
Abstract
Background
Sickle cell nephropathy is a complication of sickle cell disease characterized by functional abnormalities of the kidney and glomeruli. Our study aimed to investigate the single-nucleotide genetic variants in TGF-β-1-related genes as an early predictor of sickle cell nephropathy (SCN) risk.
Methods
Two hundred participants, 100 patients with SCD, and 100 age and sex-matched control. The study included full history taking, clinical examination, and laboratory evaluation. Renal function tests (serum urea and creatinine, microalbuminuria, albumin/ creatinine ratio, and e-GFR). Genotyping for TGF-β1 genetic variants rs1800469 and rs1800471.
Results
Twenty-one percent of patients had glomerular hyperfiltration, while 31% had reduced e-GFR. Microalbuminuria was present in 14%, and none had macroalbuminuria or edema. TGF-β1 genotyping revealed a statistically significant difference in the rs 1800471 C allele, which was more common in the control group (p 0.028). No significant correlation between the result of TGF‐ β genotyping and the albumin-to-creatinine ratio, creatinine, and e-GFR.
Conclusion
TGF-β1 rs1800469 and rs1800471 genetic variants were not associated with the risk of sickle nephropathy in children with sickle cell disease.
Similar content being viewed by others
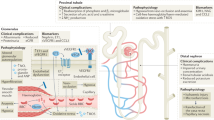


Background
Sickle cell disease (SCD) is an autosomal recessive hemoglobinopathy arising from the substitution of valine for glutamine at the sixth amino acid of the β-globin chain [1]. This point mutation affects the solubility and stability of the hemoglobin molecule, allowing the formation of rigid polymers in red blood cells upon deoxygenation [2].
SCD is a unique disease characterized by hemolytic anemia, recurrent vascular occlusions, a systemic inflammatory state substantial multiorgan disease [3].
Sickle cell nephropathy (SCN) is one of the recognized complications of SCD, causing significant morbidity and mortality in patients with end-stage renal disease (ESRD). It affects nearly 30–50% of adults with SCD [4] with asymptomatic onset in childhood and could develop into chronic kidney disease [5].
SCN could not be diagnosed at its subclinical stages as renal function biomarkers cannot detect the early deteriorating changes in renal functions, especially in children [6]. SCN invariably begins in childhood with evidence of structural changes detected as early as infancy [7].
Several genetic variants of cytokines and cytokines-related genes were found to be related to the clinical course and complications of sickle cell disease [8]. Finding genetic markers that indicate susceptibility to SCN as a profound complication of SCD is crucial for early disease management and possible preventive interaction.
There have been several cytokines implicated in the sickle cell anemia pro-inflammatory conditions, including interleukin (IL)1β, IL6, tumor necrosis factor-ɑ, and transforming growth factor β-1 (TGF-β1), which are responsible for both chronic and acute inflammatory events [9].
TGF-β is a multifunctional cytokine peptide of low molecular weight. It has three closely related isoforms TGF-β1, TGF-β2 and TGF-β3. TGF-β has dual effects, which are pro-inflammatory and anti-inflammatory, with a pronounced immunosuppressive effect [10]. TGF-β1, -β2, and -β3 share 71–80% sequence identity and signal through the same receptors [11]. Transforming growth factor β1 (TGF-β1) was found to be an indispensable immunoregulator promoting CKD progression by controlling the activation, proliferation, and apoptosis of immunocytes via both canonical and non-canonical pathways [12]. It functions in autocrine and paracrine manners to regulate cell proliferation, differentiation, apoptosis, adhesion, immunity, and extracellular matrix (ECM) turnover in the kidneys. It has fibrogenic action through enhancing the production of extracellular matrix proteins [10]. Several genetic variants have been proved to affect TGF-β1 production and functions, such as (rs1800469), which is in the promoter region of TGF-β1 gene that can alter the rate of secretion of TGF-β1 and hence the circulating levels of mature TG-β1 [10], whereas the (rs1800471) was reported to affect the level of TGF-β1 as well as be associated with inter-individual variation in levels of the TGF-β1 production in vitro and with fibrosis in lung allografts [13, 14].
