation potential of WJ-MSCs
WJ-MSC at P2 were seeded in 6-well plates (5x103/well) in 5-ml DMEM supplemented with 10% CBS and incubated in 5% CO2 for 24h. The culture media were replaced with 5ml of adipogenic, chondrogenic, or osteogenic differentiation medium. The adipogenic medium consisted of DMEM supplemented with medium 1mM dexamethasone, 100 mM indomethacin, 10mM insulin, and 500 mM isobutyl methylxanthine. Chondrogenic differentiation medium consisted of DMEM supplemented with 30 mM insulin, 10ng/ml TGF-β1, and 50nM ascorbic acid 2-phosphate. The osteogenic medium consisted of DMEM supplemented with 0.1mM dexamethasone, 10mM glycerol-2-phosphate, and 50mM ascorbate-2-phosphate. The media were changed every 3 days for 21 days. Cells were fixed with 4% formaldehyde and stained with either 2% Oil Red O, Alcian Blue, or 2 % Alizarin Red staining (all Sigma Aldrich) and examined by inverted microscope (Olympus, Japan) for evidence of differentiation. Adipogenic differentiation was indicated by the presence of droplets of fat vacuoles, chondrogenic differentiation by blue dots of glycosaminoglycans, and osteogenic differentiation by red calcium deposits in cells [22].
Flow cytometry analysis
WJ-MSCs were cultured in 6-cm dishes and incubated till confluency, were collected in 1ml of 0.05% trypsin, and centrifuged at 2000 rpm for 3 min. The cells were resuspended in 1ml PBS containing 1% FBS and were treated with 5μl of fluorescein isothiocyanate (FITC) labeled primary antibodies against (CD90, CD105, CD44, CD45, and CD34; 1:1000 dilutions, for 45 min at 4°C (all from BD Biosciences). The cells were centrifuged at 12,000 rpm for 15 min, washed twice with washing buffer before the secondary antibodies (FITC) were added 1:1000 dilutions at 4°C for 45 min in the dark. Samples were run for flow cytometry (Beckman Coulter FC500, Inc.) with 30K acquisition events per sample. Data analysis was performed with the Flowjo software.
RNA extraction
RNA was extracted from stem cells passage number P2, P4, and P6 by the Trizol method (Life Technologies, Germany). Briefly, the culture flasks were trypsinized, collected, and the cell pellet washed with ice-cold PBS. RNA was harvested in 1 ml of Trizol followed by phase separation by adding 0.2 ml of chloroform, and centrifugation at 10,000 rpm for 20 min, at 4°C. The supernatant was moved to a new micro centrifuge tube, and RNA was precipitated with 0.5 ml of isopropanol and centrifugation at 10,000 rpm for 20 min at 4°C. The RNA pellet was washed with 70% ethanol, allowed to air dry, and resuspended in 50 μl of ultrapure water. The RNA concentrations were determined by reading the RNA absorbance at 260 nm and 280 nm wavelength in 1μl of the sample (Nanodrop 2000, Thermo Fisher Scientific).
cDNA conversion
Total RNA samples were converted to cDNA using reverse transcriptase (RevertAid H Minus First-Strand cDNA Synthesis Kit Thermo Fischer, USA). Briefly, 2μg of the extracted total RNA samples was diluted to a reaction volume of 20 μl containing 1μl (200 U/μl) of reverses transcriptase and 4 μl of 5X reaction buffer containing 2μl of 10 mM dNTP mixtures. The setting of the thermal cycle was 25°C for 10 min, 42°C for 60 min, and heat inactivation at 70°C for 10 min.
RT-PCR
Table 1 summarizes the forward and the reverse, the National Center for Biotechnology Information (NCBI) reference, and the amplicon size of the primers used. The PCR reaction uses Dream Taq (Thermo Fischer, USA) to detect different CD expressions with beta-actin gene expression as a control. The PCR condition was as follows: (1) initial denaturation cycle at 95°C for 5 min; (2) 35 cycles of denaturation at 95°C for 45s, annealing at the indicated temperature (CD44 at 53°C, CD90 at 54°C, and CD105 at 55°C, for 45s), extension at 72°C for 45s; and (3) a final extension cycle at 72°C for 5 min. The PCR products were run on a 2% agarose gel electrophoresis with a 100 pb ladder (GeneDireX, Taiwan) and ethidium bromide (Carl Roth, Germany) to visualize the amplicons bands at the specific site.
