
Abstract
Background
Primary intracranial Ewing’s sarcoma (ES) is a type of primitive neuroectodermal tumour and is a rare malignant tumour in children and adolescents. The imaging features of ES overlap with other central nervous system embryonal tumours, making it difficult to pinpoint a specific diagnosis. We aim to explore the clinical, neuroimaging and differential diagnoses of this entity.
Case presentation
We describe a 6-month-old infant who presented with complaints of enlarging the head size and poor feeding. Imaging revealed a contrast-enhancing large solid-cystic mass lesion with internal calcification, focal bone erosion and haemorrhage in the posterior fossa. Histopathological examinations, immunohistochemistry, and molecular analysis confirmed ES.
Conclusions
The confirmative diagnosis of primary intracranial ES requires histological examination, immunohistochemical analysis, and genetic detection, along with radiological findings. Surgical excision followed by combined radiotherapy and chemotherapy is the treatment of choice.
Similar content being viewed by others

Background
Primary intracranial Ewing sarcoma (ES)/Primitive neuroectodermal tumour (pPNET) is malignant small round cell neoplasms, that arise from bone and soft tissue, commonly seen in children and adolescents [1, 2]. Primary intracranial extraosseous ES is extremely rare, and only a few cases have been described [3,4,5,6,7]. Diagnosis of primary intracranial ES is made by the inclusion of radiological findings, histological examinations, and immunohistochemical and molecular genetic analysis. It must be differentiated from other CNS embryonal tumours as they have different treatments and prognoses. Radiological findings cannot differentiate PNET into its subtypes and often require genetic and immunohistochemical analysis such as MIC-2 antigen expression and detection of t(11;22)(q24;q12) translocation by fluorescent in situ hybridization (FISH) [7, 8]. The latest WHO 2021 classification of brain tumours lists ES as a mesenchymal non-meningothelial malignant tumour [9]. We aimed to highlight the rare malignant CNS entity with characteristic imaging features and aware the radiologist to keep the differential diagnosis.
Case presentation
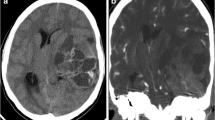
A 6-month-old infant with an uneventful perinatal history, presented with complaints of increasing head circumference, irritability, poor feeding and vomiting for one month. On examinations, the patient was alert and irritable and vitals were within normal limits. Routine blood investigations were normal. Non-contrast CT scan of the head revealed a large solid-cystic mass lesion in the posterior fossa, with a solid portion of the lesion appearing hyperdense with multiple foci of calcifications (Fig. 1a). The cystic component appeared multiloculated with thick septations. Mass effect on the cerebellum and brainstem was present with moderate supratentorial hydrocephalus. The lesion was seen infiltrating the occipital bone causing permeative destruction, predominantly involving the inner table and diploic space (Fig. 1b). No extra-calvarial soft tissue was seen.

NCCT shows a large solid-cystic lesion in posterior fossa with a hyperdense solid portion and multiple foci of calcifications (arrows in a). The lesion is infiltrating the occipital bone causing permeative destruction (arrows in b)
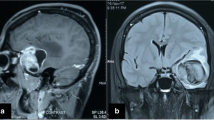
On MRI, a large extra-axial lesion (7.6 × 8.7 × 9.0 cm) was seen centred at the tentorium with cystic supratentorial and predominantly solid infratentorial components with significant compression on the cerebellum, brainstem, and fourth ventricle and hydrocephalus. The solid portion of the lesion was isointense with foci of hyperintensity on T1WIs (Fig. 2a), heterogeneously hypointense on T2WIs (Fig. 2b and c), and hyperintense on FLAIR. The mass had multiple blooming areas on SWIs (Fig. 2d) and heterogeneous areas of diffusion restriction on DWIs (Fig. 2e, f). Post-gadolinium T1-WIs showed heterogeneous post-contrast enhancement of the solid portion, with the peripheral and septal enhancement of the cystic portion (Fig. 2g, h).

Axial T1-WI (a), axial T2-WI (b), and coronal T2-WI (c) show a large solid-cystic lesion in the posterior fossa with T1 (a) and T2 (b, c) isointense solid portion and multiloculated cystic components. Hyperintense foci (arrows in a) are seen, suggesting internal haemorrhage. Extensive areas of blooming on SWI (d) are seen, suggesting calcification and internal haemorrhage. The solid portion of the lesion is bright on DWI (e) with a corresponding signal loss on ADC (f), suggesting diffusion restriction. Axial (g) and coronal (h) post-gadolinium T1-WIs show heterogeneously enhanced solid portion and enhancement of the thick septations (g, h)
Right occipital craniotomy was done; the cyst was decompressed via the Omaya reservoir system. The tumour was found to be infratentorial; tentorium was incised to enter into the cyst with the tumour. The tumour was vascular, friable, dirty white with thrombosed vessels, and CUSA (cavitron ultrasonic surgical aspirator) suckable. A gross total excision was done, and the excised tumour was sent for histopathological examination.