To our knowledge, there are no previous studies linking genetic variants of TGF-β1 related genes and sickle cell nephropathy despite being studied for the association of non-diabetic chronic end-stage kidney disease [15]. Therefore, the aim of our study was to investigate the correlations between genetic variants in TGF-β-related genes and the risk of sickle cell nephropathy as well as other clinical and laboratory data of SCD cases.
Methods
For this case–control study, a hundred pediatric confirmed SCD patients were recruited from the hematology outpatient clinic of Cairo University Children’s Hospital in December 2020. They were in steady state condition (no history of intercurrent illness such as infection, inflammation, or painful crises or hospital admission during the previous 4 weeks) [16]. Research and Ethics Committee of Cairo University approved the study protocol and the consent process SCD patients with any other chronic inflammatory condition, any metabolic or endocrinal problems, clinically evident infection, and receiving a blood transfusion in the last 3 months were excluded from the current study.
Another hundred age and gender-matched healthy children were recruited from the surgery outpatient clinic. After obtaining each participant’s signed, informed consent. blood samples were collected. Patients’ medical reports were reviewed for the following data (date of birth, age, sex, consanguinity, age at diagnosis, presenting symptoms, duration of follow-up, exchange transfusion, and history of other siblings and/or close relatives with SCD). The patient’s height, weight, and body mass index were assessed. Other findings, including jaundice, pallor, organomegaly, history of crises, and transfusion history, were recorded. Two milliliters of the EDTA tube were withdrawn for complete blood count and hemoglobin electrophoresis. Two milliliters on a plane tube were used for routine chemistry, including renal function tests. Another 2 ml of blood in the EDTA tube was withdrawn for genotyping. A mid-stream early morning urine sample in sterile cups was ordered for albuminuria and albumin/creatinine ratio analysis.
Genotyping of TGF-β1 was performed by restriction fragment length polymorphism polymerase chain reaction (RFLP-PCR) analysis as described before [17].
DNA extraction was done using a Gene JET Genomic DNA purification kit (Cat. #K0721 #KO72, Fermentas Life Sciences, Canada), and extracted DNA was stored at − 20 °C.
The used primers to investigate TGF-β1 genetic variants, enzymes used for digestion and reaction conditions are summarized in Table 1, (primers were provided by (Invitrogen, Thermo Fisher Scientific, UK).
The PCR experiments were conducted using a DNA thermal cycler (Perkin Elmer No.: 9600).
This was followed by digestion of the PCR amplicon by the specific restriction enzyme, the digested products were subjected to agarose electrophoresis and then visualized by Ethidium bromide under UV lamb Eco81I (Cat. No.: ER0372, Thermo Fisher) was used for rs1800469, while BGLI enzyme (Cat. No.: ER0071, Thermo Fisher) was used for rs 1800471.
Statistical analysis
Data were analyzed by the Statistical Package for Social Science (IBM SPSS) version 20. The qualitative data were presented as numbers and percentages, while quantitative data were presented as mean, standard deviations, and ranges when their distribution was found parametric. The comparison between two groups with qualitative data was made using the chi-square test, and the exact Fisher test was used instead of the chi-square test when the expected count was found in less than five. The comparison between two independent groups with quantitative data and parametric distribution was made using an independent t test. The comparison between more than two independent groups with quantitative data and parametric distribution was made using the one-way ANOVA test. The comparison between two independent groups with quantitative data and non-parametric distribution was made using the Mann–Whitney test. The comparison between more than two independent groups with quantitative data and non-parametric distribution was made using Kruskal–Wallis. Pearson correlation between numerical data, the confidence interval was set to 95%, and the margin of error accepted was set to 5%. So, the p value was considered significant as P < 0.05 = significant.