Karyotype of the isolated umbilical cord mesenchymal stem cell
WJ-MSCs cultured in low glucose DMEM media supplemented with 10% CBS in 10 ml flasks, incubated in 5% CO2, and 37°C until it reaches 80% confluence after 72h. Colcemid was added at a final concentration of 0.1 μg/ml and then incubated for 6h at 37°C. Cells were detached b 1ml of 0.05% trypsin-EDTA for 3 min then collected into a centrifuge tube containing 10 ml of media supplemented with 10% CBS. Cells were centrifuged at 2000 rpm for 5 min, and the supernatant discarded, followed by hypotonic treatment with 5 ml of 0.075 M KCl for 20 min, at 37°C. The cells were fixed in 10 ml of freshly prepared ice-cold Crony’s solution for 30 min at 4°C, centrifuged at 2500 rpm for 5 min, and the supernatant discarded. The cell pellet was washed twice with ice-cold Crony’s solution before slide preparation. Then, the cell suspension dropped onto the surface of a clean glass slide. The slides aged at 60°C for 16 h, immersed in 0.025 trypsin solution for 5s, then immediately in 0.5% CBS for 5s, and finally quickly rinsed in distilled water. The staining procedure was carried out by immersion in 0.5% Giemsa solution for 8 min, producing trypsin and Giemsa (GTG) bands. Karyotype performed using a light microscope (Olympus Corp., Tokyo, Japan) connected to Nikon JENOPTIK, model ProgRes MF camera using Lucia-Cytogenetics software.
Statistical analysis
Statistics were calculated using Graph-Pad Prism, version 3 (Graph-Pad Software Inc., San Diego, CA). The PDT was calculated from the growth curve by applying a nonlinear regression exponential growth equation.
The formula used to calculate the PDT is PDT = (CT × ln2) / Ln (NF/Ni). CT represents the cell culture time, Ni is the initial number of cells, and NF is the final number of cells [21].
Results
Isolation of mesenchymal stem cell from umbilical cord Wharton’s jelly explants
Several flasks of the cultured umbilical cord tissue were processed as described in the material and methods, grown in media supplemented with 10% human CBS, and followed up by serial light microscope examination. The flasks showed rounded or spindle-shaped cells at the edges of umbilical cord explants with a whirlpool appearance when they reach a confluence state after 21 days of the culture (P0) (Fig. 1A). Sub-culture of P0 cells into several passages the cells maintained the fibroblast-like morphology (Fig. 1B–F).

Established MSCs from the umbilical cord: the isolated WJ-MSCs at P0 (A) and the sub-cultured at P1 and P6. Cells showed the typical fibroblastic morphology of mesenchymal stem, which are maintained in passages 2 through 6
Growth characteristics and proliferation doubling time of the Wharton’s jelly-derived mesenchymal cells
The growth and PDT of the isolated WJ-MSCs at passage numbers P2, P4, and P6 were evaluated by trypan blue. The PDT of WJ-MSCs was 38.5 ± 1.5 h, 42.1 ± 2.9 h, 42.2 ± 2.6 h, at P2, P4, and P6, respectively (Fig. 2a). The effect of several passages on cell senescence was evaluated by senescence β-galactosidase staining. Cells at P10 and P15 were 5% and 25 % positive for β-galactosidase, indicating the occurrence of senescence in the cells in late passages (Fig. 2b).

Growth and PDT of the isolated UC WJ-MSCs. A Growth curve of cell in P2, P4, and P6 cultured in CBS; X, axis culture time in days; y, axis mean± SEM of the cell count. B WJ-MSC senescence. β-Galactosidase staining (blue) of WJ-MSCs at P2, P10, and P15. P2 cell line was used as a negative control of the β-galactosidase staining
Wharton’s jelly-derived mesenchymal cells had a tri-lineage differentiation ability
Tri-lineage differentiation was induced by culturing MSCs in 6-well plates and maintaining them for up to 21 days in the inducing media. MSC differentiation was observed by fixing the cells with 4% paraformaldehyde and staining with either Oil Red O, Alizarin Red, or Alcian Blue to detect the differentiation into adipocyte, chondrocyte, and osteocyte. Cells differentiated to adipocytes were positive for Oil Red O, indicating accumulation of lipid vacuoles. MSCs differentiated to chondrocytes showed positive blue staining of amino glycans with Alcian Blue. Cells differentiated into osteocytes showed positive staining for alizarin red, indicating the presence of calcium deposits (Fig. 3).