Histopathological sections (Fig. 3a) showed diffuse sheets of small round tumour cells, a scant amount of cytoplasm, round to oval nuclei and stippled chromatin with inconspicuous nucleoli. The tumour cells showed diffuse immunopositivity for CD99 (strong membranous) and NKX2.2 (strong nuclear) on immunohistochemistry (Fig. 3b, c). The FISH assay was performed using an EWSR1 break-apart probe showed positivity for EWSR1 rearrangement and showed typical (1 fusion, 1 green, and 1 orange) and atypical (1 fusion & 1 green, 1 fusion & 1 orange) signal patterns (Fig. 3d). The histopathology, immunohistochemistry, and the FISH assay were confirmatory for Ewing’s sarcoma.

H & E (×400) shows a malignant small round cell tumour with cells having scant cytoplasm, round to oval nuclei with finely stippled chromatin (a). Immunohistochemistry for CD99 shows strong membranous staining (b). Immunohistochemistry for NKX2.2 shows strong nuclear staining (c). EWSR1 break-apart FISH shows typical (1 fusion, 1 green and 1 orange) and atypical (1 fusion & 1 green, 1 fusion & 1 orange) signal pattern (d)
Discussion
Radiologic features of the intracranial ES are varied in different case reports. Most of them have described it as a heterogenous lobulated solid mass appearing hyperdense on CT, isointense on T1WIs and a hypointense signal on T2WIs with heterogenous post-contrast enhancement, calcification and haemorrhage [4, 10,11,12]. The large cystic component seen in our case has not been described. Most cases possibly arise from dura mater. Imaging differentials in an extra-axial lesion include metastatic tumours such as neuroblastoma, lymphoma, rhabdomyosarcoma, and primary tumours like meningeal sarcoma and solitary fibrous tumours [11]. These lesions can have a similar imaging appearance, but most have an associated extra-calvarial soft tissue component and significant bone destruction. If extra-axial versus intra-axial location remains a conundrum, differentials should include medulloblastoma, atypical teratoid/rhabdoid tumour, Embryonal tumour with multilayered rosettes, CNS tumour with BCOR internal tandem duplication, cribriform neuroepithelial tumour and CNS neuroblastoma, FOXR2-activated [11, 12].
Intracranial ES can be misdiagnosed as embryonal tumours like medulloblastoma, ATRT ( Atypical teratoid and rhabdoid tumour), or neuroblastoma due to the similarity in histologic appearance [12]. Recent advances in molecular testing have clarified these two entities. Molecular testing by RTPCR and FISH methods can depict ESR1 gene rearrangement which has high sensitivity and specificity in diagnosing ES [13]. Most common genetic aberration in ES is chromosomal translocation t(11,22) (q24; q12) followed by ESW/ERG t(21,22) (q22; q12). MIC2 gene (a pseudoautosomal gene) is commonly seen in ES [13, 14]. CD99, a cell surface glycoprotein (the gene product of MIC2), is essentially observed in all cases of ES on immunohistochemical staining [13, 14]. CNS embryonal tumours do not express MIC2 and are negative for CD99 [14]. The distinction between ES and embryonal tumours is important because their treatment is different, and they carry different prognoses. As a result of the latest advances in diagnosis and treatment, the outcome has significantly improved [15]. Intracranial ES requires surgery, chemotherapy, and field radiation therapy. Embryonal tumours also require surgery; however, chemotherapeutic and radiation protocols differ.
Conclusions
Radiologists should be aware of intracranial ES as a newly recognized entity. In our case, characteristic imaging findings were observed as similar to the PNET group, i.e. solid cystic, CT hypodensity, diffusion restriction, calcification and haemorrhage. The confirmative diagnosis of primary intracranial ES requires histological examination, immunohistochemical analysis, and genetic detection, combined with radiological findings.
Availability of data and materials
Yes, on request.