Results
Hundred SCD patients were enrolled in this study, 44% of whom were females and 56% were males. There were 69 patients with SCA, three with sickle cell trait, 26 with Hb S/B + thalassemia, and two with Hb S/B0 thalassemia. The patients in this study had a median age (IQR) of 9 (5–13) years (range 2–17.7 years). The recruited patients’ mean weight was 27.78 ± 11.67 (range 10–64) kg. Their mean height was 152.22 ± 11.67 (range 83–156) cm. Their mean BMI was 16.88 ± 2.71 (range 10.38–28.07).
The average hemoglobin (HB)of the studied patients with SCD was 8.5 ± 1.5 g/dl; range 5.4–13.8 g/dl, hematocrit 24.6 ± 6.67%; range 17.2–75.9, mean corpuscular volume 77.2 ± 1.38; range 21.6–113.6, platelets 296.37 ± 126.57 platelets/µl; range 101–759, creatinine 0.5 ± 0.14 mg/dl; range 0.2–1.1, urea 20.13 ± 3.4 mg/dl; range 4–27.9, and e-GFR 106 ± 28.7 ml/min/1.73 m2; range 57–212. The median and inter-quartile range (IQR) absolute neutrophilic count was 3332 (2258–4808), range 442–14,472, and urinary albumin and creatinine ratio 9 (4.6–18.7); range 0.6–74. The average hemoglobin A1 was 16.55%, hemoglobin A2 was 2.5%, hemoglobin F was 4.05, and hemoglobin S was 62.4%.
The primary complications of SCD in the studied patients were vaso-occlusive crisis (82%), recurrent and unpredictable episodes of acute pain in the arms, legs, joints, and back, 2% with cardiovascular disease, and 1% with acute chest pain). The vaso-occlusive crisis was significantly higher in patients with G/G polymorphism (Table 3). GG genotype also had an earlier age for presentation and that finding was not associated with a significant change in HB S level. While gall bladder stone was documented in 4% requiring cholecystectomy, and acute splenic sequestration (4%) requiring splenectomy.
Hydroxyurea, dosage with an average dose of 633.53 ± 300.06 mg/day, was prescribed in 85% of the recruited patients with SCD. Iron chelators were prescribed to 16% of our patients. The main prescribed iron chelator in our research was Deferiprone (62.5%), followed by Deferasoirox (31.3%) and Deferoxamine (6.2%). Many patients needed blood transfusions (81%).
The genotyping results of both cases and controls are illustrated in Fig. 1, and there was no statistically significant difference between them regarding rs1800469 promotor and rs 1,800,471 genetic variants (p = 0.12, 0.26), respectively. At the same time, the dominant and recessive modes comparison between the cases and controls are illustrated in Table 2 with no statistically significant difference between cases and controls.

Distribution of genotypic polymorphism of TGF-β1 in cases and controls
However, an allelic mode comparison, revealed that the C allele of rs1800471 was more common in the control group than in the SCD group.
Table 3 illustrates the difference in the clinical picture of different genotyping results. In the cases of rs 1,800,471, GG experienced early presentation and had more VOC. No difference between the different genotypes and all other clinicopathological features of both genetic variants.
There was a statistically significant negative correlation between BUN, urea, creatinine, and HBF percentage (p 0.04, 0.041, 0.0, and 0.02)and there was a statistically significant positive correlation between age, height, and e-GFR (p 0.024, 0.032), respectively, results, glomerular hyperfiltration rates characterization to normal GFR, hyperfiltration and diminished GFR with correlation to clinical and laboratory findings and genotyping results, revealed that GFR hyperfiltration was affected by age, weight, height, creatinine, and blood transfusion, as illustrated in Table 4.
Table 5 shows that there was no statistically significant difference between patients with sickle cell anemia (HBSS) and other sickle cell anemia types regarding renal functions, ACR, and molecular results of TGF-β1 genotyping.
Discussion
In our result, 21% of patients had glomerular hyperfiltration (e-GFR > 125 ml/min/1.73 m2), while 31% of them had reduced e-GFR and the rest had normal ranges from 90 to 125 ml/min/1.73 m2. Similarly, (Ghobrial et al. 2016) reported glomerular hyperfiltration (177.44 ± 35.6 mL/min/1.73 m2) in patients with SCD, so It was recommended to monitor the renal function of children with sickle cell disease, especially in homozygotic (Hb SS) patients [18].