Tri-lineage mesenchymal differentiation potential of WJ-MSC. A Differentiation into adipocytes, as evidenced by intracellular lipid droplets on Oil Red O staining. B Differentiation into chondrocytes, as evidenced by proteoglycans and glycosaminoglycans on Alcian Blue staining. C Differentiation into osteocytes, as evidenced by calcium deposition on Alizarin Red staining
Wharton’s jelly-derived mesenchymal cells express mesenchymal cell surface markers
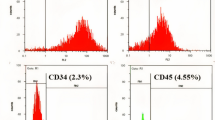
The expressions of mesenchymal cell surface markers were assessed in the isolated WJ-MSCs by flow cytometry. Cells stained with antibodies labeled with FITC against (CD34, CD44, CD45, CD90, and CD105) with IgG1-FITC antibodies utilized as a labeling control. Flow cytometry demonstrated that stem cells were positive for CD44 (98± 1.5%), CD90 (97 ± 1.8%), CD105 (97.7 ± 0.7%) and were negative for CD34 (1.8 ± 1.2%), and CD45 (1.8 ± 0.5%) (Fig. 4A). Molecular phenotyping of the extracted RNA from MSCs at passage numbers P2, P4, and P6 showed that the isolated MSCs were positive for CD90, CD105, CD44, and negative for CD34 (Fig. 4B, C) surface markers.

Expression of surface marker of the isolated UCWJ-MSCs. A Flow cytometry analysis of the surface molecule markers. Positive expression of mesenchymal stem cell markers (CD44, CD90, and CD105) and negative hematopoietic markers (CD34 and CD45) in WJ-MSC at passage 4. B Thidium bromide 2% stained agarose gel electrophoresis for amplified CD44, CD90, and CD105 expressed by MSCs at P2, P4, and P6. Lane1 DNA ladder, lanes (2–4) CD44 amplicons of 232 bp, lanes (5–7) CD105 amplicons of 254 bp, lanes (8–10) CD90 amplicons of 233 bp. GAPDH amplicon of 498bp was used as an internal control for CD expression. C Ethidium bromide 2% stained agarose gel electrophoresis for amplified CD34 expressed by MSCs at P 2, P4, and P6. Lane1 DNA ladder, lane 2, -ve con, lane 3, +ve con. Lane (4–6) CD34 amplicons of P2, P4, and P6. GAPDH amplicon of 498bp was used as an internal control
Wharton’s jelly-derived mesenchymal cells maintained a normal karyotype without any numerical or structural abnormalities
The chromosomal stability of the isolated WJ-MSCs was evaluated by conventional cytogenetic analysis at passages P2, P4, and P6. WJ-MSCs at the three passages were generally characterized as normal diploid karyotypes, showing a typical chromosomal plate consisted of 23 pairs of sister chromatids, with the classic appearance of four arms attached at the centromere (Fig. 5). Some metaphases were noticed to have arbitrary chromosome losses, probably due to the technical preparation of slides. Therefore, the isolated MSCs maintained a normal karyotype with no evidence of any numerical or structural chromosomal abnormalities.

Metaphase spreads of the cultured P2 isolated umbilical cord Wharton’s jelly MSCs. Photomicrograph of the metaphase spread viewed at the 100x objective lens showing a typical chromosomal plate consisted of 23 pairs of sister chromatids, with the classic appearance of four arms attached at the centromere
Discussion
This study described a simple method for the isolation and in vitro expansion of Wharton’s jelly-driven MSCs and their proliferative, differentiation, phenotypic, and genetic characteristics. The protocol utilized a suitable inexpensive in-house prepared CBS as a xeno-free supplement that eliminates undesirable immune responses, therefore, ensures its usage in clinical application [23, 24]. In agreement with other studies, the use of pooled CBS as alternatives to FBS maintained the proliferative capacity of the MSCs after cryopreservation, retained a fibroblast-like morphology, plastic adherence, and potential for multi-lineage differentiation [25,26,27]. Furthermore, the isolated WJ-MSC showed proliferative tri-lineage differentiation potential and corresponded well with the standard positive and negative phenotypic and molecular profile (CD44+, CD90+, CD 105+, and CD34-), indicating that the protocol used did not alter the expression of MSC surface markers during isolation and expansion period [28, 29].