Abbreviations
- ATRT:
-
Atypical teratoid and rhabdoid tumour
- CT:
-
Computed tomography
- CNS:
-
Central nervous system
- CD99:
-
Cluster of differentiation 99
- CUSA:
-
Cavitron ultrasonic surgical aspirator
- DWI:
-
Diffusion weighed sequence
- ES:
-
Ewing’s sarcoma
- EWSR1:
-
Ewing sarcoma breakpoint region 1
- FLAIR:
-
Fluid attenuated recovery sequence
- FISH:
-
Fluorescent in situ hybridization
- FOXR2:
-
Forkhead box R2
- PNET:
-
Primitive primitive neuroectodermal tumor
- MRI:
-
Magnetic Resonance imaging
- MIC-2:
-
A gene name
- NKX2.2:
-
NK2 homeobox 2
- pPNET:
-
Primary primitive neuroectodermal tumour
- RTPCR:
-
Real-time reverse transcriptase polymerase chain reaction
- SWI:
-
Susceptibility weighted imaging
- WHO:
-
World Health Organization
References
Chen J, Cheng R, Fan F et al (2019) Cranial Ewing sarcoma/peripheral primitive neuroectodermal tumors: a retrospective study focused on prognostic factors and long-term outcomes. Front Oncol 9:1023
Yim J, Lee WS, Kim SK et al (2019) Intracranial Ewing sarcoma with whole genome study. Childs Nerv Syst 35:547–552
Pekala JS, Gururangan S, Provenzale JM et al (2006) Central nervous system extraosseous Ewing sarcoma: radiologic manifestations of this newly defined pathologic entity. Am J Neuroradiol 27:580–583
Jiang Y, Zhao L, Wang Y et al (2020) Primary intracranial Ewing sarcoma/peripheral Primitive neuroectodermal tumor mimicking meningioma: a case report and literature review. Front Oncol 10:528073
Chen J, Jiang Q, Zhang Y et al (2019) Clinical features and long-term outcome of primary intracranial Ewing sarcoma/peripheral primitive neuroectodermal tumors: 14 cases from a single institution. World Neurosurg 122:e1606–e1614
Sohail AH, Sachal M, Maan MAA et al (2019) Primary intracranial extraosseous Ewing’s sarcoma. Childs Nerv Syst 35:541–545
Dedeurwaerdere F, Giannini C, Sciot R et al (2002) Primary peripheral PNET/Ewing’s sarcoma of the dura: a clinicopathologic entity distinct from central PNET. Mod Pathol Off J US Can Acad Pathol Inc 15:673–8
Kovar H (1998) Ewing’s sarcoma and peripheral primitive neuroectodermal tumors after their genetic union. Curr Opin Oncol 10:334–342
Choi SW, Ko H (2019) Primary Ewing sarcoma of the squamous temporal bone with intracranial and extracranial extension: a rare cause of sudden sensorineural hearing loss. Head Neck 41:E38-41
Huang J, Ghent F, Levingston R et al (2020) Intracranial Ewing sarcoma—a case report. Surg Neurol Int 30(11):134
Bano S, Yadav SN, Garga UC (2009) Case Report: Intracranial peripheral primitive neuroectodermal tumor—Ewing’s sarcoma of dura with transcalvarial–subgaleal extension: an unusual radiological presentation. Indian J Radiol Imaging 19:305–307
Singh AK, Srivastava AK, Pal L et al (2018) Extraosseous primary intracranial Ewing sarcoma/peripheral primitive neuroectodermal tumor: series of seven cases and review of literature. Asian J Neurosurg 13:288–296
Mobley BC, Roulston D, Shah GV et al (2006) Peripheral primitive neuroectodermal tumor/Ewing’s sarcoma of the craniospinal vault: case reports and review. Hum Pathol 37:845–853
Yang MJ, Whelan R, Madden J et al (2018) Intracranial Ewing sarcoma: four pediatric examples. Childs Nerv Syst 34:441–448
Potratz J, Dirksen U, Jürgens H et al (2012) Ewing Sarcoma: clinical state-of-the-art. Pediatr Hematol Oncol 29:1–11
Acknowledgements
Not applicable.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
SAH, BDC, SA, SD, PKS and LJDS contributed to the acquisition, analysis, conception, design, and drafting of the work. AG, along with SAH, BDC, PKS and LJDS contributed to the final draft, revisions, upload and submission of final revised work. All authors have agreed both to be personally accountable for their contributions and ensured that questions related to the accuracy or integrity of any part of the work, even ones in which one was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work has been approved by Institute Ethical Committee (IEC). Retrospective, consent waiver.
Consent for publication
Consent for publication has been obtained from the patient in writing, however, their identity is not disclosed. All the authors have approved submitting the manuscript to your esteemed journal. On behalf of all the contributors, I will act as guarantor and correspond with the journal from this point onward.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, S.A., Charan, B.D., Agarwal, S. et al. Posterior fossa primary intracranial extraosseous Ewing’s sarcoma: case report. Egypt J Radiol Nucl Med 55, 108 (2024). https://doi.org/10.1186/s43055-024-01268-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43055-024-01268-1