Furthermore, Nnaji et al. 2020 indicated that abnormally high e-GFR in children with asymptomatic sickle cell anemia was observed and assumed to progress to chronic kidney disease; therefore, regular monitoring of renal function in asymptomatic pediatric patients with sickle cell disease and implementing management protocol is crucial to avoid anemic or crises episodes and advanced kidney disease.
Glomerular damage in sickle cell anemia was suggested to be caused by the damage of the cytoskeleton of cells called podocytes that line the visceral surface of Bowman’s capsule. Such damage was assumed to be caused by chronic ischemia–reperfusion injury occurring during vaso-occlusion episodes in sickle cell disease, therefore subsequent activity of TGF-β1 could lead to further damage and apoptosis of podocytes and glomerular damage [19]. Agata et al. 2014 noted that in patients with sickle cell disease, due to the internal medulla microenvironment being hypoxic, acidic, and hyperosmolar, polymerization of deoxygenated hemoglobin S results in RBC sickling and microinfarction causing reduced medullary blood flow, but if the hypoxia deteriorates, prostaglandins are released causing marked vasodilation and glomerular hyperfiltration [20].
In the current study, microalbuminuria was diagnosed in fourteen patients (14%) and none had macroalbuminuria or edema. Such results were in agreement with Belisário et al.,2020 study that reported the earliest manifestation of renal disease in pediatric SCD is an increase in the glomerular filtration rate and the occurrence of microalbuminuria [21].
Persistent proteinuria in children with SCD usually follows the occurrence of glomerular hyperfiltration, then the GFR is reduced as the sickle nephropathy progresses [22].
Microalbuminuria was found to be a good preclinical marker of glomerular damage predicting progressive renal failure in pediatric patients with SCD [23]. Ocheke and his colleagues reported that anemia and high e-GFR are risk factors for microalbuminuria and that the glomerular filtration rate was higher in children with microalbuminuria than those who do not (p ≤ 0.01) and it was also higher in children with sickle cell disease than in control [24]. The vaso-occlusive crisis was diagnosed in 82% of patients and 96% of them (n = 75) needed hospitalization. The vaso-occlusive crisis in sickle cell disease is due to the young sticky erythrocytes containing hemoglobulin S attaching to the walls of capillary venules leading to narrowing of their lumens which leads to the decrease in blood velocity and an increase in erythrocytes transient time; therefore, HB S become deoxygenated and subsequent erythrocyte sickling occurs; moreover, necrosis of the affected vascular area and inflammatory response are initiated which produce pain [25].
A previous study indicated that reduced e-GFR occurred during the vaso-occlusive crisis [26]. Moreover, Sarray et al. observed reduced IL-10 and increased IL-6 and TNFα levels during the vaso-occlusive crisis in pediatric sickle cell disease patients [27].
Hydroxyurea was the key treatment in 85% of our patients. In a previous study, it was found to be an effective and proven medication to reduce the frequency of painful episodes by 50% in sickle cell disease, it also decreases the rate of blood transfusions by inducing the production of Hb F [28].
In our study, four patients developed gallbladder stones and needed cholecystectomy. Cholelithiasis results from the chronic accelerated rate of erythrocyte destruction in individuals with sickle cell disease leading to the formation of insoluble calcium bilirubin that precipitates to form gallstones [29]. The rs1800469 polymorphism changes codon 25 which encodes arginine into proline in the signal peptide of TGF-β1. The amino acid substitution affects signal peptide properties that may inhibit the transport of TGF-β1 into the endoplasmic reticulum and eventually decline cytokine production. The arginine substitution into proline decreased the polarity of the signal peptide for TGF-β1. The increased hydrophobicity with increased binding energy of the signal peptide for TGF-β1 to signal recognition particle and translocon of endoplasmic reticulum implies decreased protein complex stability in potentially blocking the transport of TGF-β1 into the endoplasmic reticulum. This transport retention possibly hampers the synthesis and maturation of TGF-β1 leading to decreased cytokine production [30]. Other authors have proved that higher TGF-β1 is associated with higher susceptibility to different infections, even for septicemia [31]. Both findings show that more susceptibility to infections may increase sickling attacks [32].