The PDT of WJ-MSCs ranged from 38.5 to 42h with a tendency to increase with culture passages. Chen et al. and Shaer et al. reported a PDT of WJ-MSCs ranged from 30 to 44h associated with an increase in the PDT with the passage number [30, 31]. On the contrary, other studies showed that the increase in the passage number of the cultured WJ-MSCs was associated with a decrease in the PDT [32, 33]. These variations in the doubling time across these studies may be due to the difference in the cultural protocols and the growth factors added to the culture media, which might affect the proliferative potential of WJ-MSCs. As for any other normal somatic cell, after several passages, and WJ-MSCs enter a senescent state and ultimately halt proliferating. This process is accompanied by changes in the phenotype, the gene expression patterns, and the differentiation potential that might affect stem cell usage in clinical application [21].
Karyotyping is an essential part of the initial characterization of the MSCs. The metaphase of the isolated MSCs was identified as a normal diploid karyotype. The absence of chromosomal aberrations in G-banded karyotyping indicates the maintenance of chromosomal stability of the WJ-MSCs on further cell culture passage. Despite, karyotyping can detect chromosomal stability in MSCs [34,35,36], some chromosomal abnormalities require a more sensitive method than conventional karyotyping [37].
Although protocols for isolation, expansion, and cryopreservation vary widely, yet stringent standard technical specifications are required to ensure reproducibility and minimize the heterogeneity and diversity of isolated MSCs [38]. While the applied protocol for the dissection and isolation of cells from the explants is simple, it has some pitfalls and may be challenging. It requires careful handling and sterility to avoid any source of contamination, especially during sample collection, transfer, and processing. Explant adherence to the plastic surfaces of the culture flask is a crucial point in the success of the process. Floating explants and free cell debris inhibit cellular migration and attachment and undermine the retrieval of cells.
Of clinical importance and translational value of Wharton’s jelly provides a promising alternative stem cell source. The differentiation capacity of WJ-MSCs allows them to transform into cells from all three primary germ layers under appropriate conditions. In addition to WJ-MSCs possessing the essential criteria for regular MSCs, they also express several embryonic stem cell markers that mark the differentiation capabilities. Nevertheless, these cells lack the MHC-II, which makes them suitable for allogeneic transplantations [39]. Several preclinical studies using WJ-MSCs have been accomplished to examine their possible application in various diseases. Animal studies showed that the use of WJ-MSCs may be advantageous in Parkinson’s disease, spinal cord injury, certain types of cancer, and tissue fibrosis. The proposed mechanism of these studies may be due to the differentiation of WJ-MSCs into the same kind of cells in the damaged tissues or may be due to the paracrine effects of WJ-MSCs. Clinical studies on human patients revealed that WJ-MSCs improved the clinical outcomes of some immunologic-related disorders and neurological diseases with no evidence of tumor formation or immune rejection. Therefore, WJ-MSC seems to be a great promise of regenerative medicine [39].
Conclusions
The explant method in media supplemented with human CBS provides an efficient and non-invasive method for isolating large quantities of highly proliferative, non-immunogenic WJ-MSCs. Expansion of the isolated MSCs maintained their differentiation capabilities and did not alter the phenotypic or the genotypic patterns of the MSCs. Evidence of senescence was observed at the later passage of the cell culture. The cells maintained the expression of the standard positive and negative molecular surface markers (CD44+, CD90+, CD 105+, and CD34−) and the diploid karyotype pattern.
Availability of data and materials
The datasets used during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CBS:
-
Cord blood serum
- CD:
-
Cluster of differentiation
- DMEM:
-
Dulbecco′s modified Eagle′s medium
- FBS:
-
Fetal bovine serum
- FITC:
-
Fluorescein isothiocyanate
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- MSCs:
-
Mesenchymal stem cells
- PCR:
-
Polymerase chain reaction
- PBS:
-
Phosphate-based saline
- PDT:
-
Proliferation doubling time RPMI media: Roswell Park Memorial Institute
- WJ-MSCs:
-
Wharton Jelly mesenchymal stem cells
References
Anthony DF, Shiels PG (2015) Exploiting paracrine mechanisms of tissue regeneration to repair damaged organs. Transplant Res 2(1):10
Zimran E et al (2020) Expansion and preservation of the functional activity of adult hematopoietic stem cells cultured ex vivo with a histone deacetylase inhibitor. Stem Cells Transl Med 9(4):531–542
Bieback K, Brinkmann I (2010) Mesenchymal stromal cells from human perinatal tissues: from biology to cell therapy. World J Stem Cells 2(4):81–92
Liu L et al (2013) Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep 4(1):189–204
Liau LL et al (2019) Characteristics and clinical applications of Wharton's jelly-derived mesenchymal stromal cells. Curr Res Transl Med 68(1):5–16
Velarde F et al (2020) Use of human umbilical cord and its byproducts in tissue regeneration. Front Bioeng Biotechnol 8:117
Vohra M et al (2020) Human umbilical cord-derived mesenchymal stem cells induce tissue repair and regeneration in collagen-induced arthritis in rats. J Clin Transl Res 6(6):203–216
Wang XY et al (2008) Identification of mesenchymal stem cells in aorta-gonad-mesonephros and yolk sac of human embryos. Blood 111(4):2436–2443
Bakhtyar N et al (2017) Acellular gelatinous material of human umbilical cord enhances wound healing: a candidate remedy for deficient wound healing. Front Physiol 8:200
Ren H et al (2016) Comparative analysis of human mesenchymal stem cells from umbilical cord, dental pulp, and menstrual blood as sources for cell therapy. Stem Cells Int 2016:3516574
Chen SY et al (2011) A new isolation method of human limbal progenitor cells by maintaining close association with their niche cells. Tissue Eng Part C Methods 17(5):537–548
Diaz-Prado S et al (2010) Isolation and characterization of mesenchymal stem cells from human amniotic membrane. Tissue Eng Part C Methods 17(1):49–59
Malek A, Bersinger NA (2011) Human placental stem cells: biomedical potential and clinical relevance. J Stem Cells 6(2):75–92
Can A, Karahuseyinoglu S (2007) Concise review: human umbilical cord stroma with regard to the source of fetus-derived stem cells. Stem Cells 25(11):2886–2895
Yoon JH et al (2013) Comparison of explant-derived and enzymatic digestion-derived MSCs and the growth factors from Wharton's jelly. Biomed Res Int 2013:428726
Mishra PJ et al (2009) Mesenchymal stem cells: flip side of the coin. Cancer Res 69(4):1255–1258
Sensebe L, Bourin P (2009) Mesenchymal stem cells for therapeutic purposes. Transplantation 87(9 Suppl):S49–S53
Astori G et al (2016) Platelet lysate as a substitute for animal serum for the ex-vivo expansion of mesenchymal stem/stromal cells: present and future. Stem Cell Res Ther 7(1):93
Fernandez-Rebollo E et al (2017) Human platelet lysate versus fetal calf serum: these supplements do not select for different mesenchymal stromal cells. Sci Rep 7(1):5132
Kinzebach S, Bieback K (2013) Expansion of mesenchymal stem/stromal cells under xenogenic-free culture conditions. Adv Biochem Eng Biotechnol 129:33–57
Redaelli S et al (2012) From cytogenomic to epigenomic profiles: monitoring the biologic behavior of in vitro cultured human bone marrow mesenchymal stem cells. Stem Cell Res Ther 3(6):47
Huang S et al (2015) An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J Orthop Translat 3(1):26–33
Kandoi S et al (2018) Evaluation of platelet lysate as a substitute for FBS in explant and enzymatic isolation methods of human umbilical cord MSCs. Sci Rep 8(1):12439
Shetty P, Bharucha K, Tanavde V (2007) Human umbilical cord blood serum can replace fetal bovine serum in the culture of mesenchymal stem cells. Cell Biol Int 31(3):293–298
Hassan G et al (2017) A simple method to isolate and expand human umbilical cord derived mesenchymal stem cells: using explant method and umbilical cord blood serum. Int J Stem Cells 10(2):184–192