In the current study the rs 1800471 G/G genotype had a higher incidence of vaso-occlusive attacks and earlier onset of the disease-related symptoms that was not attributed to increased HbS concentration alone, could suggest a modifying effect on the HbS polymerization, which should be further studied, As TGF-β 1 was found to be associated with hemolysis, leukocytes, platelets, and lipid metabolism, this provides evidence that this immunomarker likely modulates the inflammatory response in SCD in previous studies [33]. Previous studies described the effect of the presence of the G allele of rs 1800471 as associated with increased TGF-β1 production, this may explain a higher incidence of vaso-occlusive attacks and earlier onset of the disease-related symptoms [34].
In our study, a comparison between variants of the TGF-β1 gene between cases and controls. indicated the presence of a statistically significant difference regarding the C allele of rs1800471, that the C allele was found more in the control group.
Previous studies noted that TGF- β1 polymorphism would regulate its expression and mediate the occurrence of several diseases such as rheumatoid arthritis, colorectal carcinoma, diabetes mellitus, osteoporosis, asthma, Crohn’s disease, and fibrotic diseases of the skin and kidney [35].
El-Sherbini et al. 2013 demonstrated that when TGF‐β binds to its receptors, it exerts signals via SMAD, activating MAP kinases. Such pathway controls cell proliferation, apoptosis, and response to tissue injury, infection, bone homeostasis, endothelial growth, diabetic nephropathy, pulmonary fibrosis, inflammation, immune regulation, and extracellular matrix synthesis [36]. Moreover, Santiago et al. 2021 revealed that the increased TGF-β1 levels play essential roles in vascular remodeling, vasculopathy, angiogenesis, and inflammation in pediatric patients with sickle cell disease [33].
Regarding the hypothesis of using the urinary TGF‐β as a marker of renal dysfunction in sickle cell disease, a previous study by Ghobrial et al. 2016 [37] revealed that urinary excretion of TGF-β1 was higher in sickle cell disease patients than in control children (p < 0.001).
Contrarily Sundaram, 2011 [38] indicated that urinary TGF‐β levels did not show any relationship with albuminuria in patients with sickle cell disease. Moreover, Mohtat, 2010 [39] reported elevated urine TGF- β1 levels in patients with sickle cell disease, but there was no correlation between urinary TGF-β1 and microalbuminuria or eGFR.
Our results indicated no significant effect of TGF‐β1 rs1800469 and rs1800471 genetic variants on the albumin-to-creatinine ratio (ACR), creatinine, and e-GFR in the case group. however prolonged period of microalbuminuria precedes persistent proteinuria, which is followed by renal failure in SCD patients. Therefore, early detection of microalbuminuria may allow earlier intervention to prevent renal complications [40].
In progressive kidney failure, previous studies indicated the correlation between TGF‐β 1 genetic variants and the progression of chronic kidney failure and that TGF‐β1 single nucleotide variants are helpful as a prognostic indicator of progressive kidney disease [41].
A recent study suggested that genetic variants in the TGF-β1 and IL-4 genes rs1800469, rs1800470, rs1800471, and rs8179190 may play a role as a genetic contributor to the susceptibility of chronic kidney disease [10]. In addition, TGF β1 was found to induce renal hypertrophy and fibrosis [10].
Saraf et al. 2015 study on the genetic markers of sickle nephropathy indicated the association of APOL1 G1/G2 with kidney disease in sickle cell disease through increased risk of hemoglobinuria and associations of HMOX1 variants with kidney disease through reduced protection of the kidney from hemoglobin-mediated toxicity [42].
This study had some limitations, as not all cases had histological evidence of SCN, also, the limited literature on the two studied genetic variants in the pathogenesis of SCN necessitates more studies on different ethnic populations, and different age groups with metanalysis studies.
Conclusion
TGF-β1 rs1800469 and rs1800471 genetic variants were not associated with the risk of sickle nephropathy in children with sickle cell disease. These genetic variants also didn’t affect the susceptibility to SCD.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ACR:
-
Albumin creatinine ratio
- BMI:
-
Body mass index
- ECM:
-
Extracellular matrix
- e-GFR:
-
Estimated glomerular filtration rate
- ESRD:
-
End-stage renal disease
- HB:
-
Hemoglobin
- IL:
-
Interleukin
- IQR:
-
Inter-quartile range
- REFLP-PCR:
-
Restriction fragment length polymorphism polymerase chain reaction
- SCD:
-
Sickle cell disease
- SCN:
-
Sickle cell nephropathy
- TGF-β1:
-
Transforming growth factor β-1
References
Wastnedge E, Waters D, Patel S et al (2018) The global burden of sickle cell disease in children under five years of age: a systematic review and meta-analysis. J Glob Health 8(2):21103. https://doi.org/10.7189/jogh.08.021103
Bakr S, Khorshied M, Talha N et al (2019) Implication of HMOX1 and CCR5 genotypes on clinical phenotype of Egyptian patients with sickle cell anemia. Ann Hematol 98(8):1805–1812. https://doi.org/10.1007/s00277-019-03697-9
Hebbel RP, Belcher JD, Vercellotti GM (2020) The multifaceted role of ischemia/reperfusion in sickle cell anemia. J Clin Invest 130(3):1062–1072. https://doi.org/10.1172/JCI133639
Marouf R, Adekile AD, El-Muzaini H, Abdulla R, Mojiminiyi OA (2021) Neutrophil gelatinase–associated lipocalin as a biomarker of nephropathy in sickle cell disease. Ann Hematol 100(6):1401–1409. https://doi.org/10.1007/s00277-021-04500-4
Nnaji UM, Ogoke CC, Okafor HU, Achigbu KI (2020) Sickle cell nephropathy and associated factors among asymptomatic children with sickle cell anaemia. Int J Pediatr 2020:1286432. https://doi.org/10.1155/2020/1286432
Alvarez O, Zilleruelo G, Wright D, Montane B, Lopez-Mitnik G (2006) Serum cystatin C levels in children with sickle cell disease. Pediatr Nephrol 21(4):533–537. https://doi.org/10.1007/s00467-006-0033-6
Olaniran KO, Eneanya ND, Nigwekar SU et al (2019) Sickle cell nephropathy in the pediatric population. Blood Purif 47(1–3):205–213. https://doi.org/10.1159/000494581
Vicari P, Adegoke SA, Mazzotti DR, Cançado RD, Nogutti MAE, Figueiredo MS (2015) Interleukin-1β and interleukin-6 gene polymorphisms are associated with manifestations of sickle cell anemia. Blood Cells Mol Dis 54(3):244–249. https://doi.org/10.1016/j.bcmd.2014.12.004
Carvalho MOS, Araujo-Santos T, Reis JHO et al (2017) Inflammatory mediators in sickle cell anaemia highlight the difference between steady state and crisis in paediatric patients. Br J Haematol 182(6):933–936. https://doi.org/10.1111/bjh.14896
Mai M, Jiang Y, Wu X, Liu G, Zhu Y, Zhu W (2020) Association of TGF-β1, IL-4, and IL-10 polymorphisms with chronic kidney disease susceptibility: a meta-analysis. Front Genet 11:79. https://doi.org/10.3389/fgene.2020.00079
Huang T, Schor SL, Hinck AP (2014) Biological activity differences between TGF-β1 and TGF-β3 correlate with differences in the rigidity and arrangement of THEIR COMPONENT MONOMERS. Biochemistry 53(36):5737–5749. https://doi.org/10.1021/bi500647d
Tang PCT, Chan ASW, Zhang CB, et al (2021) TGF-β1 signaling: immune dynamics of chronic kidney diseases. Front Med (Lausanne) 8. https://www.frontiersin.org/articles/10.3389/fmed.2021.628519
Liu K, Liu X, Gu S et al (2017) Association between TGFB1 genetic polymorphisms and chronic allograft dysfunction: a systematic review and meta-analysis. Oncotarget 8(37):62463–62469. https://doi.org/10.18632/oncotarget.19516