Pelekanos RA et al (2016) Isolation and expansion of mesenchymal stem/stromal cells derived from human placenta tissue. J Vis Exp 112:54204. https://doi.org/10.3791/54204
Cooper K, SenMajumdar A, Viswanathan C (2010) Derivation, expansion and characterization of clinical grade mesenchymal stem cells from umbilical cord matrix using cord blood serum. Int J Stem Cells 3(2):119–128
Dominici M et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4):315–317
Pham H et al (2018) CD73, CD90, CD105 and cadherin-11 RT-PCR screening for mesenchymal stem cells from cryopreserved human cord tissue. Int J Stem Cells 11(1):26–38
Chen G et al (2015) Comparison of biological characteristics of mesenchymal stem cells derived from maternal-origin placenta and Wharton's jelly. Stem Cell Res Ther 6:228
Shaer A et al (2014) Isolation and characterization of human mesenchymal stromal cells derived from placental Decidua Basalis; umbilical cord Wharton's jelly and amniotic membrane. Pak J Med Sci 30(5):1022–1026
Bruder SP, Jaiswal N, Haynesworth SE (1997) Growth kinetics, self-renewal, and the osteogenic potential of purified human mesenchymal stem cells during extensive subcultivation and following cryopreservation. J Cell Biochem 64(2):278–294
Kang TJ et al (2004) Growth kinetics of human mesenchymal stem cells from bone marrow and umbilical cord blood. Acta Haematol 112(4):230–233
Ben-David U, Mayshar Y, Benvenisty N (2011) Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell 9(2):97–102
Meza-Zepeda LA et al (2008) High-resolution analysis of genetic stability of human adipose tissue stem cells cultured to senescence. J Cell Mol Med 12(2):553–563
Sensebe L et al (2012) Limited acquisition of chromosomal aberrations in human adult mesenchymal stromal cells. Cell Stem Cell 10(1):9–10 author reply 10-1
Chen KG et al (2014) Alternative cultures for human pluripotent stem cell production, maintenance, and genetic analysis. J Vis Exp 89:51519. https://doi.org/10.3791/51519
Wilson A et al (2019) Multiplicity of mesenchymal stromal cells: finding the right route to therapy. Front Immunol 10:1112
Stefanska K et al (2020) Human Wharton's jelly-cellular specificity, stemness potency, animal models, and current application in human clinical trials. J Clin Med 9(4):1102. https://doi.org/10.3390/jcm9041102
Acknowledgements
We appreciate the Department of Clinical Biochemistry and Molecular diagnostics, National Liver Institute, Menoufia University, for help with the laboratory equipment, microscopy, and the chemicals required to complete the research. We appreciate the Department of Obstetrics and Gynecology, at Menoufia University Hospital, for providing help in the collection of the umbilical cord materials.
Funding
The authors declare they do not have any financial disclosure.
Author information
Authors and Affiliations
Contributions
H.A: Umbilical cord sample collection, establish the protocol for the umbilical cord dissection, collection of the Wharton’s jelly, isolation and growth of WJ-MSC, and coordination of the study. D.K: Tissue culture, isolation of the umbilical cord stem cells, and developed the methodology for karyotype. Y.M: Cell counting and growth curve generation. A. ElF: Revised the methods and perform RT-PCR experiments. M.G: Revised the methods and helped in the manuscript preparation. A.K: Conceived and designed the experiments; revised the methods, fluorescent microscopy, immunocytochemistry, karyotype; and performed the statistical analysis and writing of the manuscript. The authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research ethics committees of the National Liver Institute (IRB00003418), Menouffia University, approved the research proposal and the protocols to fulfill the national research guidelines. Patients provided informed written consent for tissue usage in the research.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abouelnaga, H., El-Khateeb, D., Moemen, Y. et al. Characterization of mesenchymal stem cells isolated from Wharton’s jelly of the human umbilical cord. Egypt Liver Journal 12, 2 (2022). https://doi.org/10.1186/s43066-021-00165-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43066-021-00165-w