Zhu ML, Wang M, Shi TY et al (2013) No association between TGFB1 polymorphisms and late radiotherapy toxicity: a meta-analysis. PLoS ONE 8(10):e76964–e76964. https://doi.org/10.1371/journal.pone.0076964
Smyth LJ, Cañadas-Garre M, Cappa RC (2019) No Title. BMJ Open 9(null):e026777
Ballas SK (2012) More definitions in sickle cell disease: steady state v base line data. Am J Hematol 87(3):338. https://doi.org/10.1002/ajh.22259
Behboudi Farahbakhsh F, Nazemalhosseini Mojarad E, Azimzadeh P et al (2017) TGF-β1 polymorphisms -509 C>T and +915 G>C and risk of pancreatic cancer. Gastroenterol Hepatol Bed Bench 10(1):14–20
de Paula RP, Nascimento AF, Sousa SMB, Bastos PRV, Barbosa AAL (2013) Glomerular filtration rate is altered in children with sickle cell disease: a comparison between Hb SS and Hb SC. Rev Bras Hematol Hemoter 35(5):349–351. https://doi.org/10.5581/1516-8484.20130107
Ansari J, Gavins FNE (2019) Ischemia-reperfusion injury in sickle cell disease: from basics to therapeutics. Am J Pathol 189(4):706–718. https://doi.org/10.1016/j.ajpath.2018.12.012
Ataga KI, Derebail VK, Archer DR (2014) The glomerulopathy of sickle cell disease. Am J Hematol 89(9):907–914. https://doi.org/10.1002/ajh.23762
Belisário AR, Vieira ÉLM, de Almeida JA et al (2020) Evidence for interactions between inflammatory markers and renin-angiotensin system molecules in the occurrence of albuminuria in children with sickle cell anemia. Cytokine 125:154800
Lebensburger JD, Aban I, Pernell B et al (2019) Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol 94(4):417–423. https://doi.org/10.1002/ajh.25390
Mishra N, Meher S, Khamari D, Nayak B (2020) Diagnostic accuracy of microalbuminuria among sickle cell children with nephropathy. J Pediatr Crit Care 7(2):73. https://doi.org/10.4103/jpcc.jpcc_2_20
Ocheke IE, Mohamed S, Okpe ES, Bode-Thomas F, McCullouch MI (2019) Microalbuminuria risks and glomerular filtration in children with sickle cell anaemia in Nigeria. Ital J Pediatr 45(1):143. https://doi.org/10.1186/s13052-019-0720-0
Ilesanmi OO (2010) Pathological basis of symptoms and crises in sickle cell disorder: implications for counseling and psychotherapy. Hematol Rep 2(1):e2–e2. https://doi.org/10.4081/hr.2010.e2
Anigilaje EA, Adeniyi A, Adedoyin OT (2013) Effect of sickle cell crises on glomerular filtration rate in children with sickle cell disease in Ilorin. Nigeria Indian J Nephrol 23(5):354–357. https://doi.org/10.4103/0971-4065.116320
Sarray S, Saleh LR, Lisa Saldanha F, Al-Habboubi HH, Mahdi N, Almawi WY (2015) Serum IL-6, IL-10, and TNFα levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine 72(1):43–47. https://doi.org/10.1016/j.cyto.2014.11.030
Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B (2014) Hydroxyurea in sickle cell disease: drug review. Indian J Hematol Blood Transfus 30(2):91–96. https://doi.org/10.1007/s12288-013-0261-4
Vasavda N, Menzel S, Kondaveeti S et al (2007) The linear effects ofα-thalassaemia, theUGT1A1andHMOX1polymorphisms on cholelithiasis in sickle cell disease. Br J Haematol 138(2):263–270. https://doi.org/10.1111/j.1365-2141.2007.06643.x
Susianti H, Gunawan A, Putri JF, Purnomo BB, Handono K, Kalim H (2014) The potential effect of G915C polymorphism in regulating TGF-β1 transport into endoplasmic reticulum for cytokine production. Bioinformation 10(8):487–490. https://doi.org/10.6026/97320630010487
Zheng R, Fu Z, Zhao Z (2021) Association of transforming growth factor β1 gene polymorphisms and inflammatory factor levels with susceptibility to sepsis. Genet Test Mol Biomarkers 25(3):187–198. https://doi.org/10.1089/gtmb.2020.0143
Cannas G, Merazga S, Virot E (2019) Sickle cell disease and infections in high- and low-income countries. Mediterr J Hematol Infect Dis 11(1):e2019042–e2019042. https://doi.org/10.4084/MJHID.2019.042
Santiago RP, Carvalho MOS, Figueiredo CVB et al (2021) Associations between TGF-β1 levels and markers of hemolysis, inflammation, and tissue remodeling in pediatric sickle cell patients. Mediators Inflamm 2021:4651891. https://doi.org/10.1155/2021/4651891
Hadj-Ahmed M, Ghali RM, Bouaziz H et al (2019) Transforming growth factor beta 1 polymorphisms and haplotypes associated with breast cancer susceptibility: a case-control study in Tunisian women. Tumor Biology 41(8):1010428319869096. https://doi.org/10.1177/1010428319869096
El-Sherbini SM, Shahen SM, Mosaad YM, Abdelgawad MS, Talaat RM (2013) Gene polymorphism of transforming growth factor- 1 in Egyptian patients with type 2 diabetes and diabetic nephropathy. Acta Biochim Biophys Sin (Shanghai) 45(4):330–338. https://doi.org/10.1093/abbs/gmt003
Ziyadeh FN (2004) Mediators of diabetic renal disease: the case for TGF- as the major mediator. J Am Soc Nephrol 15(90010):55S – 57. https://doi.org/10.1097/01.asn.0000093460.24823.5b
Ghobrial EE, Abdel-Aziz HA, Kaddah AM, Mubarak NA (2016) Urinary transforming growth factor β-1 as a marker of renal dysfunction in sickle cell disease. Pediatr Neonatol 57(3):174–180. https://doi.org/10.1016/j.pedneo.2015.05.001
Sundaram N, Bennett M, Wilhelm J et al (2011) Biomarkers for early detection of sickle nephropathy. Am J Hematol 86(7):559–566. https://doi.org/10.1002/ajh.22045
Mohtat D, Thomas R, Du Z et al (2010) Urinary transforming growth factor beta-1 as a marker of renal dysfunction in sickle cell disease. Pediatr Nephrol 26(2):275–280. https://doi.org/10.1007/s00467-010-1677-9
Alzahrani YA, Algarni MA, Alnashri MM et al (2020) Prevalence and risk factors for microalbuminuria in children with sickle cell disease at King Abdulaziz University Hospital: A retrospective cross-sectional study. Cureus 12(1):e6638
Khalil MS, El Nahas AM, Blakemore AIF (2005) Transforming growth factor-β1 SNPs: genetic and phenotypic correlations in progressive kidney insufficiency. Nephron Exp Nephrol 101(2):e31–e41. https://doi.org/10.1159/000086227
Saraf SL, Zhang X, Shah B et al (2015) Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 100(10):1275–1284. https://doi.org/10.3324/haematol.2015.124875
Acknowledgements
Not applicable
Funding
Self-funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript. MH and IS outlined the research idea and study design. HR and YR collected the samples and clinical data of cases. FA helped in the interpretation of molecular results. YR and FA wrote the manuscript, and then finally revised it by MH and IS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Pediatric Department of Kasr Alainy Faculty of Medicine and by REC of Kasr Alainy.
Consent for publication
Not applicable. No identifying data are present in the manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamdy, M., Shaheen, I., Ramadan, H. et al. Transforming growth factor-β1: relation between its single-nucleotide genetic variants and sickle cell nephropathy. Egypt Pediatric Association Gaz 72, 43 (2024). https://doi.org/10.1186/s43054-024-00283-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43054-024-00283-1